

Abstract
The innate immune system uses pattern recognition receptors such as RIG-I and NLRP3 to sense pathogen invasion and other danger signals. Activation of these receptors induces robust signal transduction cascades that trigger the production of cytokines important for host protection. MAVS and ASC are essential adaptor proteins downstream of RIG-I and NLRP3, respectively, and both contain N-terminal domains belonging to the death domain superfamily. Recent studies suggest that both MAVS and ASC form functional prion-like fibers through their respective death domains to propagate downstream signaling. Here, we review these findings, and in this context discuss the emerging concept of prion-like polymerization in signal transduction. We further examine the potential benefits of this signaling strategy, including signal amplification, host evolutionary advantage, and molecular memory.
Keywords: prion-like polymerization, signaling, pattern recognition receptors, signal transduction
Beneficial prions
Two decades ago, the discovery of the first prion, the mammalian prion protein (PrP), elucidated the mechanistic basis of a group of infectious neurodegenerative diseases – the misfolding of PrP from a soluble α-helical conformation (PrPC) to β-sheet rich aggregates (PrPSC) that bind and convert other soluble PrPC into PrPSC [1]. Following this initial discovery, the characterization of other prion-like proteins ensued, and prions are now well accepted as proteins that adopt distinct conformations, at least one of which is self-propagating and heritable upon cell division [2,3]. In yeast and fungi, over a dozen prion determinants have been described, some as potential adaptive mechanisms of phenotypic diversification during environmental fluctuations [4–6]. Two notable examples include the essential yeast translation termination factor SUP35 and the fungal protein HET-s. SUP35 contains a prion domain (NM) at its N terminus followed by an enzymatic domain (SUP35C) that carries out its translation termination function. Following prion conversion, SUP35 forms large, insoluble aggregates that compromise its enzymatic activity, resulting in stop codon read-through and translation of novel RNAs that may be beneficial during stress. HET-s prion, by contrast, mediates cell fusion-induced cell death by converting its allelic variant, HET-S protein (big-S), into a membrane permeabilizing toxin that kills the fused cell; this has been suggested to be a possible mechanism of self-defense [7].
In higher eukaryotes, functional prions have been described in Aplysia and Drosophila, where members of the CPEB protein family undergo prion conversion that facilitates memory formation [8,9]. In mammals, however, prion-like proteins have been, until recently, invariably associated with diseases of protein aggregation [10–12]. Biochemical, structural, and genetic studies have now revealed that the adaptor proteins MAVS and ASC propagate respective downstream signals through prion conversion, and that this signal amplification strategy is crucial for the initiation of the innate immune response to pathogens detected by the RIG-I and inflammasome pathways. These findings suggest a beneficial role for prions in mammals and highlight a highly conserved function for prion proteins in host defense. We discuss these recent findings here and propose that prion-like polymerization is a conserved mechanism of signal transduction that mediates host defense. We first review evidence leading to the identification of prion-like properties in MAVS and ASC. We then discuss the significance of these properties in signal transduction and their broad conservation in light of homology with a cell death pathway in fungi. We conclude by examining the advantages of signaling through prion-like polymerization and highlight important outstanding questions in the field.
Identification of MAVS as a functional mammalian prion
RIG-I-like receptors (RLRs), which include RIG-I, MDA5, and LGP2, survey the cytoplasm and are essential for the detection of RNA viruses [13]. RIG-I specifically binds viral RNAs bearing 5′ triphosphates (5′ppp) or diphosphates (5′pp), which distinguish them from cellular RNAs that normally contain a 5′-cap or other modifications [14–16]. Our group has estimated that as few as 20 5′ppp–RNA molecules can trigger a robust antiviral response through RIG-I, suggesting an ultrasensitive mechanism of signal transduction [17].
RIG-I contains N-terminal tandem caspase activation and recruitment domains (CARDs) [RIG-I(N)], a middle helicase domain, and a C-terminal domain (CTD). Following activation by 5′ppp–RNA, RIG-I oligomerizes through the binding of RIG-I(N) to lysine 63 (K63)-linked polyubiquitin chains that are not anchored to any cellular proteins [17,18]. Oligomerized RIG-I then activates the adaptor protein MAVS. MAVS is a mitochondrial tail anchored protein and also contains an N-terminal CARD (MAVS-CARD), which is essential for MAVS activation through interactions with RIG-I CARDs [19]. Activated MAVS then signals downstream to the kinases IKK and TBK1, which activate transcription factors, nuclear factor-κB (NF-κB) and interferon regulatory factor 3 (IRF3), respectively, to induce antiviral cytokine expression [20] (Figure 1).
Figure 1.

Model of RIG-I-dependent MAVS activation. Following virus infection, 5′ppp–RNA is generated in the cytoplasm and recognized by RIG-I, bringing multiple RIG-I proteins into proximity and releasing its N-terminal tandem CARDs from autoinhibition. Free RIG-I CARDs can then bind to unanchored K63-linked polyubiquitin, which induces the formation of an active RIG-I tetramer. The active RIG-I tetramer then interacts with the mitochondrial adaptor protein MAVS, through CARD–CARD interactions, and converts MAVS into its prion form. The initial MAVS prion seeds then rapidly convert other native MAVS proteins into the filamentous prion form, eventually leading to the activation of NF-κB and IRF3 for antiviral interferon production. Abbreviations: 5′ppp, 5′ triphosphates; CARD, caspase activation and recruitment domain; K63, lysine 63; NF-κB, nuclear factor-κB; IRF3, interferon regulatory factor 3.
RIG-I and MAVS belong to the death domain (DD) superfamily of proteins, which play diverse and important roles in immune, inflammatory, and cell death signaling [21]. Members of the DD superfamily share a common six α-helical structural fold and contain characteristic domains (CARD, DED, DD, and PYRIN), which further distinguish these proteins into subfamilies. The DDs largely function through homotypic interactions. However, while some DD-containing proteins signal through limited oligomers, others have been observed to form much larger polymers [22–24].
MAVSCARD is a bona fide prion domain
Studies on the RIG-I signaling pathway led to the identification of the adaptor protein MAVS (also known as IPS-1, VISA, and CARDIF) [19,25–27]. In addition to its unique mitochondrial localization, MAVS was also found to be resistant to detergent extraction after viral infection, suggesting active MAVS underwent significant biochemical changes [19]. Indeed, subsequent analyses revealed that virus infection led to the aggregation of MAVS into very large particles that were resistant to 2% SDS, as revealed by semi-denaturing detergent agarose gel electrophoresis (SDD-AGE), a commonly used assay to detect prionparticles [28]. Surprisingly, recombinant MAVS proteins formed prion-like fibers with a protease resistant core composed of MAVSCARD. Cell free assays revealed that substoichiometric amounts of MAVSCARD fibers (which lack the signaling domain) were capable of converting endogenous full-length MAVS into SDS-resistant polymers; importantly, these newly converted polymers were functional in triggering downstream signaling [28]. Subsequent super-resolution imaging of virus-infected cells revealed rod-shaped MAVS clusters on the mitochondria, providing evidence for MAVS fiber formation in cells [29]. From these results, we proposed that MAVS propagates antiviral response through functional prion conversion.
To test if MAVS had other prion-like properties, we took a genetic approach using the well-established yeast SUP35-based prion assay, where a candidate prion domain is tested for its ability to functionally replace SUP35NM, a bona fide yeast prion domain [30]. In yeast strains expressing MAVSCARD–SUP35C in place of endogenous SUP35 (NM–SUP35C), MAVSCARD faithfully reconstituted the key functions and properties of the SUP35 prion domain [31]. As with all prions, transient overexpression of MAVS-CARD greatly enhanced the frequency of MAVSCARD polymerization, resulting in phenotypes that were stably inherited over numerous cell divisions. The MAVSCARD prion state, [MAVSCARD+], was also dominant, cytoplasmically inherited, and dependent on the MAVSCARD protein. As with all other prion domains, MAVSCARD also exhibited a two-step nucleation and polymerization-dependent prion conversion. Thus, from these biochemical and genetic studies, MAVS harbors key hallmarks of prions [28,31].
RIG-I mediated MAVS polymerization
RIG-I(N) directly nucleates MAVS prion conversion
Key to fully comprehending the mechanisms mediating MAVS activation is understanding how active RIG-I leads to MAVS prion conversion. In yeast, which lack a RIG-I-like pathway, the expression of RIG-I(N) is sufficient to induce MAVSCARD–SUP35C prion conversion, suggesting a direct interaction between the CARDs of RIG-I and MAVS for signal transduction [31]. Point mutations in MAVSCARD that abolished RIG-I-dependent antiviral signaling and MAVS polymerization in mammalian cells also abrogated RIG-I(N)-induced MAVSCARD prion conversion in yeast, indicating that the yeast assay faithfully recapitulated RIG-I-dependent MAVS activation and that MAVS prion conversion is necessary for signaling [20,31].
K63 polyubiquitin-mediated oligomerization activates RIG-I
While the fully active RIG-I complex awaits structural elucidation, accumulating evidence suggests that unlike the previously proposed monomer or dimer [32], the active RIG-I complex is rather an oligomer, which would be more conducive to its role as a MAVS nucleation factor. Specifically, an oligomeric RIG-I seed would be more amenable to recruiting multiple MAVS molecules for their polymerization.
The binding to 5′ppp–RNA was initially thought to be sufficient for downstream signaling by releasing RIG-I from its autoinhibited state, where RIG-I(N) and its helicase domain interact in a closed conformation [33–35]. However, in vitro reconstitution of RIG-I signaling revealed that in addition to activation by RNA, the binding of unanchored K63 polyubiquitin to the released RIG-I(N) is essential for RIG-I activation [17]. Specifically, in the presence of RNA and ATP, the binding to K63 polyubiquitin induced RIG-I to form active tetramers – the first evidence suggesting the active RIG-I complex to be more than a dimer [18]. Indeed, free RIG-I(N) alone requires polyubiquitin binding to form a tetrameric complex that can then activate MAVS both in vitro and in cells [17,36,37]. While RIG-I ubiquitin-binding mutants are defective in signaling, artificial oligomerization of a RIG-I(N) ubiquitin-binding mutant can rescue its signaling activity, further indicating an essential role for polyubiquitin-mediated RIG-I oligomerization [36]. In addition, incubation of RIG-I with RNA and K63–Ub4 resulted in SDS-resistant MAVS polymers similar to those observed following virus infection, providing direct evidence in support of a requirement for ubiquitin binding for RIG-I-mediated MAVS nucleation and prion conversion [28].
Recent biophysical and structural studies have further supported RLR activation through oligomerization and RIG-I(N) as an ubiquitin sensor. Both RIG-I and MDA5 were observed to form filaments in vitro when incubated with RNA [36,38–40]. However, despite an essential role for ATP hydrolysis in RIG-I and MDA5 activation, ATP was found to promote RIG-I filament formation while destabilizing MDA5 filaments. In addition, the formation of both RIG-I and MDA5 filaments were independent of their N-terminal CARDs, which as the effector domains are essential for downstream signaling. These observations suggest that while formation of filaments in vitro correlates with the ability of receptors to polymerize MAVS, these filaments may not represent the fully active forms of the receptors in cells, where additional ubiquitin binding and stabilization is required. Supporting an essential role for K63 polyubiquitin, the in vitro MAVS polymerization activity of full-length RIG-I filaments was greatly enhanced (by over 10-fold) in the presence of K63 polyubiquitin, suggesting polyubiquitin is required to correctly orient RIG-I(N) for MAVS binding [36]. Genetic deletion studies have shown that the ubiquitin E3 ligases, TRIM25, RIPLET, and MEX3C, are each essential for the activation of the RIG-I pathway, indicating that ubiquitination is important for RIG-I activation in vivo [41–43]. It has been proposed that these E3 ligases ubiquitinate RIG-I at different lysine residues, but further work is needed to clarify why each of these E3s has an indispensable role in the RIG-I pathway.
RIG-I(N) as a K63 polyubiquitin sensor
Consistent with previous mechanistic studies, the recent crystal structure of RIG-I(N) in a tetrameric complex with K63–Ub2 (K63-linked diubiquitin) provides further evidence for RIG-I CARDs functioning as a ubiquitin sensor, and suggests a likely mechanism for the activation of full-length RIG-I. Specifically, the binding to K63 polyubiquitin induces RIG-I(N) to form a one-turn helical tetramer, where the seam of the ‘lock washer’ is displaced by half its thickness or one CARD subunit [37]. Mutation of the top, but not the bottom surface of the RIG-I(N):Ub2 helix abolished MAVSCARD polymerization in vitro, suggesting MAVSCARD may interact with the top of the helical RIG-I tetramer to polymerize into its prion conformation. However, additional studies are required to understand the mechanism of MAVSCARD nucleation by the active RIG-I–RNA–polyubiquitin complex or the RIG-I(N)–polyubiquitin tetramer. For instance, the RIG-I nucleus could directly template MAVSCARD polymerization or simply serve as a scaffold to recruit multiple MAVSCARDs, which then take on a different helical structure, as discussed below.
Structural insights into MAVS fiber formation
Recent structural studies have begun to shed light on potential mechanisms for MAVS polymerization. X-ray crystallography studies of MAVSCARD monomer revealed a six α-helical bundle structure that is shared by the death domain superfamily of proteins [44]. However, understanding the mechanism of RIG-I nucleated MAVS polymerization requires examination of MAVSCARD filaments and MAVSCARD in complex with active RIG-I.
To this end, two recent cryo-EM structural models of MAVS filaments have emerged. Consistently, both studies did not reveal any major conformational change of MAVSCARD from its monomer to its filament form (i.e., no α-helix to β-sheet transition, as seen for PrPC to PrPSC conversion) [29,45]. Specifically, the two studies suggested that the MAVSCARD fiber forms a left-handed helix, in which each individual MAVSCARD monomer maintains its native, six α-helical fold without undergoing any β-sheet conversion [29,45]. However, while the model presented by Xu et al. suggested a three-start helix, Wu et al. obtained a single-stranded left-handed helix that conforms to the structure of the helical RIG-I(N):Ub2 complex described earlier [37]. It is possible that active MAVS forms different kinds of filaments, a phenomenon known as strain variation that has been observed for most prions identified to date [46,47]. In each case, different prion strains form as a result of structural variations during nucleation. For instance, RIG-I induced MAVS filaments may be different from spontaneously formed MAVS filaments, which were prepared differently and used in the above structural studies.
To address the mechanism of RIG-I nucleated MAVS prion conversion, a mutant RIG-I(N)-MAVSCARD fusion protein (to increase its solubility) was crystallized for structural analysis, which revealed a helical complex where four MAVSCARDs extended the tetrameric RIG-I(N) helix by one turn [45]. The structure suggests a possible mechanism of direct RIG-I nucleated MAVS prion conversion, where MAVS polymerization follows the trajectory set by the helical ring of RIG-I(N):Ub2 complex.
While the studies described above made significant contribution to our understanding of MAVS prion conversion, subsequent studies should be aimed at understanding the structural features of MAVS fibers in cells and structure of active RIG-I:RNA:K63Ub [or RIG-I(N):K63Ub] in complex with MAVSCARD. Importantly, the structure of active RIG-I caught in the act of nucleating MAVSCARD polymerization could be informative to our understanding of heterotypic prion nucleation in general (i.e., between NLRP3 and ASC or NWD2 and HET-S/s).
A model for RIG-I and MAVS activation
We propose the following model for RIG-I-induced MAVS polymerization, which reconciles the key conclusions from the studies described earlier (Figure 1): (i) the binding of 5′ppp–RNA to RIG-I CTD and helicase domain frees RIG-I(N) from its autoinhibited conformation; (ii) in the presence of ATP, RIG-I translocates along the RNA, potentially allowing it to form unstable oligomers that transiently bring multiple RIG-I CARDs into proximity; and (iii) the binding to K63 polyubiquitin stabilizes the freed RIG-I CARDs, leading to the formation of a signaling competent RIG-I tetramer that then rapidly nucleates MAVS prion conversion. In the absence of polyubiquitin binding and stabilization, RIG-I CARDs are unlikely to form a properly structured seed needed for efficient MAVS polymerization, even upon RNA binding and ATP hydrolysis. The active complex of RIG-I with 5′ppp–RNA, ATP, and K63 polyubiquitin remains to be structurally determined.
ASC is a functional prion in mammalian inflammasome signaling
DDs are also present in inflammasomes, protein complexes that function in host defense by propagating an inflammatory response through activation of pattern recognition receptors, such as AIM2 and NLRP3, each of which contains an N-terminal PYRIN domain [48,49]. Following activation by cytoplasmic DNA and a myriad of cellular damage signals, respectively, AIM2 and NLRP3 individually activate the adaptor protein ASC, a bipartite PYRIN (N terminus) and CARD-containing protein. After activation through PYRIN–PYRIN interactions with upstream sensors, ASC recruits and activates caspase-1, which then cleaves its substrates pro-interleukin (IL)-1β and pro-IL-18 into active cytokines that can be secreted by the cell.
The unexpected prion-like behavior of MAVSCARD prompted our group to examine if other DD-containing proteins could also exhibit prion switching. Using the SUP35-based prion assay that faithfully recapitulated MAVSCARD prion activity in yeast, we identified the PYRIN domain of the inflammasome adaptor ASC as the top hit from a candidate-based prion screen of 18 DDs [31]. Similar to MAVSCARD, ASCPYD exhibited classic hallmarks of prions including the ability to form SDS-resistant, self-perpetuating fibers that allowed cells to take on distinctive and heritable states. Transient expression of AIM2 or NLRP3PYD induced robust ASCPYD and full-length ASC prion conversion in yeast, suggesting direct interactions between PYRINs of ASC and upstream sensors [31]. Consistently, in vitro polymerization assays indicated that substoichiometric amounts of AIM2 and NLRP3PYD–NBD robustly induced formation of ASCPYD fibers, as observed by EM, where each nucleation factor localized to one end of a filament [50]. Mutations that abolished ASC prion conversion in yeast also blocked its signaling in mammalian cells, indicating its prion activity is essential for its function [31].
In addition to NLRP3 and AIM2, the inflammasome contains a number of other receptors, such as NLRC4, which has been proposed to activate ASC [51]. Unlike NLRP3 and AIM2, NLRC4 does not contain a PYRIN domain but instead harbors an N-terminal CARD in its place. Interestingly, studies of ASC prion conversion in yeast indicated that NLRC4CARD is able to convert full-length ASC into a prion through NLRC4CARD–ASCCARD interactions, but ASC prion switch is strictly dependent on its ASCPYD-mediated polymerization [31]. By contrast, despite its ability to interact with ASCCARD, Caspase-1CARD was unable to convert ASC into a prion. These results further support a nucleation model proposed earlier for RIG-I and MAVS, whereby the receptor serves as a scaffold to recruit multiple adaptor proteins for subsequent proximity-induced prion conversion, rather than as a direct template. However, the proposed model of prion nucleation by a heterologous upstream protein needs to be further investigated.
Structural analysis of ASCPYD fibers
High-resolution cryo-EM analysis revealed that ASCPYD fibers also form a helix composed of three strands of polymerized ASCPYD monomers [50], which is overall similar to the cryo-EM MAVSCARD filament structure presented earlier [29]. In the ASCPYD fiber assembly, each monomer maintained its native α-helical conformation (i.e., no β-sheet conversion), but with noticeable conformational changes in the α2–α3 loop, and the α3-helix as compared with ASCPYD in the monomer state. Interestingly, as opposed to the left-handed helix of MAVSCARD fibers, ASCPYD fiber helices are right-handed, suggesting a unique chirality possibly induced by the upstream nucleating pattern recognition receptor. Nonetheless, structural and biophysical analyses of both MAVS and ASC indicate their respective prion conversion involves an all-or-none transition from soluble, α-helical monomers to polymers composed of similarly structured monomeric subunits organized in a helical fiber, each nucleated at one end by their respective upstream signaling protein.
Prion properties of MAVS and ASC
Overall, MAVS and ASC exhibit classic hallmarks of prions. For instance, both proteins can take on at least two distinctive conformations: an inactive, soluble form and a self-perpetuating prion-like fiber. The prion forms of the proteins endow the host cell with novel, heritable traits that are dominant and inherited cytoplasmically; these features distinguish MAVS and ASC from other fiber-forming proteins, such as actin and microtubules. Specifically, MAVSCARD and ASCPYD undergo energetically irreversible polymerization, a property that seems appropriate for their function as on and off switches within innate immune signaling cascades. These prion fibers could, in principle, perpetuate indefinitely in the absence of other signaling regulators. By extension, a cell expressing MAVS or ASC is bistable and can exist in either the native (prion minus) or the prion state (prion plus). Furthermore, a population of cells that propagated from a prion plus cell can again be separated into a prion plus and a prion minus population (Figure 2).
Figure 2.

Distinguishing features of MAVS and ASC prion fibers. Cells expressing MAVS or ASC can exist in at least two possible states: a soluble or prion state, which allows a population to take on a bimodal distribution. Unlike microtubule fibers, MAVS or ASC fibers endow heritable states to their host cells. Specifically, when prion fiber-containing cells are serially passaged, a large proportion of the daughter cells will inherit the prion phenotype and the population is again characterized by a bimodal distribution. In sharp contrast to this, when sorted by their average microtubule fiber lengths, cells distribute in one continuous unimodal population, characterized by the relative sizes of their microtubules. Following serial passages, cells initially containing larger microtubule fibers will again produce a unimodal population of cells containing heterogeneous microtubule sizes. Abbreviations: ASC, apoptosis-associated speck-like protein containing a caspase activation and recruitment domain; MAVS, mitochondrial antiviral signaling.
Polymerization of nonprion proteins, by contrast, is dynamic and reversible, a property inherent to their functions. For example, microtubules and actin rely on their dynamic cycles of polymerization and depolymerization to carry out essential cellular processes. Their polymerization state is energetically unstable and defined by the relative affinity of each monomeric subunit for the polymer based on its nucleotide binding and hydrolysis. No polymerization state of microtubule or actin is heritable, as each individual cell and its progeny share a similar distribution of soluble and polymerized forms of the protein (Figure 2).
Prion properties unique to MAVS and ASC
Despite sharing the classic properties of prions, MAVS and ASC also exhibit several features that stand in contrast to those of other prions identified to date (Figure 3). First, both proteins turn into gain-of-function prions, while most of the currently identified prions display loss-of-function phenotypes. Point mutations that abolished the prion conversion of MAVS and ASC also abrogated their function. For example, cells expressing MAVS proteins containing mutations that inhibited polymerization essentially displayed the same phenotype following viral infection as mavs-deficient cells. Second, the prion domains of both proteins are structurally distinct from those of other known prions. In their soluble states, MAVSCARD and ASCPYD adopt well-folded six α-helical bundle structures, and these domains are not rich in glutamine and asparagine, amino acids often found in other disordered prion domains. β-Amyloid conversion is not observed in their prion states: both MAVSCARD and ASCPYD subunits maintain an α-helical conformation within their respective fibers, with only minor structural changes. Consistently, MAVS and ASC fibers do not stain with thioflavin T (ThT), a dye that fluoresces upon binding to β-amyloids, and their prion states are propagated independently of the Hsp104 chaperone that is required for the maintenance of all other β-amyloid prions in yeast. Nonetheless, MAVS and ASC can maintain their prion states in yeast following numerous cell divisions, suggesting a unique mechanism of fiber fragmentation and propagation. Third, higher eukaryotic prion-like proteins described to date all reside in postmitotic or rarely dividing resident tissues. MAVS and ASC, however, serve as signaling adaptors in immune as well as nonimmune cells with rapid turnover and wide dissemination throughout the body. Lastly, in contrast to other proteins that undergo stochastic prion conversion, MAVS and ASC undergo polymerization nucleated by heterologous upstream sensors, the activation of which is tightly regulated.
Figure 3.
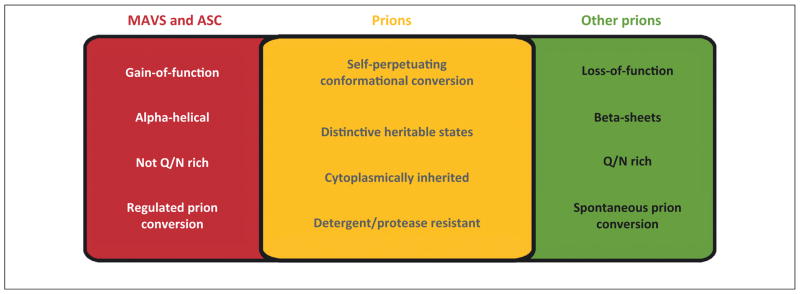
Shared and unique features of MAVS and ASC prions. While MAVS and ASC share key properties with other prions (middle box), they also contain unique differences (left and right boxes). Abbreviations: ASC, apoptosis-associated speck-like protein containing a caspase activation and recruitment domain; MAVS, mitochondrial antiviral signaling.
Conserved fungal inflammasome pathway that signals through a gain-of-function prion
MAVS and ASC are unlikely to be the only gain-of-function prions that undergo polymerization regulated by heterologous nucleation factors. The most striking similarities to MAVS and ASC are found in the filamentous fungal prion HET-s, an observation further supported by recent bioinformatic and functional studies (Figure 4) [31,52]. In addition to its unique gain-of-function phenotype, HET-s prion also does not stain with ThT or require Hsp104 for its propagation [7]. In its prion state, HET-s propagates a cell death reaction induced by spontaneous fungal cell fusion. Remarkably, recent bioinformatic analysis revealed that NWD2, encoded by a gene immediately adjacent to the HET-S/s locus, displays similarities to mammalian NOD-like receptors (NLRs) such as NLRP3 [52]. NWD2 contains a C-terminal WD40 repeat and a middle NACHT domain, similar to NLRP3, which also contains a middle NACHT domain in addition to C-terminal leucine-rich repeats (LRRs). Furthermore, NWD2 contains an N-terminal effector domain (NWD2N30) that is predicted to take on a similar structural fold as the HET-s prion domain (HET-sPrD) [52], reminiscent of the structure–function relationship between NLRP3PYD and ASCPYD (Figure 4).
Figure 4.

Functional prions in immune signaling. In mammals, receptors such as RIG-I and NLRP3 induce the prion conversion of their respective adaptors MAVS and ASC, which then rapidly polymerize leading to protective cytokine secretion and cell death. In filamentous fungi, a homologous pathway depends on the NWD2 protein, which likely converts the downstream HET-S/s protein into a prion to induce cell death. The fungal NWD2/HET-S/s pathway shares remarkable similarities to the mammalian NLRP3/ASC inflammasome in function and organization. Abbreviations: ASC, apoptosis-associated speck-like protein containing a caspase activation and recruitment domain; CARD, caspase activation and recruitment domain; CTD, C-terminal domain; LLR, leucine-rich repeat; MAVS, mitochondrial antiviral signalling; NLRP3, NLR family, pyrin domain containing 3; RIG-I, retinoic acid-inducible gene I.
The striking similarity between the fungal NWD2 and HET-s pair and mammalian NLRP3 and ASC suggests evolutionary conservation of an inflammasome-like pathway, which is supported by recent functional studies [31]. Specifically, we found that NWD2N30 induces polymerization of the HET-s prion domain (HET-sPrD). Moreover, NWD2N30 and HET-sPrD can together functionally replace the PYRINs of NLRP3 and ASC in mammalian inflammasome signaling [31]. Hence, the regulated prion conversion of signaling adaptors (i.e., HET-S/s and ASC) by their respective upstream receptors (i.e., NWD2 and NLRP3) appears to be an evolutionarily conserved mechanism of signal transduction from fungi to mammals (Figure 4). In addition, studies in mammalian cells indicate that both pathways signal through prion conversion of an intermediate protein to initiate cell death, highlighting a common function between the two homologous pathways [31]. However, additional studies are needed to understand the mechanism, regulation, and function of NWD2-mediated HET-S/s activation in the context of a fungal host. While the NLRP3 inflammasome has been the subject of intense investigation, we suggest that studies of the fungal NWD2–HET-S/s pathway could contribute important insights to our understanding of mammalian inflammasomes, from the mechanism of their activation to their execution of pyroptosis.
Prion-like polymerization as a unique mechanism of signal transduction
Forming irreversible prion-like fibers to protect oneself from pathogen invasion and other danger signals seems counterintuitive at first. Irreversible protein aggregates are usually associated with disease states. However, the advantages of using functional prions to mediate essential cellular functions are numerous, including robust signal amplification, formation of an active signalosome, evolutionary advantage to the host, and possible memory formation.
Robust signal amplification
Our cells are constantly bombarded by an array of extra-cellular and intracellular signals and must evolve mechanisms to properly control signal transduction, minimizing noise while responding quickly to urgent calls. In this regard, innate immune signaling is critical for our protection and survival. While other signal amplification mechanisms exist (i.e., the formation of defined, cooperative oligomeric complexes such as MyDDosome that bring signaling components into proximity) [21], a self-perpetuating mechanism of signal amplification in response to a stimulation threshold ensures an all-or-none commitment to the appropriate response. Interestingly, receptors such as RIG-I and NLRP3 are usually expressed at very low levels in the cell. The activation of RIG-I, for example, induces the secretion of interferon and other cytokines that further induce the expression of RIG-I, NLRP3, and other genes. In this case, the sensitive and robust switch of MAVS to its prion state perpetuates a feed-forward cycle of host defense that ultimately protects the organism.
Mutations in receptors such as RIG-I and NLRP3 may tip the balance and destabilize the system, resulting in inappropriate cytokine secretion. For instance, single amino acid mutations in NLRP3 are known to cause a family of autoinflammatory diseases known as cryopyrin-associated periodic syndrome (CAPS) [53]. While some CAPS-associated NLRP3 mutations may result in only a slightly over-sensitive NLRP3, the downstream all-or-none response mediated through ASC will ensure robust cytokine secretion as long as the signaling threshold is reached. In other words, a small change in analog signal amplitude results in a full-scale digital response, this time, to the detriment of the host.
Recent studies have suggested that activated ASC polymers are secreted by cells and taken up by neighboring cells to amplify inflammasome signaling [54,55]. These infectious ASC prion particles take on fiber-like structures in cells and in vitro. It remains to be seen if MAVS prion particles can also be released by virus-infected cells and enter uninfected cells through phagocytosis or other means to amplify antiviral defense. If so, understanding how these self-perpetuating signaling pathways are downregulated is an important area of future studies (see concluding remarks section).
Signalosomes for recruitment and activation of downstream components
Signal transduction through the formation of fibrous particles offers cells not only temporal but also spatial control over cellular signaling. Notably, activated MAVS and ASC both form distinct punctate structures in cells and sediment to high-molecular weight fractions in sucrose gradients [28,51]. Consistent with the idea that these prion polymers serve as signalosomes for recruitment of downstream signaling molecules, mutations abolishing MAVS aggregation prevented the recruitment of TRAF proteins necessary for signal propagation, essentially exhibiting the same deficiency as mavs−/− cells in response to viral infection [20]. Similarly, the ASC polymerization mutant was also defective in recruiting procaspase-1 [31].
Altruistic prions
Intriguingly, virus infection and inflammatory signaling are usually detrimental to the affected cell, often resulting in cell death, which protects the host from further damage. The evolution of irreversible signaling mechanisms through prion switches therefore may benefit the organism as a whole at the expense of individual cells. The sacrifice of compromised cells to protect the host would increase the overall evolutionary fitness of the organism. Hence, it may not be surprising that signal transduction through altruistic prions has only been observed in multicellular organisms, such as filamentous fungi and mammals, despite the discovery of over two dozen prions in unicellular yeast.
Memory of previous infections
In filamentous fungi, cell fusion will lead to cell death only if one cell containing the prion form of HET-s fuses with another expressing soluble HET-S, the cell death executioner [56]. By itself, the HET-s prion will stably propagate without causing any cellular damage, suggesting its prion state could potentially serve as a memory of an earlier infection or insult. The mammalian innate immune prions could serve similar functions. For instance, mechanisms that desensitize mammalian cells to constant stimulation by pathogens and danger signals have been described [57]. Negative regulatory mechanisms that prevent constant signaling from MAVS and ASC fibers would allow recovered cells to serve as memory of previous host infections through continuous prion propagation. Interestingly, cytosolic extracts of virus-infected cells were refractory to further activation by MAVS fibers in a cell free assay, suggesting the existence of mechanisms to turn off antiviral signaling downstream of MAVS [28]. The persistence of MAVS prions in these desensitized cells would allow them to serve as remnants of a previous infection, in other words, a novel mechanism of innate immune memory, a concept that has mainly been associated with the adaptive immune system to date.
Concluding remarks
Studies of the RIG-I and inflammasome pathways have uncovered a surprisingly conserved mechanism of innate immune signaling, which relies on autocatalytic conformational conversions of adaptor proteins, much like prions, to propagate signal transduction. Such a mechanism offers the innate immune system exquisite sensitivity and robustness to rapidly respond to pathogens and other danger signals.
The idea that self-catalytic protein conformational changes govern innate immune signaling bridges the divergent fields of prion regulation and signal transduction and offers many exciting new areas of investigation. To fully understand MAVS and ASC regulation, we need to not only study their activation but also how their signaling is turned off, which would be necessary to prevent autoimmunity and autoinflammatory diseases. For instance, humans and mice infected with a virus do not continuously produce type I interferons indefinitely. It seems unlikely that MAVS and ASC prion propagation would be mitigated by dilution as for yeast prions or by complete refolding of converted fibers into soluble monomers, an energetically unfavorable process. Other cellular processes that mediate the clearance of these prion fibers or desensitize downstream signaling must exist to prevent their constitutive signaling, which would otherwise be detrimental to the host.
In addition to fiber polymerization through monomer addition, prion propagation also requires continuous fiber fragmentation and seeding [58,59]. In contrast to yeast prions that depend on Hsp104 for fiber fragmentation [60], the mechanism of MAVS and ASC fragmentation and seeding is Hsp104-independent in yeast and remains unclear in mammalian cells. In addition, MAVS and ASC function in higher eukaryotes that must orchestrate more complex responses to outside stimuli to maintain homeostasis. Hence, the propagation of MAVS and ASC fibers in cells and in vivo likely depends on other factors and mechanisms that remain to be identified.
While examples of transmissible phenotypes mediated by distinctive protein conformations remain relatively rare, especially in mammals, it is likely that prion-like polymerization is employed by other cellular pathways. The identification and characterization of other functional mammalian prions would contribute to our understanding of protein regulation and further establish this new paradigm of signal transduction (Box 1).
Box 1. Questions for future research.
-
Structural determination of the following proteins to support current models:
RIG-I:RNA:K63 polyubiquitin complex,
Active RIG-I in complex with MAVSCARD (mechanism of MAVSCARD nucleation).
Investigation of mechanisms that remove or degrade MAVS/ASC fibers in cells or block their signaling to prevent autoimmune and autoinflammatory diseases.
Identification of mammalian cellular factors essential for MAVS/ ASC fiber propagation (i.e., fiber nucleation or fragmentation factors).
Investigation of possible mechanisms and consequences of MAVS and ASC fiber propagation between cells.
-
In-depth characterization of the NWD2–HET-S/s pathway in fungi:
Demonstration of direct NWD2-mediated HET-S/s activation for fungal cell death induction,
Identification of NWD2 ligand or activation signals in fungi,
Characterization of NWD2-like pathways in fungi and other organisms.
Identification of prion-like switches in other mammalian signaling pathways.
Acknowledgments
We thank Nicole Varnado and Siqi Liu for critically reading the manuscript. Research in the Chen laboratory is supported by grants from the National Institutes of Health (RO1-GM63692 and RO1-63967), the Cancer Prevention and Research Institute of Texas (CPRIT; RP120718), and the Welch Foundation (I-1389). X.C. is supported by an international student fellowship from the Howard Hughes Medical Institute (HHMI). Z.J.C. is an HHMI investigator.
References
- 1.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tuite MF, Serio TR. The prion hypothesis: from biological anomaly to basic regulatory mechanism. Nat Rev Mol Cell Biol. 2010;11:823–833. doi: 10.1038/nrm3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 5.Alberti S, et al. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137:146–158. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suzuki G, et al. A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science. 2012;336:355–359. doi: 10.1126/science.1219491. [DOI] [PubMed] [Google Scholar]
- 7.Saupe SJ. The [Het-s] prion of Podospora anserina and its role in heterokaryon incompatibility. Semin Cell Dev Biol. 2011;22:460–468. doi: 10.1016/j.semcdb.2011.02.019. [DOI] [PubMed] [Google Scholar]
- 8.Si K, et al. Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell. 2010;140:421–435. doi: 10.1016/j.cell.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 9.Majumdar A, et al. Critical role of amyloid-like oligomers of Drosophila Orb2 in the persistence of memory. Cell. 2012;148:515–529. doi: 10.1016/j.cell.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 10.Kim HJ, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramaswami M, et al. Altered ribostasis: RNA–protein granules in degenerative disorders. Cell. 2013;154:727–736. doi: 10.1016/j.cell.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoneyama M, Fujita T. RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev. 2009;227:54–65. doi: 10.1111/j.1600-065X.2008.00727.x. [DOI] [PubMed] [Google Scholar]
- 14.Hornung V, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 15.Pichlmair A, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- 16.Goubau D, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature. 2014;514:372–375. doi: 10.1038/nature13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeng W, et al. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang X, et al. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity. 2012;36:959–973. doi: 10.1016/j.immuni.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seth RB, et al. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 20.Liu S, et al. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife. 2013;2:e00785. doi: 10.7554/eLife.00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrao R, Wu H. Helical assembly in the death domain (DD) superfamily. Curr Opin Struct Biol. 2012;22:241–247. doi: 10.1016/j.sbi.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park HH, et al. Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell. 2007;128:533–546. doi: 10.1016/j.cell.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiao Q, et al. Structural architecture of the CARMA1/Bcl10/ MALT1 signalosome: nucleation-induced filamentous assembly. Mol Cell. 2013;51:766–779. doi: 10.1016/j.molcel.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun L, et al. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell. 2004;14:289–301. doi: 10.1016/s1097-2765(04)00236-9. [DOI] [PubMed] [Google Scholar]
- 25.Meylan E, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 26.Kawai T, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 27.Xu LG, et al. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol Cell. 2005;19:727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 28.Hou F, et al. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu H, et al. Structural basis for the prion-like MAVS filaments in antiviral innate immunity. Elife. 2014;3:e01489. doi: 10.7554/eLife.01489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alberti S, et al. Biochemical, cell biological, and genetic assays to analyze amyloid and prion aggregation in yeast. Methods Enzymol. 2010;470:709–734. doi: 10.1016/S0076-6879(10)70030-6. [DOI] [PubMed] [Google Scholar]
- 31.Cai X, et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell. 2014;156:1207–1222. doi: 10.1016/j.cell.2014.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cui S, et al. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol Cell. 2008;29:169–179. doi: 10.1016/j.molcel.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 33.Jiang F, et al. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479:423–427. doi: 10.1038/nature10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo D, et al. Structural insights into RNA recognition by RIG-I. Cell. 2011;147:409–422. doi: 10.1016/j.cell.2011.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kowalinski E, et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147:423–435. doi: 10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 36.Peisley A, et al. RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol Cell. 2013;51:573–583. doi: 10.1016/j.molcel.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 37.Peisley A, et al. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature. 2014;509:110–114. doi: 10.1038/nature13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berke IC, Modis Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 2012;31:1714–1726. doi: 10.1038/emboj.2012.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peisley A, et al. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc Natl Acad Sci USA. 2011;108:21010–21015. doi: 10.1073/pnas.1113651108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu B, et al. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 2013;152:276–289. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- 41.Kuniyoshi K, et al. Pivotal role of RNA-binding E3 ubiquitin ligase MEX3C in RIG-I-mediated antiviral innate immunity. Proc Natl Acad Sci USA. 2014;111:5646–5651. doi: 10.1073/pnas.1401674111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gack MU, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007;446:916–920. doi: 10.1038/nature05732. [DOI] [PubMed] [Google Scholar]
- 43.Oshiumi H, et al. The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe. 2010;8:496–509. doi: 10.1016/j.chom.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 44.Potter JA, et al. Crystal structure of human IPS-1/MAVS/ VISA/Cardif caspase activation recruitment domain. BMC Struct Biol. 2008;8:11. doi: 10.1186/1472-6807-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu B, et al. Molecular imprinting as a signal-activation mechanism of the viral RNA sensor RIG-I. Mol Cell. 2014;55:511–523. doi: 10.1016/j.molcel.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toyama BH, et al. The structural basis of yeast prion strain variants. Nature. 2007;449:233–237. doi: 10.1038/nature06108. [DOI] [PubMed] [Google Scholar]
- 47.Frederick KK, et al. Distinct prion strains are defined by amyloid core structure and chaperone binding site dynamics. Chem Biol. 2014;21:295–305. doi: 10.1016/j.chembiol.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martinon F, et al. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 49.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 50.Lu A, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156:1193–1206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Broz P, et al. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med. 2010;207:1745–1755. doi: 10.1084/jem.20100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daskalov A, et al. Genomic clustering and homology between HET-S and the NWD2 STAND protein in various fungal genomes. PLoS ONE. 2012;7:e34854. doi: 10.1371/journal.pone.0034854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dinarello CA. Interleukin-1β and the autoinflammatory diseases. N Engl J Med. 2009;360:2467–2470. doi: 10.1056/NEJMe0811014. [DOI] [PubMed] [Google Scholar]
- 54.Baroja-Mazo A, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15:738–748. doi: 10.1038/ni.2919. [DOI] [PubMed] [Google Scholar]
- 55.Franklin BS, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol. 2014;15:727–737. doi: 10.1038/ni.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seuring C, et al. The mechanism of toxicity in HET-S/HET-s prion incompatibility. PLoS Biol. 2012;10:e1001451. doi: 10.1371/journal.pbio.1001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 58.Collins SR, et al. Mechanism of prion propagation: amyloid growth occurs by monomer addition. PLoS Biol. 2004;2:e321. doi: 10.1371/journal.pbio.0020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Knowles TP, et al. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326:1533–1537. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- 60.Chernoff YO, et al. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science. 1995;268:880–884. doi: 10.1126/science.7754373. [DOI] [PubMed] [Google Scholar]