

Abstract
Elastin is predominantly comprised of crosslinked tropoelastin. For many years elastin was considered to serve a solely structural role but is now being increasingly identified as causal in cell signaling, development and repair. We introduced tropoelastin into an in vitro model in which airway smooth muscle cells (ASMCs) were stimulated with transforming growth factor (TGF)-β1 to examine the modulatory effect of this modular elastin sequence on release of angiogenic factors and matrix metalloproteinases (MMPs). Human ASMCs were presented to surfaces coated with tropoelastin or collagen and controls, then stimulated with TGF-β1. Transcript levels of vascular endothelial growth factor (VEGF) and connective tissue growth factor (CTGF) were quantified 4 and 24 h after TGF-β1 stimulation. Protein VEGF release from cells and CTGF sequestered at cell surfaces were measured by ELISA at 24 and 48 h. TGF-β1 increased VEGF mRNA 2.4 fold at 4 h and 5 fold at 24 h, accompanied by elevated cognate protein release 3 fold at 24 h and 2.5 fold at 48 h. TGF-β1 stimulation increased CTGF mRNA 6.9 fold at 4 h and 11.8 fold at 24 h, accompanied by increased sequestering of its protein counterpart 1.2 fold at 24 h and 1.4 fold at 48 h. Pre-incubation of cells with tropoelastin did not modulate VEGF or CTGF mRNA expression, but combined with TGF-β1 stimulation it led to enhanced VEGF release 5.1-fold at 24 h and 4.4-fold at 48 h.Pre-incubation with tropoelastin decreased CTGF sequestering 0.6-fold at 24 and 48 h, and increased MMP-2 production. Collagen pre-incubation under the same conditions displayed no effect on TGF-β1 stimulation apart from a slightly decreased (0.9 fold) sequestered CTGF at 48 h. As CTGF is known to anchor VEGF to the matrix and inhibit its angiogenic activity, a process which can be reversed by digestion with MMP-2, these findings reveal that elastin sequences can disrupt the balance of angiogenic factors, with implications for aberrant angiogenesis. The results suggest a model of molecular crosstalk and support an active role for elastin in vascular remodeling.
Keywords: Connective Tissue Growth Factor, Elastin, Electrospinning, Transforming Growth Factor β1, Tropoelastin, Vascular Endothelial Growth Factor A
1. Introduction
As a structural protein, elastin is responsible for the strength and elasticity of elastic tissues, such as lungs, arteries and skin. Elastin is predominantly composed of a soluble precursor, tropoelastin, which is released by elastogenic cells during early development and cross-linked following oxidation by one or more members of the lysyl oxidase family (Mithieux and Weiss, 2005). Elastogenesis can be restarted to repair damaged elastic fibers although this often results in disorganized elastin (Wagenseil and Mecham, 2007). Elastin is damaged in diseases such as emphysematous chronic obstructive pulmonary disease (COPD) (Shapiro and Ingenito, 2005; Black et al., 2008), cutis laxa (Ringpfeil, 2005; Hu et al., 2006) and asthma (Mauad et al., 1999; Reddel et al., 2012). Disorganized and damaged elastin can contribute to the pathology of disease not only through structural changes (Kelleher et al., 2005) but also molecular effects of exposed elastin sequences (Senior et al., 1980; Fulop et al., 1998; Duca et al., 2004; Turino et al., 2011).
The specific interaction of elastin sequences with cells, supporting an overall inflammatory profile and having a causal role in disease, is becoming increasingly well recognized. Fragments of elastin, for example, are central in the progression of a mouse model of emphysema through chemotaxis of monocytes, a process which is blocked by administration of an anti-elastin antibody (Houghton et al., 2006). The elastin receptor (ELR) is a mediator of these molecular effects of elastin (Robert, 2005), which include chemotaxis of multiple inflammatory cells (Senior et al., 1980; Tajima et al., 1997) and changes in the activities of mesenchymal-derived cells including adhesion to elastic fibers (Hornebeck et al., 1986), increase of cellular proliferation (Tajima et al., 1997; Mochizuki et al., 2002), migration (Senior et al., 1982; Skeie and Mullins, 2008; Shiratsuchi et al., 2010) and increase in expression and secretion of various pro-matrix metalloproteinases (MMPs), especially -1, -2 and -3 (Cozlin et al., 2006; Brassart et al., 2001; Heinz et al., 2010), and a decrease in tissue inhibitor of metalloproteinases (TIMP)-1 and -2 (Cozlin et al., 2006).
It is therefore reasonable to propose that cellular interactions of damaged elastin may play a role in inflammatory processes such as wound healing and tissue remodeling. Transforming growth factor β1 (TGF-β1) stimulation may be used in vitro as a model of inflammation and remodeling (Burgess et al., 2006a), due to its central role in these processes (Douglas, 2010) and in this context upregulates and increases production of vascular endothelial growth factor (VEGF) and connective tissue growth factor (CTGF) (Burgess et al., 2006a), which are important for angiogenesis (Ferrara and Davis-Smyth, 1997; Ferrara et al., 2003; Shimo et al., 1999). CTGF sequesters VEGF, negatively regulating its angiogenic activity (Inoki et al., 2002; Hashimoto et al., 2002). We therefore challenged ASMCs with both TGF-β1 and tropoelastin or collagen as a matrix comparison, and measured expression and production of VEGF and CTGF to explore the role of elastin sequences in inflammation and wound healing.
2. Results
2.1 Airway smooth muscle cells physically interact with tropoelastin
By using electrospun tropoelastin or collagen fibers, we were able to observe the physical interaction of airway smooth muscle (ASM) cells with elastin-based substrata. Electrospinning for short time periods directly on glass coverslips produced a thin layer of fibers analogous to biological structures which were stable in culture conditions. Cells were viable and proliferated for at least 2 weeks in culture on these surfaces during which time cells physically attached to tropoelastin or collagen fibers (Fig. 1A–D). Actin-containing cellular projections adhered to and organized around tropoelastin fibers. On non-fibrous surfaces coated with soluble tropoelastin or collagen, cells attached and cytoskeletal organization occurred (Fig. 1E–F).
Fig. 1.

Physical contacts of airway smooth muscle cells with elastin or collagen fibers. Images are representative of 5 experimental repeats. (A) Cells from patient 12 were seeded onto electrospun crosslinked tropoelastin fibers, stained with rhodamine phalloidin (red) after 24 h and visualized by confocal microscopy. (B) The same procedure was carried out with cells from patient 13, stained 8 h after seeding. Cells from patient 9 were seeded onto electrospun (C) tropoelastin or (D) collagen fibers and coated (E) tropoelastin or (F) collagen respectively, stained with rhodamine phalloidin and DAPI (blue) after 6 h and visualized.
2.2 Tropoelastin but not collagen attachment increases TGF-β1 mediated VEGF protein but not mRNA expression
mRNA levels for VEGF were investigated at 4 and 24 h after stimulation with TGF-β1, where cells were presented to either tropoelastin or collagen coated surfaces (Fig. 2). At 4 h, but not 24 h, TGF-β1 increased mRNA production of VEGF (p<0.05). In TGF-β1 stimulated samples, VEGF mRNA expression trended higher on tropoelastin (fold change compared to TGF-β1 negative no-protein control 4.0 ± 3.5) and collagen (fold change 4.0 ± 2.4) than on the no-protein control (fold change 2.4 ± 0.1) at 4 h.
Fig. 2.

VEGF mRNA expression after exposure to tropoelastin or collagen, in the presence or absence of TGF-β1, after (A) 4 h or (B) 24 h. Data were expressed as fold change relative to TGF-β1 negative time-matched samples. *Significant difference due to TGF-β1 by two-way ANOVA (n=4).
The amount of VEGF released into media by ASM cells after stimulation with TGF-β1 in the presence of coated tropoelastin or collagen was assessed at 24 and 48 h (Fig. 3). TGF-β1 enhanced VEGF release at 24 h (p<0.05) and 48 h (p<0.01). When stimulated with TGF-β1, tropoelastin caused a further increase in VEGF release by 24 h, as more VEGF was released by cells on tropoelastin (fold change compared to TGF-β1 negative no-protein control 5.1 ± 4.1) than either collagen (fold change 2.8 ± 1.0) or no-protein control (fold change 3.0 ± 1.4) (both p<0.05). The effect was maintained at 48 h where VEGF release on tropoelastin (fold change 4.4 ± 2.7) was again higher than either collagen (fold change 2.6 ± 1.1) or no-protein control (fold change 2.5 ± 1.1) (both p<0.01).
Fig. 3.

VEGF release after exposure to tropoelastin or collagen in the presence or absence of TGF-β1 stimulation on after (A) 24 h or (B) 48 h. Data were expressed as fold change relative to TGF-β1 negative time-matched samples. * Significant difference due to TGF-β1 and Δ significant difference due to tropoelastin by two-way ANOVA (n=6).
2.3 Tropoelastin but not collagen attachment decreases TGF-β1 mediated CTGF protein but not mRNA expression
mRNA levels for CTGF were investigated at 4 and 24 h after stimulation with TGF-β1, where cells were presented to either tropoelastin or collagen coated surfaces (Fig. 4). TGF-β1 increased mRNA production of CTGF at 4 h, (p<0.05) and 24 h TGF-β1 (p<0.01). In TGF-β1 stimulated samples, CTGF expression was not significantly higher for tropoelastin (fold change 17.4 ± 12.3) than the no-protein control (fold change 11.8 ± 4.5) at 24 h.
Fig. 4.

CTGF mRNA expression after exposure to tropoelastin or collagen in the presence or absence of TGF-β1 after (A) 4 h or (B) 24 h. Data were expressed as fold change relative to TGF-β1 negative time-matched samples. * Significant difference due to TGF-β1 by two-way ANOVA (n=4).
Irrespective of TGF-β1 stimulation, tropoelastin caused a decrease in CTGF sequestering at 24 h and 48 h (Fig. 5). This was particularly marked at 48 h where unstimulated cells exposed to tropoelastin displayed half the levels of sequestered CTGF of that found with the TGF-β1 negative no-protein control (p<0.01), and was also lower than cells on collagen (fold change 1.2 ± 0.4, p<0.001). TGF-β1 stimulated cells on tropoelastin displayed a fold change of 0.6 ± 0.3, which was lower than stimulated cells on no-protein control (fold change 1.4 ± 0.4, p<0.001) but not collagen (fold change 0.9 ± 0.2), which was also lower than no-protein control (p<0.01).
Fig. 5.

CTGF sequestering after exposure to tropoelastin or collagen in the presence or absence of TGF-β1 stimulation after (A) 24 h or (B) 48 h. Data were expressed as fold change relative to TGF-β1 negative time-matched samples. *Significant difference due to tropoelastin, Δsignificant difference due to tropoelastin and #significant difference due to collagen by two-way ANOVA (n=7).
2.4 Tropoelastin, but not collagen attachment increases MMP-2 production
Release of MMP-2, -9 and -12 was investigated in ASMCs after pre-incubation with surface protein coating. MMP-2 was significantly elevated after exposure to tropoelastin (550.6 ± 254.2 pg/mL) compared with collagen (309.3 ± 150.0 pg/mL) or no-protein control (288.0 ± 110.2 pg/mL, p<0.05 for both), and this increase did not persist over time (Figure 6). Under these conditions, there was no detectable production of MMP-9 or -12 from the ASMCs.
Fig. 6.
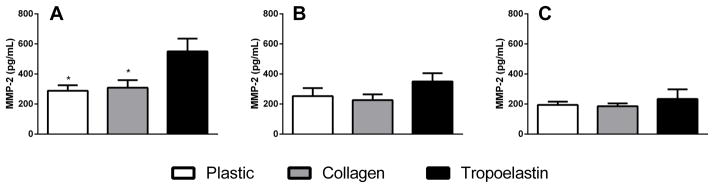
MMP-2 release (A) after pre-incubation with tropoelastin or collagen, (B) after 24 h and (C) after 48 h. *Significant difference due to tropoelastin by one-way ANOVA (n=3).
3. Discussion
A combination of TGF-β1 stimulation and tropoelastin exposure was found to increase the release of VEGF, while tropoelastin increased release of MMP-2 and decreased the sequestration of CTGF. This molecular crosstalk supports a role for elastin sequences in wound healing and angiogenesis, where these factors are typically found (Ferrara and Davis-Smyth, 1997; Ferrara et al., 2003; Shimo et al., 1999).
Angiogenesis is an important part of normal wound healing and aberrant angiogenesis plays a role in tumor development and diseases involving tissue remodeling such as asthma. The growth factor TGF-β1 plays a significant role in wound healing and angiogenesis (Douglas, 2010; Pardali et al., 2010), and in this study, TGF-β1 was used to stimulate ASMCs. We confirmed that TGF-β1 increases the release of soluble VEGF (Burgess et al., 2006a). VEGF is one of the most important factors inducing angiogenesis in physiology and pathology (Ferrara and Davis-Smyth, 1997; Ferrara et al., 2003). CTGF, while having innate angiogenic activity (Shimo et al., 1999), anchors VEGF in the matrix in various cell culture models, including ASMCs, and thereby modulates its release (Inoki et al., 2002; Burgess et al., 2006a). The model is robust as demonstrated by upregulation of CTGF by TGF-β1 and would be expected to maintain the regulatory balance of these factors (Igarashi et al., 1993; Burgess et al., 2006a; Burgess et al., 2006b).
When the cells were exposed to tropoelastin prior to addition of TGF-β1, the levels of sequestered CTGF decreased, while TGF-β1-dependent release of soluble VEGF further increased. These effects are in accord with a model where the normal balance of VEGF release is disrupted by elastin, leading to aberrant angiogenesis. Our findings support the concept that elastin participates in the regulatory control of angiogenesis and so enhances understanding of the underlying mechanism. These findings help to explain why in a chick chorio-allantoic membrane, the application of elastin-derived peptides directly increases the microvessel density (Robinet et al., 2005) and in a model of aberrant angiogenesis (abdominal aortic aneurysm in rats), elastin-derived peptides increase vessel diameter and wall thickness (Nackman et al., 1997).
We found that enhanced CTGF levels following elastin sequence exposure was independent of TGF-β1, as cells not stimulated with TGF-β1 decreased CTGF sequestration if they were pre-exposed to tropoelastin, and this was accompanied by no change in VEGF levels. Similarly, 48 h exposure to collagen reduced the amount of sequestered CTGF, but had no effect on VEGF release levels. Neither elastin nor collagen affected mRNA levels, implying these effects are post-transcriptional. In addition, tropoelastin alone, in the absence of TGF-β1 stimulation, caused an increase in release of MMP-2. This effect was observed immediately following the pre-incubation stage, and did not persist at further timepoints. We have previously found MMP-2 plays a role in digesting CTGF and releasing VEGF from matrix deposited by ASMCs (Burgess et al., 2006a). These new data suggest a possible mechanism for VEGF release and provide further support for our theoretical model in which elastin exposure promotes aberrant remodelling.
A model representing the potential role of elastin in regulating angiogenesis is presented in Fig. 7, where exposed elastin sequences increase levels of MMP-2, contributing to digestion of CTGF and, in combination with TGF-β1, increase of free VEGF, as well as further degradation of the ECM, including elastin. We cannot exclude the possibility that there was a contributory post-transcriptional down-regulation of CTGF.
Fig. 7.
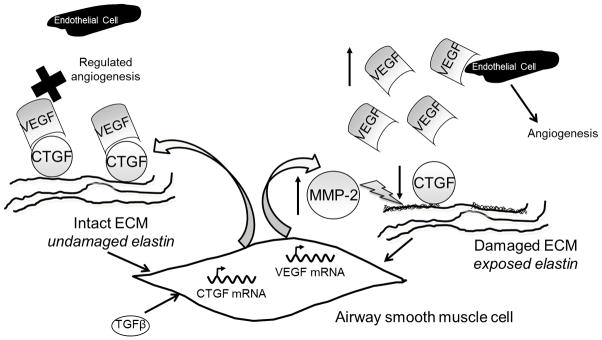
Schematic representing the proposed effect of elastin on angiogenic factors. In a normal airway, a balance between CTGF and VEGF regulates angiogenesis (indicated by a cross). In a damaged or diseased airway, exposure of elastin fragments increases MMP-2 production and thereby disrupts this inherent braking mechanism within the angiogenic process, increasing release of VEGF and leading to further degradation of the matrix.
Our findings strengthen the case for a specific network of effects of elastin sequences (Duca et al., 2004; Turino et al., 2011). The outcome of imbalanced factors known to regulate angiogenesis, as demonstrated here, could theoretically lead to aberrant angiogenesis and/or vascular remodeling. In diseases where elastin is damaged, structural effects should be appreciated as only part of the story.
4. Experimental Procedures
4.1 Proteins
Recombinant human tropoelastin isoform SHELΔ26A (Synthetic Human Elastin without domain 26A), corresponding to amino acid residues 27–724 of GenBank entry AAC98394 (gi 182020), was purified from bacteria on a multigram scale, as previously described (Martin et al., 1995) (Wu et al., 1999). Gelatin was from porcine skin (Type A; Sigma). Rat tail collagen I was from Becton & Dickinson and TGF-β1 from R&D Systems.
4.2 Cell culture
ASM cells were obtained from isolated human airway tissue from explanted/resected lungs or deep endobronchial biopsies obtained by flexible bronchoscopy as previously described (Burgess et al., 2003). Patient characteristics are presented in Table 1. Approval for all experiments with human lung was provided by the Human Ethics Committee of the University of Sydney and the Sydney South West Area Health Service, and all patients provided written informed consent. Cells were cultured using Dulbecco’s modified Eagle’s medium (DMEM) with 10% (v/v) fetal bovine serum (FBS), and 1% (v/v) Penicillin-Streptomycin (Gibco BRL). ASMCs were used between passages 4–8 for all experiments.
Table 1.
| Patient | Age | Gender | Disease |
|---|---|---|---|
| 1 | 45 | M | Cystic Fibrosis |
| 2 | 59 | M | Cancer |
| 3 | 44 | F | Emphysema |
| 4 | 39 | M | Rheumatoid pulmonary fibrosis |
| 5 | 57 | M | Pulmonary Fibrosis |
| 6 | 38 | M | Asthma |
| 7 | 30 | M | Cystic fibrosis |
| 8 | 77 | M | NSCCa (non small cell Ca) |
| 9 | 53 | M | Bronchiectasis |
| 10 | 71 | M | SCCa(small cell Ca), COPD |
| 11 | 64 | M | Pulmonary Fibrosis |
| 12 | 66 | F | Non squamous cell carcinoma |
| 13 | 60 | M | Cancer |
4.3 Confocal microscopy
Glass coverslips were coated with tropoelastin or gelatin by soluble protein coating, or by a thin layer of fibers formed by electrospinning. For electrospinning, each protein was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFP) at 20% (w/v), solutions were loaded into a 1 ml plastic syringe equipped with a blunt 18 gauge needle, and flow rate of 1 ml/h was applied using a syringe pump (World Precision Instruments). The needle was connected to the positive output of a high voltage power supply at 20 kV with an air gap distance between the needle and collector of ~20 cm. Fibers were cross-linked for 4 h in a solution of 10% (v/v) 4,4′-diisocyanato-methylenedicyclohexane (HMDI, Sigma) in isopropanol (Univar). Following cross-linking, the HMDI solution was removed by sequential washing in decreasing ratios of isopropanol:phosphate buffered saline (PBS) then washed three times in sterile PBS and allowed to equilibrate in PBS overnight. ASM cells were seeded at 1 – 5 × 103 cells/cm2 in 5% (v/v) FBS in DMEM.
6 to 24 h after cell seeding onto scaffolds, media were aspirated and samples were fixed with 3% (w/v) formaldehyde (Sigma) in PBS at room temperature for 20 min, then washed 3 times in PBS and quenched with 0.2 M glycine (Ajax Finechem) for > 20 min at room temperature. Cells were permeabilized by addition of 0.2% (v/v) Triton-X-100 (Sigma) for 6 min, and samples were washed twice in PBS before being blocked with 5% (w/v) BSA (Sigma) for 1 h at room temperature. Samples were washed three times in PBS then incubated in TRITC labeled Phalloidin Conjugate stain (phalloidin tetramethylrhodamine B isothiocyanate, Sigma) 1:1000 in 3% (w/v) BSA for 40 min to visualize F-actin. Samples were briefly rinsed with PBS, stained with 300 nM DAPI stain (Sigma) in water for 5 min to visualize cell nuclei, then washed and mounted onto glass slides using Fluoromount Aqueous Mounting Medium (Sigma).
Images were taken at the Australian Centre for Microscopy and Microanalysis, University of Sydney, using an Olympus Fluoview FV1000 confocal laser scanning biological microscope. Tropoelastin autofluorescence was utilized with excitation at 488 nm (Loria et al., 1979), and a spectral curve taken to assess optimal emission wavelengths. The following wavelengths were utilized: DAPI stain was visualized with an excitation wavelength of 405 nm and an emission range of 425 – 465 nm, TRITC was visualized with an excitation wavelength of 559 nm and an emission range of 575 – 675 nm, partial 3D tropoelastin fibers were visualized with an excitation wavelength of 488 nm and emission range of 520 – 540 nm.
4.4 Stimulation with TGF-β1
Six- or 96-well tissue culture plates were coated with 300 μL/cm2 saturating solutions of 12.5 μM tropoelastin or 40 μg/mL rat tail collagen I or left uncoated, then incubated at 4°C overnight. Coating solutions were then removed, and wells were rinsed with sterile PBS. ASM cells were seeded on these treated surfaces at 2 – 4 × 103 cells/cm2 in 5% (v/v) FBS in DMEM and allowed to attach for 24 h. To avoid the influence of serum on expression and production of factors, cells were synchronized in 0.1% BSA (w/v) in DMEM for 24 h (Satoh et al., 1997) then stimulated for up to 48 h with 10 ng/mL TGF-β1 in 0.1% (w/v) BSA in DMEM. Unstimulated cells were maintained in 0.1% (w/v) BSA in DMEM.
4.5 Real-time reverse transcription-polymerase chain reaction
RNA was extracted using the RNeasy kit (Qiagen). The treated cells were lyzed in 6-well plates with Qiagen RNeasy RLT buffer with β-mercaptoethanol, frozen at −80°C, then thawed for total RNA extraction according to the manufacturer’s instructions. cDNA synthesis used an Invitrogen SuperScript III First-Strand Synthesis with random primers (New England Biolabs).
Real-time assays for VEGF (Hs00903129) and CTGF (Hs00170014) and endogenous ribosomal RNA control (18S rRNA) were from PE Applied Biosystems. Amplification was carried out in an ABI Prism 7000 Sequence Detection System (PE Applied Biosystems) with initial stages of 50°C for 2 min and 95°C for 10 min followed by 40 cycles of amplification (95°C for 15 s followed by 60°C for 1 min) in patients 1, 2, 4 and 9. TaqMan assays of 18S rRNA to normalize and negative controls were included. Cycle thresholds (CTs) were assessed with Applied Biosystems 7000 system sequence detection software (v.1.2.3).
4.6 Protein assays
Levels of VEGF165 in conditioned media collected from 6-well plates were measured with a R&D Systems ELISA kit in patients 1–3 and 9–11. For analysis, measured levels were normalized to cell count determined by hemocytometer.
For detection of CTGF sequestered to the cell surface in patients 1–8, supernatants were removed from the 96-well plates containing treated cells and the plates were allowed to dry overnight at room temperature, then washed with 0.05% (v/v) Tween-20 (Sigma) in PBS (Tween-PBS), blocked with 1% BSA (w/v) in PBS for 60 min at room temperature, and washed again. Primary antibody goat anti-CTGF was added to half the wells, while to the other half an IgG control was added (Santa Cruz Biotechnology), both at 1 μg/mL in 1% BSA (w/v) in PBS, and allowed to bind overnight at 4°C. The wells were subsequently washed and the secondary antibody (polyclonal rabbit anti-goat horseradish peroxidase, Dako) was added at 1:2000 in 1% BSA (w/v) in Tween-PBS for 1 h. Wells were washed and TMB Single Solution (Zymed) added. After 10 to 30 min reactions were stopped by the addition of an equal volume of 1 M phosphoric acid (R&D Systems) and absorbances were read at 450 nm.
Total MMP-2, MMP-9 and MMP-12 were measured in patients 3, 10 and 11 with Fluorokine MAP human MMP kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. Briefly, ASM supernatants were 5-fold diluted and incubated with MMPs microparticle mixture for 2 hours at room temperature followed by incubation with Biotin antibody and Streptavidin-PE respectively. Concentration of MMPs (pg/ml) was read by Luminex 100/200 analyzer (Luminex, Brisbane, AUS).
4.7 Statistical analyses
All VEGF and CTGF results were normalized to the corresponding TGF-β1 negative time-matched no-protein control surface by calculating fold change, and two-way ANOVA using Bonferroni post-tests were performed. MMP-2 results were analyzed by one-way ANOVA at each time using Bonferroni post-tests. All data were assessed as mean ± standard deviation and statistical analyses performed using GraphPad Prism v5.0 for Windows. Significant differences were accepted when p<0.05.
Acknowledgments
We acknowledge the collaborative effort of the cardiopulmonary transplant team and the pathologists at St Vincent’s Hospital, Sydney and the thoracic physicians and pathologists at Royal Prince Alfred Hospital, Concord Hospital and Strathfield Private Hospital and Rhodes Pathology, Sydney. We also acknowledge Joanne Thompson for excellent technical assistance.
Grants
CJR was funded by the National Health and Medical Research Council under a Biomedical Postgraduate Scholarship #358851. ASW acknowledges funding from the Australian Research Council, Australian Defense Health Foundation, National Health & Medical Research Council, National Heart Foundation and National Institutes of Health NIH EB014283. ASW is the Scientific Founder of Elastagen Pty Ltd. JKB is supported by a National Health and Medical Research Council, Australia Career Development Fellowship #1032695.
Abbreviations
- ASMC
Airway smooth muscle cell
- TGF
transforming growth factor
- VEGF
vascular endothelial growth factor
- CTGF
connective tissue growth factor
Footnotes
Author contributions:
C.J. Reddel: study design, data collection, statistical analysis, data interpretation, manuscript preparation
D. Cultrone: data collection, statistical analysis, manuscript preparation
J. Rnjak-Kovacina: data collection, manuscript preparation
A.S. Weiss: study design, data interpretation, manuscript preparation
J.K. Burgess: study design, data interpretation, manuscript preparation
Contributor Information
Caroline J. Reddel, Email: creddel@anzac.edu.au.
Daniele Cultrone, Email: danielcultrone@gmail.com.
Jelena Rnjak-Kovacina, Email: jelena.rnjak@tufts.edu.
Anthony S. Weiss, Email: anthony.weiss@sydney.edu.au.
Janette K. Burgess, Email: janette.burgess@sydney.edu.au.
References
- Black PN, Ching PS, Beaumont B, Ranasinghe S, Taylor G, Merrilees MJ. Changes in elastic fibres in the small airways and alveoli in COPD. Eur Respir J. 2008;31:998–1004. doi: 10.1183/09031936.00017207. [DOI] [PubMed] [Google Scholar]
- Brassart B, Fuchs P, Huet E, Alix AJ, Wallach J, Tamburro AM, Delacoux F, Haye B, Emonard H, Hornebeck W, Debelle L. Conformational dependence of collagenase (matrix metalloproteinase-1) up-regulation by elastin peptides in cultured fibroblasts. J Biol Chem. 2001;276:5222–5227. doi: 10.1074/jbc.M003642200. [DOI] [PubMed] [Google Scholar]
- Burgess JK, Ge Q, Poniris MH, Boustany S, Twigg SM, Black JL, Johnson PR. Connective tissue growth factor and vascular endothelial growth factor from airway smooth muscle interact with the extracellular matrix. Am J Physiol Lung Cell Mol Physiol. 2006a;290:L153–161. doi: 10.1152/ajplung.00287.2005. [DOI] [PubMed] [Google Scholar]
- Burgess JK, Johnson PR, Ge Q, Au WW, Poniris MH, McParland BE, King G, Roth M, Black JL. Expression of connective tissue growth factor in asthmatic airway smooth muscle cells. Am J Respir Crit Care Med. 2003;167:71–77. doi: 10.1164/rccm.200205-416OC. [DOI] [PubMed] [Google Scholar]
- Burgess JK, Oliver BG, Poniris MH, Ge Q, Boustany S, Cox N, Moir LM, Johnson PR, Black JL. A phosphodiesterase 4 inhibitor inhibits matrix protein deposition in airways in vitro. J Allergy Clin Immunol. 2006b;118:649–657. doi: 10.1016/j.jaci.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Cozlin A, Barthelemy S, Garnotel R, Antonicelli F, Kaplan H, Hornebeck W, Lorimier S. Elastolysis induces collagenolysis in a gingival lamina propria model. J Dent Res. 2006;85:745–750. doi: 10.1177/154405910608500811. [DOI] [PubMed] [Google Scholar]
- Douglas HE. TGF-β in wound healing: a review. J Wound Care. 2010;19:403–406. doi: 10.12968/jowc.2010.19.9.78235. [DOI] [PubMed] [Google Scholar]
- Duca L, Floquet N, Alix AJ, Haye B, Debelle L. Elastin as a matrikine. Crit Rev Oncol Hematol. 2004;49:235–244. doi: 10.1016/j.critrevonc.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Fulop T, Jr, Jacob MP, Khalil A, Wallach J, Robert L. Biological effects of elastin peptides. Pathol Biol (Paris) 1998;46:497–506. [PubMed] [Google Scholar]
- Hashimoto G, Inoki I, Fujii Y, Aoki T, Ikeda E, Okada Y. Matrix metalloproteinases cleave connective tissue growth factor and reactivate angiogenic activity of vascular endothelial growth factor 165. J Biol Chem. 2002;277:36288–36295. doi: 10.1074/jbc.M201674200. [DOI] [PubMed] [Google Scholar]
- Heinz A, Jung MC, Duca L, Sippl W, Taddese S, Ihling C, Rusciani A, Jahreis G, Weiss AS, Neubert RH, Schmelzer CE. Degradation of tropoelastin by matrix metalloproteinases--cleavage site specificities and release of matrikines. FEBS J. 2010;277:1939–1956. doi: 10.1111/j.1742-4658.2010.07616.x. [DOI] [PubMed] [Google Scholar]
- Hornebeck W, Tixier JM, Robert L. Inducible adhesion of mesenchymal cells to elastic fibers: elastonectin. Proc Natl Acad Sci U S A. 1986;83:5517–5520. doi: 10.1073/pnas.83.15.5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, Mecham RP, Senior RM, Shapiro SD. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116:753–759. doi: 10.1172/JCI25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Reymond JL, Pinel N, Zabot MT, Urban Z. Inflammatory destruction of elastic fibers in acquired cutis laxa is associated with missense alleles in the elastin and fibulin-5 genes. J Invest Dermatol. 2006;126:283–290. doi: 10.1038/sj.jid.5700047. [DOI] [PubMed] [Google Scholar]
- Igarashi A, Okochi H, Bradham DM, Grotendorst GR. Regulation of connective tissue growth factor gene expression in human skin fibroblasts and during wound repair. Mol Biol Cell. 1993;4:637–645. doi: 10.1091/mbc.4.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki I, Shiomi T, Hashimoto G, Enomoto H, Nakamura H, Makino K, Ikeda E, Takata S, Kobayashi K, Okada Y. Connective tissue growth factor binds vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. FASEB J. 2002;16:219–221. doi: 10.1096/fj.01-0332fje. [DOI] [PubMed] [Google Scholar]
- Kelleher CM, Silverman EK, Broekelmann T, Litonjua AA, Hernandez M, Sylvia JS, Stoler J, Reilly JJ, Chapman HA, Speizer FE, Weiss ST, Mecham RP, Raby BA. A functional mutation in the terminal exon of elastin in severe, early-onset chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2005;33:355–362. doi: 10.1165/rcmb.2005-0206OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria RM, Kos WL, Campbell AE, Madge GE. Suppression of aortic elastic tissue autofluorescence for the detection of viral antigen. Histochemistry. 1979;61:151–155. doi: 10.1007/BF00496527. [DOI] [PubMed] [Google Scholar]
- Martin SL, Vrhovski B, Weiss AS. Total synthesis and expression in Escherichia coli of a gene encoding human tropoelastin. Gene. 1995;154:159–166. doi: 10.1016/0378-1119(94)00848-m. [DOI] [PubMed] [Google Scholar]
- Mauad T, Xavier AC, Saldiva PH, Dolhnikoff M. Elastosis and fragmentation of fibers of the elastic system in fatal asthma. Am J Respir Crit Care Med. 1999;160:968–975. doi: 10.1164/ajrccm.160.3.9809088. [DOI] [PubMed] [Google Scholar]
- Mithieux SM, Weiss AS. Adv Protein Chem. Academic Press; 2005. Elastin; pp. 437–461. [DOI] [PubMed] [Google Scholar]
- Mochizuki S, Brassart B, Hinek A. Signaling pathways transduced through the elastin receptor facilitate proliferation of arterial smooth muscle cells. J Biol Chem. 2002;277:44854–44863. doi: 10.1074/jbc.M205630200. [DOI] [PubMed] [Google Scholar]
- Nackman GB, Karkowski FJ, Halpern VJ, Gaetz HP, Tilson MD. Elastin degradation products induce adventitial angiogenesis in the Anidjar/Dobrin rat aneurysm model. Surgery. 1997;122:39–44. doi: 10.1016/s0039-6060(97)90262-2. [DOI] [PubMed] [Google Scholar]
- Pardali E, Goumans MJ, ten Dijke P. Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol. 2010;20:556–567. doi: 10.1016/j.tcb.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Reddel CJ, Weiss AS, Burgess JK. Elastin in asthma. Pulm Pharmacol Ther. 2012;25:144–153. doi: 10.1016/j.pupt.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Ringpfeil F. Selected disorders of connective tissue: pseudoxanthoma elasticum, cutis laxa, and lipoid proteinosis. Clin Dermatol. 2005;23:41–46. doi: 10.1016/j.clindermatol.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Robert L. Cell-elastin interaction and signaling. Pathol Biol (Paris) 2005;53:399–404. doi: 10.1016/j.patbio.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Robinet A, Fahem A, Cauchard JH, Huet E, Vincent L, Lorimier S, Antonicelli F, Soria C, Crepin M, Hornebeck W, Bellon G. Elastin-derived peptides enhance angiogenesis by promoting endothelial cell migration and tubulogenesis through upregulation of MT1-MMP. J Cell Sci. 2005;118:343–356. doi: 10.1242/jcs.01613. [DOI] [PubMed] [Google Scholar]
- Satoh H, Togo M, Hara M, Miyata T, Han K, Maekawa H, Ohno M, Hashimoto Y, Kurokawa K, Watanabe T. Advanced glycation endproducts stimulate mitogen-activated protein kinase and proliferation in rabbit vascular smooth muscle cells. Biochem Biophys Res Commun. 1997;239:111–115. doi: 10.1006/bbrc.1997.7424. [DOI] [PubMed] [Google Scholar]
- Senior RM, Griffin GL, Mecham RP. Chemotactic activity of elastin-derived peptides. J Clin Invest. 1980;66:859–862. doi: 10.1172/JCI109926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senior RM, Griffin GL, Mecham RP. Chemotactic responses of fibroblasts to tropoelastin and elastin-derived peptides. J Clin Invest. 1982;70:614–618. doi: 10.1172/JCI110654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro SD, Ingenito EP. The pathogenesis of chronic obstructive pulmonary disease: advances in the past 100 years. Am J Respir Cell Mol Biol. 2005;32:367–372. doi: 10.1165/rcmb.F296. [DOI] [PubMed] [Google Scholar]
- Shimo T, Nakanishi T, Nishida T, Asano M, Kanyama M, Kuboki T, Tamatani T, Tezuka K, Takemura M, Matsumura T, Takigawa M. Connective tissue growth factor induces the proliferation, migration, and tube formation of vascular endothelial cells in vitro, and angiogenesis in vivo. J Biochem (Tokyo) 1999;126:137–145. doi: 10.1093/oxfordjournals.jbchem.a022414. [DOI] [PubMed] [Google Scholar]
- Shiratsuchi E, Ura M, Nakaba M, Maeda I, Okamoto K. Elastin peptides prepared from piscine and mammalian elastic tissues inhibit collagen-induced platelet aggregation and stimulate migration and proliferation of human skin fibroblasts. J Pept Sci. 2010;16:652–658. doi: 10.1002/psc.1277. [DOI] [PubMed] [Google Scholar]
- Skeie JM, Mullins RF. Elastin-mediated choroidal endothelial cell migration: possible role in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2008;49:5574–5580. doi: 10.1167/iovs.08-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima S, Wachi H, Uemura Y, Okamoto K. Modulation by elastin peptide VGVAPG of cell proliferation and elastin expression in human skin fibroblasts. Arch Dermatol Res. 1997;289:489–492. doi: 10.1007/s004030050227. [DOI] [PubMed] [Google Scholar]
- Turino GM, Ma S, Lin YY, Cantor JO, Luisetti M. Matrix Elastin: A Promising Biomarker for COPD. Am J Respir Crit Care Med. 2011 doi: 10.1164/rccm.201103-0450PP. [DOI] [PubMed] [Google Scholar]
- Wagenseil JE, Mecham RP. New insights into elastic fiber assembly. Birth Defects Res C Embryo Today. 2007;81:229–240. doi: 10.1002/bdrc.20111. [DOI] [PubMed] [Google Scholar]
- Wu WJ, Vrhovski B, Weiss AS. Glycosaminoglycans mediate the coacervation of human tropoelastin through dominant charge interactions involving lysine side chains. J Biol Chem. 1999;274:21719–21724. doi: 10.1074/jbc.274.31.21719. [DOI] [PubMed] [Google Scholar]