

Abstract
RNases H are involved in the removal of RNA from RNA/DNA hybrids. Type I RNases H are thought to recognize and cleave the RNA/DNA duplex when at least four ribonucleotides are present. Here we investigated the importance of RNase H type I encoding genes for model organism Mycobacterium smegmatis. By performing gene replacement through homologous recombination, we demonstrate that each of the two presumable RNase H type I encoding genes, rnhA and MSMEG4305, can be removed from M. smegmatis genome without affecting the growth rate of the mutant. Further, we demonstrate that deletion of both RNases H type I encoding genes in M. smegmatis leads to synthetic lethality. Finally, we question the possibility of existence of RNase HI related alternative mode of initiation of DNA replication in M. smegmatis, the process initially discovered in Escherichia coli. We suspect that synthetic lethality of double mutant lacking RNases H type I is caused by formation of R-loops leading to collapse of replication forks. We report Mycobacterium smegmatis as the first bacterial species, where function of RNase H type I has been found essential.
Introduction
RNases H are ubiquitous enzymes present is eukaryotes, prokaryotes, archaeons and viruses. They are involved in the removal of RNA from RNA/DNA hybrids during DNA replication, repair and transcription. Based on the differences in their amino acid sequences, RNases H have been divided into two types and three classes. Prokaryotic RNase HI or eukaryotic H1 represent RNases H type 1, while prokaryotic RNases HII and HIII or eukaryotic RNases H2 represent RNases H type 2.
Type 1 RNases H require at least four ribonucleotides as a substrate for a cleavage to occur [1,2] and they do not prefer any consensus sequence to perform this reaction [1]. In vitro studies showed that introducing various modifications that decrease DNA flexibility in RNA/DNA duplex abrogates cleavage by RNase H1 [3]. Thus, the enzyme must recognize both RNA and DNA strands. This observation was confirmed by obtaining co-crystal structure of RNase HI of Bacillus halodurans in complex with RNA/DNA [4]. In contrast, RNases H type II recognize and cleave single ribonucleotides within RNA/DNA duplex and are thought to recognize the transition from deoxiribonucleotides to ribonucleotides on a single strand [5,6] (Fig 1).
Fig 1. Schematic representation of a mode of action of RNases H.
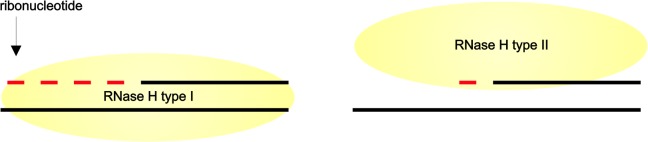
RNases H cleave RNA from the RNA/DNA duplex. RNases H type I recognize two strands of the heteroduplex and cleave RNA when at least four ribonucleotides are present. In contrast, RNases H type II cleave even single ribonucleotides and recognize transition from DNA to RNA on a single strand.
RNases H type I recognize two substrate types. The first type is RNA primers generated during DNA synthesis. The second type is R-loops: a three strand nucleic acid structures consisting of an RNA/DNA hybrid and a displaced DNA strand. It is thought that such loops occur as a consequence of transcription, when a nascent transcript anneals to the matrix DNA. The presence of R-loops has been reported in several processes, including DNA replication [7–9], transcription termination [10], regulation of gene expression [11,12] and other processes specific to eukaryotes [13–15]. R-loops can achieve considerable length, as they for example exceed 1 kbp at immunoglobulin class switch region [15]. In fact, RNases H type I are thought to be likely to act on relatively stable, transcription associated R-loops [16,17].
To date, higher eukaryotes have been shown to possess one RNase H type 1 and one RNase H type 2 (composed of three subunits). Both have been shown essential for survival, even at the stage of embryogenesis [18,19]. Saccharomyces cerevisiae also possesses two RNases H–one RNase H type 1 and one RNase H type 2 composed of three subunits–but both are dispensable for growth [20]. In contrast, the number of RNases H encoded in bacterial genomes is more variable [21], and more complicated in terms of being essential for viability. For example, Escherichia coli possesses one RNase HI and one RNase HII [22], while the genome of Bacillus subtilis contains two RNase H type 2- RNase HII and RNase HIII [23]. In both of these species, there are contradictory reports regarding essentiality of RNase H enzymes [24–31], however, most recent studies show that they are dispensable under certain conditions [25–27,30,31]. RNase HI has also been shown dispensable in Haemophilus influenzae [32]. The genome of Mycobacterium tuberculosis contains one gene encoding an RNase H type II, rnhB [33], and one gene encoding a bifunctional protein, Rv2228c. Its N-terminal domain is homologous with eukaryotic and prokaryotic RNases H type I, while C-terminal domain is homologous with alpha ribazole phosphatase (CobC) involved in cobalamin (vitamin B12, B12) biosynthesis [34]. Recombinant protein expression confirmed the activity of both domains in vitro (in an artificially set up reaction between the enzyme and the substrate) [34]. In turn, the genome of Mycobacterium smegmatis seems to encode four RNases H. Two of them belong to RNases H type I. The first one is encoded by rnhA and the RNase HI activity of the derived protein has been confirmed in vitro [35]. The second is a homolog of Rv2228c of M. tuberculosis, MSMEG4305. Further, the genome of M. smegmatis encodes an RNase H type II, through gene rnhB. Additionally, the gene MSMEG5849 of M. smegmatis encodes a protein which presents RNase HII activity through domain Duf429 (until recently referred to as a domain of unknown function) [36]. A protein with limited homology to Duf429 can be found in M. tuberculosis. The summary of presumable RNase H encoding genes in E. coli, M. tuberculosis and M. smegmatis is presented in Table 1.
Table 1. Summary of presumable RNases H of E. coli K12_MG1655, M. tuberculosis H37Rv and M. smegmatis mc2 155.
| Type of RNase H | E. coli K12_MG1655 | M. tuberculosis H37Rv | M. smegmatis mc2 155 |
|---|---|---|---|
| I | RnhA | Homolog is absent | RnhA |
| I | Homolog with RNase HI domain is absent; CobC (homology with acid phosphatase domain) | Rv2228c | MSMEG4305 |
| II | RnhB | RnhB | RnhB |
| II | Homolog is absent | MT0800 (homology with Duf429 domain) | MSMEG5849 |
The aim of this study was to understand the importance of RNases H type I for M. smegmatis. M. smegmatis is a model organism to study Mycobacterium genus, which includes M. tuberculosis. We demonstrate that deletion of both RNases H type I encoding genes leads to synthetic lethality. Further, we demonstrate that deletion of each RNase H type I did not alter growth rates of the mutants. Finally, we question the possibility of existence of RNase HI related alternative mode of initiation of DNA replication in M. smegmatis, the process initially discovered in E. coli. We suspect that RNases H type I may be essential for the survival of M. smegmatis due to formation of R-loops, which when unresolved, collapse replication forks.
Materials and Methods
In silico analysis
Genes presumably encoding RNases H in M. smegmatis mc2 155, E. coli K12_MG1655 and M. tuberculosis H37Rv were identified and obtained from National Center for Biotechnology Information (NCBI) database. The span of the domains was defined using Simple Modular Architecture Research Tool (SMART) [37]. The homology between the domains was estimated using Basic Local Alignment Search Tool (BLAST) of NCBI. The alignments were visualized using MultiAlin [38] and ESPript 3.0 [39].
Bacteria and culture conditions
Cultures of E. coli T10 were carried out at 37°C for 18–20 h in liquid Luria-Bertani broth, and when necessary supplemented with antibiotics or other supplements or both at the following concentrations: kanamycin (Bioshop) 50 μg/ml; ampicillin (Bioshop) 100 μg/ml; X-gal (Sigma) 40 μg/ml, IPTG 0.4 mM (Sigma). Cultures of M. smegmatis during gene replacement were carried out in nutrient broth (NB) (Difco) or 7H9 broth (Becton, Dickinson and Company) with or without oleic albumin dextrose catalase growth supplement (OADC) (Becton- Dickinson) and 0.05% Tween 80 (Sigma) at 37°C or 28°C. When necessary, media were supplemented with antibiotics or other supplements or both at the following concentrations: kanamycin (Sigma) 25 μg/ml; X-gal (Sigma) 40 μg/ml, 0.4% succinate (Sigma), sucrose 2% (Sigma), vitamin B12 10 μg/ml (Sigma). A list of M. smegmatis strains used in this study is presented in Table 2.
Table 2. List of M. smegmatis strains used in this study.
| Strain | Characteristics |
|---|---|
| M. smegmatis mc2 155 | Reference strain |
| ∆rnhA | rnhA deletion mutant of M. smegmatis mc2 155 |
| ∆4305 | MSMEG4305 deletion mutant of M. smegmatis mc2 155 |
| ∆rnhA/∆4305attB::Pami-rnhA | derivative of M. smegmatis mc2 155 carrying deletions within rnhA and MSMEG4305 complemented with full rnhA gene under acetamide promoter at attB site, HygR |
| ∆rnhA/∆4305attB::Pami-4305 | derivative of M. smegmatis mc2 155 carrying deletions within rnhA and MSMEG4305 complemented with full MSMEG4305 gene under acetamide promoter at attB site, HygR |
| ∆rnhA/∆dnaAattB::dnaA | derivative of M. smegmatis mc2 155 carrying deletions within rnhA and dnaA complemented with full dnaA gene under natural promoter at attB site, GmR, HygR |
For determination of growth rates bacterial cells were transferred to fresh NB medium and cultured until the cultures reached OD600 between 0.6 and 0.9. Aliquots of these seed cultures were inoculated in fresh 7H9 broth supplemented with OADC at starting OD600 = 0.05. The cultures were incubated at 37°C with vigorous shaking for 48 h. At desired intervals of time samples of cultures were harvested and analyzed using spectrophotometer (Pharmacia Biotech Ultrospec 2000). To assess the number of the colony forming units, samples were serially diluted in fresh NB broth and plated on non-selective NB medium, which were incubated at 37°C until obtaining visible colonies. Each experiment was performed at least in triplicate. For determination of cell length drops, the cultures harvested from 24h cultures were placed on glass slides, fixed in flames and analyzed on Nikon Eclipse TE2000 microscope.
M. smegmatis mutants
The mutants were obtained by using gene replacement protocol as previously described [40–42]. The procedure required construction of gene replacement plasmids and complementation plasmids. Primers used for obtaining the mutants are listed in Table 3.
Table 3. Primers used in this study.
| Construction of plasmids used for gene replacement | |||||
| Name | Sequence | Primer pair | Template | intro-duced res-triction sites | Purpose |
| MsGR1rnhA | TCGAGGGCAAGCTGCGCGAC | MsGR2rnhA | mc2 155 | - | gene replacement rnhA |
| MsGR2rnhA | CGTAGCACCGCACCCCAGCC | MsGR1rnhA | mc2 155 | - | gene replacement rnhA |
| MsGR3rnhA | CGGGATCCGTGCGCGCGCCACCAGGTC | MsGR4rnhA | mc2 155 | BamHI | gene replacement rnhA |
| MsGR4rnhA | GAAGCTTCCGCGAGGGGCCGAACACC | MsGR3rnhA | mc2 155 | HindIII | gene replacement rnhA |
| GR1MsRnhAII-KpnI | GGTACCCCGCCGACGATGATGCTGTC | GR2MsRnhAII-BamH | mc2 155 | KpnI | gene replacement MSMEG4305 |
| GR2MsRnhAII-BamH | CAAGCGGCGCAACGGGATC | GR1MsRnhAII-KpnI | mc2 155 | - | gene replacement MSMEG4305 |
| GR3MsRnhAII-BamH | CGGGATCCTACACAACCGCGCCGTAGCC | GR4MsRnhAII-Hind | mc2 155 | BamHI | gene replacement MSMEG4305 |
| GR4MsRnhAII-Hind | GCAAGCTTTCGCTGCTGGGTGCCGTGAC | GR3MsRnhAII-BamH | mc2 155 | HindIII | gene replacement MSMEG4305 |
| GmBstBs | CTTCGAAGGCTGACGGAATTTATGCCTCTTC | GmBstBr | pINT3 | BstBI | gentamycin resistance cassette |
| GmBstBr | CTTCGAACAGGAATCGAATGCAACCGG | GmBstBs | pINT3 | BstBI | gentamycin resistance cassette |
| MsA1-5562-BglIIs | CAGATCTGTGAACCACCGGCACCACGCC | MsA1-5562-XbaI | mc2 155 | BglII | complementation rnhA |
| MsA1-5562-XbaI | CTCTAGATGGTGGTCGGCCTGGCGGG | MsA1-5562-BglIIs | mc2 155 | XbaI | complementation rnhA |
| MsrnhAIIPace-sBglII | CAGATCTGTGAAGGTTCTCGTCGAGGCCGAC | MsrnhAII-rev-Xba | mc2 155 | BglII | complementation MSMEG4305 |
| MsrnhAII-rev-Xba | CTCTAGATGCACTCGTGAGCTACAGGTACGC | MsrnhAIIPace-sBglII | mc2 155 | XbaI | complementation MSMEG4305 |
| MsdnaA+PrHindIIInat | CTGTCGATCAGACGCGCCCAC | MsrnhAII-rev-Xba | mc2 155 | - | complementation dnaA |
| MsdnaA+PrXbaIrev | CTCTAGATCTCCGAGCTCAGCGTTTGGC | MsdnaA+PrHindIIInat | mc2 155 | XbaI | complementation dnaA |
| Construction of plasmid for recombinant protein expression | |||||
| Name | Sequence | Primer pair | Template | intro-duced res-triction sites | purpose |
| RnhA-f | CGAATTCGTGAACCACCGGCACCACGCC | RnhA-r | mc2 155 | EcoRI | recombinant protein |
| RnhA-r | CAAGCTTGGTGGTCGGCCTGGCGGG | RnhA-f | mc2 155 | HindIII | recombinant protein |
| Southern blotting | |||||
| gene name | primer name | Sequence | primer pair | restriction enzyme | |
| rnhA before comple-menta-tion | coDCOH1s | GTGAACCACCGGCACCACGCC | coDCOH1r | PvuI | |
| rnhA before comple-menta-tion | coDCOH1r | GGCGGGCAACAAGCTCAACGG | coDCOH1s | PvuI | |
| rnhA after comple-menta-tion | MsGR3rnhA | CGGGATCGGTGCGCGCGCCACCAGGTC | MsRnhA1probe-r-delikom | BamHI, PvuI, XbaI | |
| rnhA after comple-menta-tion | MsRnhA1probe-r-delikom | TGTGGGCCGGTGCGGTGG | MsGR3rnhA | BamHI, PvuI, XbaI | |
| MSMEG4305 | Ms4305-probe-s | GACAACGACGCCAGGTCCAGG | Ms4305-probe-r | PvuI | |
| MSMEG4305 | Ms4305-probe-r | GTGAAGGTTCTCGGTCGAGGCCG | Ms4305-probe-s | PvuI | |
| dnaA | Msdnaprobe-s | GCAAGAAGGCGCAGATGGATCG | MsdnaAprobe-r | ClaI, HindIII | |
| dnaA | MsdnaAprobe-r | GCGGATCTTCTTCTCGGCGTACATC | Msdnaprobe-s | ClaI, HindIII | |
Briefly, for gene replacement purpose, the sequences flanking desired deletion were amplified by PCR and consecutively introduced into p2NIL plasmid, followed by introduction of PacI suicidal cassette excised from pGOAL17 vector. For complementation under an acetamide promoter, a native copy of the gene of interest was amplified by PCR and introduced into pJAM2. The gene was next excised from the pJAM2 together with an acetamide promoter and introduced into pMV306Hyg. All cloning was performed in E. coli Top10 cells (Invitrogen). A plasmid allowing deletion in dnaA, previously used in [43], was modified by addition of gentamycin resistance cassette within BstBI restriction site. Plasmids were introduced into mycobacterial cells, which were further subjected to multistep selection process allowing detection of the mutants. Presence of intact copy of the dnaA gene within the ∆rnhA/∆dnaAattB::dnaA strain at the attB site was confirmed by sequencing.
Exchange of complementation vectors with genetic cassettes for an empty vector
Conditional mutants carrying a copy of a presumably essential gene were electroporated with an empty pMV306Km plasmid. For exchange of complementation vectors with genetic cassettes for an empty vector (ExEV) on rich medium, bacteria after transformation were inoculated in NB broth and cultivated overnight at 37°C with vigorous shaking. Afterwards, bacteria were plated on NB selective medium supplemented with Km and cultivated up to one week at 37°C. For ExEV on minimal medium, bacteria after transformation were inoculated in 7H9 broth without the addition of OADC and cultivated overnight at 28°C with vigorous shaking. Afterwards, bacteria were plated on 7H10 medium without the addition of OADC supplemented with Km and cultivated for up to three weeks at 28°C. In case of ExEV on cassettes containing MSMEG4305, an additional variant of the experiment was performed which included supplementation of both rich and poor media with B12. Clones growing on Km containing media were screened in search of an intact version of the gene of interest. Encountering a clone devoid of an intact version of the gene would mean that the gene is non-essential. On the other hand, inability to find such clones would confirm the essentiality of the gene.
Southern blotting
Primers used for production of hybridization probes and restriction enzymes used for digestion of genomic DNA are listed in Table 3.
Recombinant RnhA expression and purification
Sequence encoding RnhA, identified in NCBI database, was amplified by PCR (primers listed in Table 3) and introduced into pHIS2 expression plasmid. Cloning was performed in E. coli Top10 cells (Invitrogen). The plasmid was further introduced into E. coli BL21. Expression of the protein was performed at 37°C overnight in the presence of 0.4 mM IPTG. The pellet was resuspended in 10 ml of Binding Buffer (50 mM Tris-HCl pH 8 (Sigma); 6–8M urea (Sigma)) and sonicated in short bursts (Bioblock Scientific Vibracell). The addition of urea was necessary to denature and solubilize otherwise insoluble protein. Next, the sample was incubated for 2 h with mild shaking. The sample was centrifuged at 17000 x g at 12°C for 30 min. The supernatant was transferred through filtered syringe (0.45 μm, Millex) and placed on affinity column containing Ni-NTA resin (ThermoScientific). Following binding of the protein to the Ni-NTA resin, the column was washed with Binding Buffer and Wash Buffer (60 mM imidazole (Sigma), 0,4 M NaCl (Sigma), 20 mM Tris- HCl pH 8 (Sigma)). Next, the recombinant protein was washed out with Elution Buffer (1M imidazole (Sigma), 0.5 M NaCl (Sigma), 20 mM Tris- HCl pH 8 (Sigma)). The sample containing recombinant protein was concentrated on a Novagen concentrator until achieving the final concentration of 1 mg/ml. The protein was used to immunize rabbits and for production of polyclonal antibodies.
Production of anti- RnhA antibodies
Laboratory New Zealand rabbits were raised under standard conventional conditions in the approved by Polish Ministry of Science and Higher Education animal facility of the Institute Microbiology, Biotechnology and Immunology, Faculty of Biology and Environmental Protection, University of Lodz and were used for the immunization experiments. The experimental procedures were approved and conducted according to guidelines of the appropriate Polish Local Ethics Commission for Experiments on Animals No. 9 in Lodz (Agreement 54/ŁD1/2011).
Western blotting
Bacteria were cultured in NB medium until OD600 reached between 0.6 and 0.9. Aliquots of these seed cultures were inoculated in fresh 7H9 broth starting OD600 = 0.05. The cultures were incubated at 37°C with vigorous shaking overnight. The following morning 5 ml of each culture were spun down at 4°C at 8000 x g and the pellet was resuspended in 1 ml TE (10 mM Tris (Sigma), 1 mM EDTA; pH 8) with the addition of 100 mM phenylmethylsulfonyl fluoride (Sigma) and 2% SDS (Sigma). The sample was then transferred to MP tube and homogenized using FastPrep-24 MP homogenizer. Subsequently the sample incubated for 30 min at 55°C and centrifuged at 14000 x g at room temperature for 15 min. The supernatant containing proteins was transferred to a new Eppendorf tube and sample was immediately used.
Cell extracts or recombinant protein solutions were subjected to acrylamide electrophoresis. Following protein electrophoresis, the gel (12% Mini-Protean Precast Gel BioRad) was placed in a plastic container and covered with PVDF membrane (ThermoScientific) washed beforehand in methanol and Transfer Buffer (0,2 M glycine (Sigma); 25 mM Tris (Sigma); 20% methanol (Sigma)). The transfer was performed overnight at 4°C at 50 mA in an electrophoresis container filled with transfer buffer. Next the membrane was blocked for 1.5 h at room temperature in PBS (137 mM NaCl (Sigma); 2.7 mM KCl (Sigma); 10 mM Na2HPO4 • 2 H2O (Sigma); 2M KH2PO4 (Sigma) pH 7.4) containing 10% milk (Gostynin, fat- free powdered milk). Subsequently, the membrane was incubated in PBS containing 5% milk, 0.05% Tween 20 (Sigma) and primary antibodies (rabbit anti- RnhA, obtained at Department of Immunoparasitology, University of Lodz or rabbit anti- LigA, described previously [44]) at 4°C overnight. Afterwards, the membrane was washed three times in PBS with 0.05% Tween 20 (Sigma). The membrane was then placed in PBS containing 5% milk and a secondary antibody (anti-rabbit goat IgG conjugated with peroxidase (Sigma)). The membrane was incubated for 1 h at room temperature and washed three times with PBS containing 0.05% Tween 20. The membrane was then placed in a dry plastic container and covered with mixture of ECL reagent. Following a 2 minutes incubation the membrane was covered in Saran wrap and exposed to an X- ray film (ThermoScientific) for 2 minutes. The film was developed using Kodak Medical X-ray Processor.
Statistical analysis
In order to determine growth rates of the analysed strains, we fitted logistic or quadratic curves to the collected measurements of optical density of the liquid cultures. We fitted logistic curves of the form OD = A/[1 + B*exp(-KT)], where OD is optical density at the time T, A is an asymptotic value, B is a constant of integration, and K is the growth rate constant. Parameter K from the fitted curves was used as an indicator of growth rate for each strain. A T-test was used to test for the differences in growth rates between the strains. All statistical analyses were performed with Statistica 10.0 (StatSoft, Tulsa, OK, USA).
Results and Discussion
The genome of M. smegmatis encodes two predicted RNases H type I
Through performing an in silico analysis we identified two genes of M. smegmatis mc2 155 which encode proteins containing RNase HI domain- rnhA and MSMEG4305. BLAST analysis of RNase HI domains of RnhA and MSMEG4305 of M. smegmatis mc2 155 revealed that with 50% of query cover of RnhA, they share 36% of protein sequence identity. Next, MSMEG4305 was shown to be homologous to Rv2228c of M. tuberculosis H37Rv. Both proteins share 72% sequence identity with 100% query cover of MSMEG4305. RNase HI domains of MSMEG4305 and Rv2228c share 72% identity with 97% of MSMEG4305 query cover, while acid phosphatase domains share 75% sequence identity with 100% query cover of MSMEG4305. The alignment between protein sequences between RNases H type I of E. coli K12_MG1655, M. smegmatis mc2 155 and M. tuberculosis H37Rv is presented in Fig 2.
Fig 2. Comparison of protein sequences of RNases H type I.

Sequence alignment between RNases H type I of E. coli K12_MG1655 (RnhA), M. smegmatis mc2 155 (MSMEG4305 and RnhA) and M. tuberculosis H37Rv (Rv2228c) was performed using MultiAlin and visualized with ESPript 3.0. Highly similar or identical residues between protein sequences are written in bold. Identical residues across all analyzed sequences are shown in white on a black background. Similarities between protein sequences are marked by framing. The span of RNase H domains in each protein sequence, as defined by SMART, is highlighted in yellow.
A possible reason for the coexistence of two RNase H type I encoding genes within the genome of M. smegmatis is the neofunctionalization of MSMEG4305. Apart from encoding RNase HI domain it also encodes CobC domain involved in vitamin B12 biosynthesis. Such neofunctionalization is thought to increase retention of RNase H type I genes [21]. The coexistence of MSMEG4305 and rnhA might be expected to lead to future inactivation of RNase HI domain in one of them and generation of a pseudogene [45]. In fact, the genome of M. tuberculosis, which is thought to have undergone genome reduction when compared with free-living mycobacteria, contains only a homolog of MSMEG4305.
Essentiality of RNases H type I in M. smegmatis
We used the technique of gene replacement through homologous recombination to construct a series of genetic mutants with large deletions introduced within the sequence of predicted RNase H type I encoding genes. We were able to identify single mutants of both rnhA and MSMEG4305 (Fig 3). The ability to obtain the mutants signifies that none of these genes itself is essential for survival of M. smegmatis.
Fig 3. Southern blots confirming deletions in single RNase H type I mutants of M. smegmatis.

We used gene replacement through homologous recombination to obtain mutants deficient in either rnhA or MSMEG4305. Briefly, recombinant plasmids containing genomic regions of either rnhA or MSMEG4305 with large deletions within each gene were introduced into M. smegmatis mc2 155 cells. Following multistep selection we were able to identify clones where the native version of each gene has been replaced with manipulated sequence. Intermediate steps of gene replacement procedure are denoted SCO. For more information regarding plasmid construction and gene replacement procedure please refer to the text. Schematic representation of analyzed genomic regions, including enzymes used for digestion, size of restriction fragments following digestion and the site of hybridization of hybridization probe, is presented in the upper part of the figure. Photographic films presenting results of Southern blot analysis are presented in lower part of the figure. Bands corresponding to wild type genotype (wt) and mutant genotype (mut) are marked on the right side of each photograph.
We were unable to identify a mutant deficient in ∆rnhA/∆4305. Therefore, we introduced a functional copy of either MSMEG4305 or rnhA at the attB site of ∆rnhA/SCOMSMEG4305 or SCOrnhA/∆MSMEG4305, respectively. Following selection, we generated two strains ∆rnhA/∆4305attB::Pami-rnhA and ∆rnhA/∆4305attB::Pami-4305 deficient for both native versions of RNase HI encoding genes, but complemented with either rnhA or MSMEG4305 at the attB site (Fig 4). We used these strains to perform ExEV. This experiment greatly increases the number of analyzed cells. It is therefore used as a final confirmation of the essentiality of the gene [46–48]. It also limits drawbacks of gene replacement technique. In our case it was the enrichment of the medium by the substances used as selection markers. Enrichment of medium might be a source of confusion between contradictory reports regarding RNase HI essentiality in E. coli [29–31]. The authors that have obtained RNase HI deficient mutants stated that the mutants were rich broth sensitive [30,31]. This is thought to be related to high speed of metabolism. High metabolism requires efficient transcription, which leads to generation of many R-loops. R-loops, in turn, cannot be efficiently resolved in the absence of RNase HI and lead to replication fork collapse. In minimal medium, however, the accumulation of R-loops is limited by a slowed down metabolism. Hence they can be either tolerated in the genome or efficiently removed by proteins other than RNase HI [49]. Even after performing ExEV we did not identify any clones devoid of both genes encoding RNases H type I.
Fig 4. Southern blots confirming deletions in double, complemented RNase H type I mutants of M. smegmatis.

We used gene replacement through homologous recombination to obtain mutants deficient in both rnhA and MSMEG4305. We were unable to identify a mutant ∆rnhA/∆4305. Therefore, we introduced a functional copy of either MSMEG4305 or rnhA at the attB site of ∆rnhA/SCOMSMEG4305 or SCOrnhA/∆MSMEG4305, respectively. Following selection, we generated two strains ∆rnhA/∆4305attB::Pami-rnhA and ∆rnhA/∆4305attB::Pami-4305 deficient for both native versions of RNase HI encoding genes, however complemented with either rnhA or MSMEG4305 at the attB site. For more information regarding plasmid construction, gene replacement procedure and complementation please refer to the text. Schematic representation of analyzed genomic regions, including enzymes used for digestion, size of restriction fragments following digestion and the site of hybridization of hybridization probe, is presented in the upper part of the figure. Photographic films presenting results of Southern blot analysis are presented in lower part of the figure. Bands corresponding to wild type genotype (wt), mutant genotype (mut) and complementation genotype (compl) are marked on the right side of each photograph.
The difference between the essentiality of RNase H type I in M. smegmatis and E. coli may be explained by genetic background of these species. In E. coli, rnhA encoding RNase H type I has been shown to be synthetically lethal with a number of genes, namely polA [50], recB [51], recC [51] and recG [52]. These genes can be found in mycobacterial genome. However, the mutation in rnhA has also been shown to be synthetically lethal with mutation in rep [53], a homolog of which is absent in the genome of M. smegmatis. Rep is a helicase involved in restarting DNA replication forks [54], facilitates reannealing of the parental strands during replication [55] and has been shown essential for efficient replication across highly transcribed regions [56,57]. The precise role of Rep remains unknown. Sandler suggested that Rep might be involved in R-loop metabolism and this function, due to synthetic lethality, may be connected with the function of RNase HI [53]. R-loops are linked with transcription and other helicases of E. coli- RecG [52] and Cas3 [58] are involved in R-loop removal. R-loops, if unresolved, may block DNA replication in several ways. First, unrepaired lesions in displaced DNA strand may become a source of double strand ends and consequent replication fork collapse. Second, RNA hybridized to the DNA may be an obstacle for replication fork progression. Paused replication forks can be cleaved by endonucleases known to cleave recombination intermediates again creating double strand ends [59]. Finally, transcription complex linked to an R-loop might become attached to the hybrid and collide with progressing replication fork. All of these scenarios would result in replication fork collapse and could potentially lead to subsequent cell death [60]. Although the idea that RNase H substrates other than R-loops might be responsible for the lethal phenotype of ∆rnhA/∆MSMEG4305 mutant cannot be entirely excluded, it seems unlikely. In other bacteria Okazaki primers have been shown to be removed by other proteins, which are also present within M. smegmatis, PolI [61–63] and RNase HII [25].
The essentiality of RNase HI domain in M. smegmatis suggests that similar phenomenon might be present in M. tuberculosis. Data obtained by high density transposon mutagenesis in M. tuberculosis seem to confirm this hypothesis [64]. In studies assessing the essentiality of mycobacterial genes by high-density transposon mutagenesis the authors observed a small level of transposon insertions within the gene encoding a homolog of MSMEG4305- Rv2228c. The number of insertions was significantly smaller from what would be expected. Perhaps this observation could be related to insertions within CobC domain encoded by the gene and lethal phenotype in the case of insertions within the region encoding RNase HI domain.
RNase HI mutants- RnhA level, growth rates and constitutive stable DNA replication
We wanted to investigate how deletion of MSMEG4305 influences the level of RnhA. Therefore, we obtained recombinant His-tagged RnhA protein expressed in E. coli. We used this protein to immunize a rabbit and to obtain polyclonal anti-RnhA antibodies. These antibodies were used to perform detection of RnhA in protein extracts isolated from M. smegmatis strains used in this study. Further, we quantitated the amount of RnhA in M. smegmatis mc2 155 and compared it with the amount of LigA, NAD+ dependent ligase [44]. We observed that the level of RnhA in wild type strain was low (Fig 5). There were 200 ng of LigA detected in approximately 30 μg of total protein extract, while 10 ng of RnhA was identified in approximately 200 μg of total protein extract. Therefore we estimate that LigA is approximately 133 times more abundant than RnhA.
Fig 5. Western blots presenting quantitative analysis of the RnhA protein in protein extracts of M. smegmatis.

The upper part of the figure presents detection of either LigA or RnhA in protein extracts isolated from mutants used in this study. The lower part of the figure presents quantitative analysis of the amount of RnhA in M. smegmatis mc2 155. Briefly, we detected known concentrations of either recombinant LigA or recombinant RnhA. Further, we compared the intensities of the obtained bands with those obtained from various concentrations of total protein extracts of M. smegmatis mc2 155. We were able to calculate, that RnhA is approximately 133 times less abundant that LigA.
Since the level of RnhA was low, we wanted to see whether deletion of one RNase HI genes influenced growth rate of the mutants. For this purpose, we assessed growth rates by measuring optical density of the liquid cultures at determined intervals of time and fitted appropriate growth curves [65]. We did not observe any differences in growth rates or asymptotical values between the ∆rnhA and M. smegmatis mc2 155 on 7H9 medium supplemented with OADC (t = 1.90, df = 5, p = 0.12; t = 0.35, df = 5, p = 0.74, respectively, data presented in S1 Table) or ∆4305 and M. smegmatis mc2 155 on 7H9 medium supplemented with OADC and vitamin B12 (t = 0.84, df = 4, p = 0.84; t = 0.49, df = 4, p = 0.65, respectively, data presented in S2 Table) (Fig 6A and 6B). The morphology of the mutant cells grown in before mentioned conditions, in terms of cell length was not altered (t = 1.83, df = 198, p = 0.07 for ∆rnhA and M. smegmatis mc2 155, data presented in S3 Table; t = 1.22, df = 198, p = 0.22 for ∆4305 and M. smegmatis mc2 155, data presented in S4 Table) (Fig 7A and 7B).
Fig 6. Growth rates of RNase H type I deficient mutants.

We analyzed the growth rate of RNase H type I deficient mutants ∆rnhA and ∆4305 and compared them with wild type M. smegmatis mc2 155. The analysis involved measuring optical density of the liquid cultures at determined intervals of time. The cultures were performed on 7H9 medium with the addition of OADC and Tween80. For the comparison between ∆4305 and M. smegmatis mc2 155 media were additionally supplemented with vitamin B12. Each experiment was performed at least in triplicate. We observed no differences in growth rates based on optical densities of the cultures. (Box- SE, whiskers- 0.95 CI).
Fig 7. Morphology of the cells of RNase H type I deficient mutants.

We harvested cells grown in liquid cultures for 24 hours and analyzed them on Nikon Eclipse TE2000 microscope. The cultures were performed on 7H9 medium with the addition of OADC and Tween80. For the comparison between ∆4305 and M. smegmatis mc2 155 media were additionally supplemented with vitamin B12. We observed no differences in cell lengths between mutant strains and the wild type. (Box- SE, whiskers- 0.95 CI).
In summary, these results suggest that mycobacterial cells may produce more RNase H type I than would be sufficient for optimal growth, as deletion of one of the genes encoding this enzyme did not seem to affect the viability of the mutant. A similar phenomenon was observed for other replication related proteins of mycobacteria- namely LigA [44] or DnaG [48].
In E. coli R-loops have been shown to be responsible for alternative pathway of initiation of DNA replication. The phenomenon was termed constitutive stable DNA replication (cSDR). cSDR was identified in thermosensitive E. coli mutants with inactivated gene encoding RNase HI [66]. Unlike the classical pathway, cSDR initiates from regions termed oriK instead of oriC [67]. The initial strand opening involves RecA dependent hybridization of the RNA transcript to dsDNA [68] (Fig 8A and 8B). The resulting R-loop is not resolved by RNase HI and therefore RNA persists on DNA strand and serves as a primer for elongation by PolI [50] (Fig 8C). When the loop opens sufficiently, primosome is loaded on the leading strand. As replication continues, PolI removes persisting RNA transcript and primosome is loaded on the lagging strand [50] (Fig 8D and 8E). cSDR in E. coli is independent of DnaA and oriC. Hence, even though dnaA and oriC are essential for viability of wild type E. coli, it is possible to obtain double mutants bearing mutations inactivating either dnaA and rnhA or oriC and rnhA [69].
Fig 8. Constitutive stable DNA replication of E. coli ∆rnhA mutants.

An alternative mode of initiation of DNA replication was discovered in E. coli mutants lacking RNase H type I encoded by rnhA. (A,B) The initial strand opening involves RecA dependent hybridization of the RNA transcript to dsDNA, (C) The resulting R-loop is not resolved by RNase HI and therefore RNA persists on DNA strand and serves as a primer for elongation by PolI, (D,E) When the loop opens sufficiently, primosome is loaded on the leading strand. As replication continues, PolI removes persisting RNA transcript and primosome is loaded on the lagging strand.
We wanted to investigate whether unaltered growth rate in RNase HI mutants of M. smegmatis was a consequence of induced cSDR. In order to investigate the presence of cSDR in M. smegmatis we used the technique of gene replacement through homologous recombination. We introduced gene replacement plasmid for dnaA, with Gm resistance cassette cloned within the sequence of the gene to facilitate screening, into ∆rnhA and ∆4305. In order to differentiate cell’s metabolism and hence potential level of R-loop formation, we intended to remove the native version of dnaA gene on rich medium at 37°C and on minimal medium at 28°C. We were not able to identify a mutant deficient for dnaA. To confirm that the plasmid that we used for gene replacement was capable of creating a deletion within the native sequence of dnaA, we introduced the complete version of the dnaA gene under a native promoter at the attB site of ∆rnhA strain. We generated a strain, ∆rnhA/dnaASCOattB::dnaA, which was further subjected to gene replacement protocol. We were therefore able to generate a strain deficient for a native copy of dnaA, however possessing a copy of the dnaA gene at the attB site, ∆rnhA/∆dnaAattB::dnaA (Fig 9). Finally, we used the latter strain for ExEV experiment and confirmed that dnaA cannot be removed from ∆rnhA strain.
Fig 9. Southern blot confirming deletion of native dnaA gene in ∆rnhA/∆dnaAattB::dnaA mutant strain of M. smegmatis.

We used gene replacement through homologous recombination to obtain mutants deficient in rnhA. Further, we used the same procedure to generate ∆rnhA/dnaASCO strain where SCO signifies an intermediate step of gene replacement procedure. Next, through complementation procedure, we introduced an additional version of dnaA gene at the attB site of mycobacterial genome. Finally, we removed the native version of dnaA, thereby generating a mutant ∆rnhA/dnaASCOattB::dnaA. For more information regarding plasmid construction and gene replacement procedure please refer to the text. Schematic representation of analyzed genomic region, including enzymes used for digestion, size of restriction fragments following digestion and the site of hybridization of hybridization probe, is presented in the upper part of the figure. Photographic film presenting results of Southern blot analysis is presented in the lower part of the figure. Bands corresponding to wild type genotype (wt) and mutant genotype (mut) are marked on the right side of the photograph.
The results of this study confirmed that the dnaA gene is essential in M. smegmatis cells in which the formation of R-loops is limited by the presence of both RNases HI, and suggest that dnaA gene may be required in cells with an increased tendency to form these loops. However, it cannot be entirely excluded that cSDR is not present under the conditions of our experiment. Further decrease in RNase HI level could affect the ability to remove dnaA. Therefore, the existence of cSDR within mycobacteria needs to be further evaluated.
RNases HI as a new antimycobacterial target?
The products of the genes that are essential for survival of the pathogen are potential targets for novel antibiotics. The possibility that RNase HI might be essential for survival of M. tuberculosis is particularly interesting as inhibitors of RNase H are currently intensively studied as potential antiviral drugs in HIV therapy [70]. As reported by the World Health Organization, TB is the major cause of death among people living with HIV. Though the idea that one compound might be limiting for both M. tuberculosis and HIV is wonderful, it is rather utopian. Suboptimal concentrations of RNase H inhibitors of HIV have been shown to decrease susceptibility to antiretroviral drug used for the treatment of HIV, zidovudine [71], and therefore they may be questioned as a novel component of HIV therapy. Finding an RNase H inhibitor that would inhibit both viral and mycobacterial RNase H, without affecting human RNase H, though theoretically possible, does not seem achievable. As an advantage, we observed that in M. smegmatis RNase HI level was low. Therefore, perhaps even a small amount of an inhibitor could be sufficient to effectively kill these cells. This is particularly important in the case of mycobacteria due to their intracellular life niche (in the case of M. tuberculosis) and composition of their unusual and thick cell wall. Therefore the question of whether mycobacterial RNases HI may be used for drug development remains open for future research.
Supporting Information
Bacterial cultures were grown on 7H9 media supplemented with OADC and Tween80. At determined intervals of time optical density of the cultures was measured by spectrophotometer (wave length 600 nm). Results were transformed into growth curves.
(XLSX)
Bacterial cultures were grown on 7H9 media supplemented with OADC, Tween80 and vitamin B12. At determined intervals of time optical density of the cultures was measured by spectrophotometer (wave length 600 nm). Results were transformed into growth curves.
(XLSX)
Bacteria were grown for 24 hours in 7H9 medium supplemented with OADC and Tween80. Cell lengths were measured on Nikon Eclipse TE2000 microscope. Lengths are presented in (nm).
(XLSX)
Bacteria were grown for 24 hours in 7H9 medium supplemented with OADC, Tween80 and vitamin B12. Cell lengths were measured on Nikon Eclipse TE2000 microscope. Lengths are presented in (nm).
(XLSX)
Acknowledgments
We would like to express our gratitude to Prof. Richard Bowater and Anonymous Reviewer for valuable comments on earlier draft of this manuscript. We would like to thank Rebecca Schneider for improving the use of English in this manuscript.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
The study was supported by POIG.01.01.02-10-107/09 project implemented under Innovative Economy Operational Programme, years 2007–2013 "Studies of the molecular mechanisms at the interface the human organism - the pathogen - environmental factors" and by grant of Polish National Center of Science 2011/01/N/NZ6/04186 “Identification of a novel mechanism of initiation of DNA replication in Mycobacterium smegmatis”.
References
- 1. Lima WF, Rose JB, Nichols JG, Wu H, Migawa MT, Wyrzykiewicz TK, et al. The positional influence of the helical geometry of the heteroduplex substrate on human RNase H1 catalysis. Mol Pharmacol. 2007;71: 73–82. 10.1124/mol.106.025429 [DOI] [PubMed] [Google Scholar]
- 2. Shen Y, Koh KD, Weiss B, Storici F. Mispaired rNMPs in DNA are mutagenic and are targets of mismatch repair and RNases H. Nat Struct Mol Biol. 2012;19: 98–104. 10.1038/nsmb.2176 [DOI] [PubMed] [Google Scholar]
- 3. Lima WF, Rose JB, Nichols JG, Wu H, Migawa MT, Wyrzykiewicz TK, et al. Human RNase H1 discriminates between subtle variations in the structure of the heteroduplex substrate. Mol Pharmacol. 2007;71: 83–91. 10.1124/mol.106.025015 [DOI] [PubMed] [Google Scholar]
- 4. Nowotny M, Gaidamakov SA, Crouch RJ, Yang W. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;121: 1005–1016. 10.1016/j.cell.2005.04.024 [DOI] [PubMed] [Google Scholar]
- 5. Murante RS, Henricksen LA, Bambara RA. Junction ribonuclease: An activity in Okazaki fragment processing. Proc Natl Acad Sci U S A. 1998;95: 2244–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eder PS, Walder RY, Walder JA. Substrate specificity of human RNase H1 and its role in excision repair of ribose residues misincorporated in DNA. Biochimie. 1993;75: 123–126. [DOI] [PubMed] [Google Scholar]
- 7. Nossal NG, Dudas KC, Kreuzer KN. Bacteriophage T4 proteins replicate plasmids with a preformed R loop at the T4 ori(uvsY) replication origin in vitro. Mol Cell. 2001;7: 31–41. [DOI] [PubMed] [Google Scholar]
- 8. Itoh T, Tomizawa J. Formation of an RNA primer for initiation of replication of ColE1 DNA by ribonuclease H. Proc Natl Acad Sci U S A. 1980;77: 2450–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee DY, Clayton DA. Initiation of mitochondrial DNA replication by transcription and R-loop processing. J Biol Chem. 1998;273: 30614–30621. [DOI] [PubMed] [Google Scholar]
- 10. Leela JK, Syeda AH, Anupama K, Gowrishankar J. Rho-dependent transcription termination is essential to prevent excessive genome-wide R-loops in Escherichia coli. Proc Natl Acad Sci U S A. 2013;110: 258–263. 10.1073/pnas.1213123110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ginno PA, Lim YW, Lott PL, Korf I, Chédin F. GC skew at the 5’ and 3’ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res. 2013;23: 1590–1600. 10.1101/gr.158436.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anupama K, Leela JK, Gowrishankar J. Two pathways for RNase E action in Escherichia coli in vivo and bypass of its essentiality in mutants defective for Rho-dependent transcription termination. Mol Microbiol. 2011;82: 1330–1348. 10.1111/j.1365-2958.2011.07895.x [DOI] [PubMed] [Google Scholar]
- 13. Pfeiffer V, Lingner J. TERRA promotes telomere shortening through exonuclease 1-mediated resection of chromosome ends. PLoS Genet. 2012;8: e1002747 10.1371/journal.pgen.1002747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Balk B, Maicher A, Dees M, Klermund J, Luke-Glaser S, Bender K, et al. Telomeric RNA-DNA hybrids affect telomere-length dynamics and senescence. Nat Struct Mol Biol. 2013;20: 1199–1205. 10.1038/nsmb.2662 [DOI] [PubMed] [Google Scholar]
- 15. Yu K, Chedin F, Hsieh C-L, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol. 2003;4: 442–451. 10.1038/ni919 [DOI] [PubMed] [Google Scholar]
- 16.Chon H, Sparks JL, Rychlik M, Nowotny M, Burgers PM, Crouch RJ, et al. RNase H2 roles in genome integrity revealed by unlinking its activities. Nucleic Acids Res. 2013; gkt027. 10.1093/nar/gkt027 [DOI] [PMC free article] [PubMed]
- 17. Skourti-Stathaki K, Proudfoot NJ. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014;28: 1384–1396. 10.1101/gad.242990.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hiller B, Achleitner M, Glage S, Naumann R, Behrendt R, Roers A. Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J Exp Med. 2012;209: 1419–1426. 10.1084/jem.20120876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cerritelli SM, Crouch RJ. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 2009;276: 1494–1505. 10.1111/j.1742-4658.2009.06908.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arudchandran A, Cerritelli S, Narimatsu S, Itaya M, Shin DY, Shimada Y, et al. The absence of ribonuclease H1 or H2 alters the sensitivity of Saccharomyces cerevisiae to hydroxyurea, caffeine and ethyl methanesulphonate: implications for roles of RNases H in DNA replication and repair. Genes Cells Devoted Mol Cell Mech. 2000;5: 789–802. [DOI] [PubMed] [Google Scholar]
- 21. Kochiwa H, Tomita M, Kanai A. Evolution of ribonuclease H genes in prokaryotes to avoid inheritance of redundant genes. BMC Evol Biol. 2007;7: 128 10.1186/1471-2148-7-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277: 1453–1462. [DOI] [PubMed] [Google Scholar]
- 23. Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature. 1997;390: 249–256. 10.1038/36786 [DOI] [PubMed] [Google Scholar]
- 24. Itaya M, Omori A, Kanaya S, Crouch RJ, Tanaka T, Kondo K. Isolation of RNase H genes that are essential for growth of Bacillus subtilis 168. J Bacteriol. 1999;181: 2118–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fukushima S, Itaya M, Kato H, Ogasawara N, Yoshikawa H. Reassessment of the in vivo functions of DNA polymerase I and RNase H in bacterial cell growth. J Bacteriol. 2007;189: 8575–8583. 10.1128/JB.00653-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yao NY, Schroeder JW, Yurieva O, Simmons LA, O’Donnell ME. Cost of rNTP/dNTP pool imbalance at the replication fork. Proc Natl Acad Sci U S A. 2013;110: 12942–12947. 10.1073/pnas.1309506110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2: 2006.0008 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gerdes SY, Scholle MD, Campbell JW, Balázsi G, Ravasz E, Daugherty MD, et al. Experimental Determination and System Level Analysis of Essential Genes in Escherichia coli MG1655. J Bacteriol. 2003;185: 5673–5684. 10.1128/JB.185.19.5673-5684.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kanaya S, Crouch RJ. The rnh gene is essential for growth of Escherichia coli. Proc Natl Acad Sci U S A. 1984;81: 3447–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Horiuchi T, Maki H, Sekiguchi M. RNase H-defective mutants of Escherichia coli: a possible discriminatory role of RNase H in initiation of DNA replication. Mol Gen Genet MGG. 1984;195: 17–22. [DOI] [PubMed] [Google Scholar]
- 31. Torrey T, Atlung T, Kogoma T. dnaA suppressor (dasF) mutants of Escherichia coli are stable DNA replication (sdrAlrnh) mutants. Mol Gen Genet MGG. 1984;196: 350–355. [DOI] [PubMed] [Google Scholar]
- 32. Bayliss CD, Sweetman WA, Moxon ER. Destabilization of tetranucleotide repeats in Haemophilus influenzae mutants lacking RnaseHI or the Klenow domain of PolI. Nucleic Acids Res. 2005;33: 400–408. 10.1093/nar/gki180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393: 537–544. 10.1038/31159 [DOI] [PubMed] [Google Scholar]
- 34. Watkins HA, Baker EN. Structural and functional characterization of an RNase HI domain from the bifunctional protein Rv2228c from Mycobacterium tuberculosis. J Bacteriol. 2010;192: 2878–2886. 10.1128/JB.01615-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dawes SS, Crouch RJ, Morris SL, Mizrahi V. Cloning, sequence analysis, overproduction in Escherichia coli and enzymatic characterization of the RNase HI from Mycobacterium smegmatis. Gene. 1995;165: 71–75. [DOI] [PubMed] [Google Scholar]
- 36. Murdeshwar MS, Chatterji D. MS_RHII-RSD, a Dual-Function RNase HII-(p)ppGpp Synthetase from Mycobacterium smegmatis. J Bacteriol. 2012;194: 4003–4014. 10.1128/JB.00258-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc Natl Acad Sci U S A. 1998;95: 5857–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16: 10881–10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42: W320–W324. 10.1093/nar/gku316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parish T, Stoker NG. Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiol Read Engl. 2000;146 (Pt 8): 1969–1975. [DOI] [PubMed] [Google Scholar]
- 41. Korycka-Machala M, Brzostek A, Rozalska S, Rumijowska-Galewicz A, Dziedzic R, Bowater R, et al. Distinct DNA repair pathways involving RecA and nonhomologous end joining in Mycobacterium smegmatis. FEMS Microbiol Lett. 2006;258: 83–91. 10.1111/j.1574-6968.2006.00199.x [DOI] [PubMed] [Google Scholar]
- 42. Dziadek J, Rajagopalan M, Parish T, Kurepina N, Greendyke R, Kreiswirth BN, et al. Mutations in the CCGTTCACA DnaA box of Mycobacterium tuberculosis oriC that abolish replication of oriC plasmids are tolerated on the chromosome. J Bacteriol. 2002;184: 3848–3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Greendyke R, Rajagopalan M, Parish T, Madiraju MVVS. Conditional expression of Mycobacterium smegmatis dnaA, an essential DNA replication gene. Microbiol Read Engl. 2002;148: 3887–3900. [DOI] [PubMed] [Google Scholar]
- 44. Korycka-Machala M, Rychta E, Brzostek A, Sayer HR, Rumijowska-Galewicz A, Bowater RP, et al. Evaluation of NAD+-Dependent DNA Ligase of Mycobacteria as a Potential Target for Antibiotics. Antimicrob Agents Chemother. 2007;51: 2888–2897. 10.1128/AAC.00254-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Petrov DA, Hartl DL. Pseudogene evolution and natural selection for a compact genome. J Hered. 2000;91: 221–227. [DOI] [PubMed] [Google Scholar]
- 46. Amin AG, Goude R, Shi L, Zhang J, Chatterjee D, Parish T. EmbA is an essential arabinosyltransferase in Mycobacterium tuberculosis. Microbiol Read Engl. 2008;154: 240–248. 10.1099/mic.0.2007/012153-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Williams A, Güthlein C, Beresford N, Böttger EC, Springer B, Davis EO. UvrD2 is essential in Mycobacterium tuberculosis, but its helicase activity is not required. J Bacteriol. 2011;193: 4487–4494. 10.1128/JB.00302-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kuron A, Korycka-Machala M, Brzostek A, Nowosielski M, Doherty A, Dziadek B, et al. Evaluation of DNA primase DnaG as a potential target for antibiotics. Antimicrob Agents Chemother. 2014;58: 1699–1706. 10.1128/AAC.01721-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kogoma T. Stable DNA replication: interplay between DNA replication, homologous recombination, and transcription. Microbiol Mol Biol Rev MMBR. 1997;61: 212–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kogoma T, Maldonado RR. DNA polymerase I in constitutive stable DNA replication in Escherichia coli. J Bacteriol. 1997;179: 2109–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Itaya M, Crouch RJ. A combination of RNase H (rnh) and recBCD or sbcB mutations in Escherichia coli K12 adversely affects growth. Mol Gen Genet MGG. 1991;227: 424–432. [DOI] [PubMed] [Google Scholar]
- 52. Hong X, Cadwell GW, Kogoma T. Escherichia coli RecG and RecA proteins in R-loop formation. EMBO J. 1995;14: 2385–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sandler SJ. Requirements for replication restart proteins during constitutive stable DNA replication in Escherichia coli K-12. Genetics. 2005;169: 1799–1806. 10.1534/genetics.104.036962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sandler SJ. Multiple genetic pathways for restarting DNA replication forks in Escherichia coli K-12. Genetics. 2000;155: 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mahdi AA, Buckman C, Harris L, Lloyd RG. Rep and PriA helicase activities prevent RecA from provoking unnecessary recombination during replication fork repair. Genes Dev. 2006;20: 2135–2147. 10.1101/gad.382306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boubakri H, de Septenville AL, Viguera E, Michel B. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J. 2010;29: 145–157. 10.1038/emboj.2009.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Baharoglu Z, Lestini R, Duigou S, Michel B. RNA polymerase mutations that facilitate replication progression in the rep uvrD recF mutant lacking two accessory replicative helicases. Mol Microbiol. 2010;77: 324–336. 10.1111/j.1365-2958.2010.07208.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Howard JAL, Delmas S, Ivančić-Baće I, Bolt EL. Helicase dissociation and annealing of RNA-DNA hybrids by Escherichia coli Cas3 protein. Biochem J. 2011;439: 85–95. 10.1042/BJ20110901 [DOI] [PubMed] [Google Scholar]
- 59. Seigneur M, Bidnenko V, Ehrlich SD, Michel B. RuvAB acts at arrested replication forks. Cell. 1998;95: 419–430. [DOI] [PubMed] [Google Scholar]
- 60. Aguilera A, García-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol Cell. 2012;46: 115–124. 10.1016/j.molcel.2012.04.009 [DOI] [PubMed] [Google Scholar]
- 61. Ogawa T, Okazaki T. Function of RNase H in DNA replication revealed by RNase H defective mutants of Escherichia coli. Mol Gen Genet MGG. 1984;193: 231–237. [DOI] [PubMed] [Google Scholar]
- 62. Okazaki R, Arisawa M, Sugino A. Slow Joining of Newly Replicated DNA Chains in DNA Polymerase I-Deficient Escherichia coli Mutants*. Proc Natl Acad Sci U S A. 1971;68: 2954–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Olivera BM, Bonhoeffer F. Replication of Escherichia coli requires DNA polymerase I. Nature. 1974;250: 513–514. 10.1038/250513a0 [DOI] [PubMed] [Google Scholar]
- 64. Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7: e1002251 10.1371/journal.ppat.1002251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zwietering MH, Jongenburger I, Rombouts FM, van ‘t Riet K. Modeling of the Bacterial Growth Curve. Appl Environ Microbiol. 1990;56: 1875–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ogawa T, Pickett GG, Kogoma T, Kornberg A. RNase H confers specificity in the dnaA-dependent initiation of replication at the unique origin of the Escherichia coli chromosome in vivo and in vitro. Proc Natl Acad Sci U S A. 1984;81: 1040–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. De Massy B, Fayet O, Kogoma T. Multiple origin usage for DNA replication in sdrA(rnh) mutants of Escherichia coli K-12. Initiation in the absence of oriC. J Mol Biol. 1984;178: 227–236. [DOI] [PubMed] [Google Scholar]
- 68. Kogoma T, Barnard KG, Hong X. RecA, Tus protein and constitutive stable DNA replication in Escherichia coli rnhA mutants. Mol Gen Genet MGG. 1994;244: 557–562. [DOI] [PubMed] [Google Scholar]
- 69. Kogoma T, von Meyenburg K. The origin of replication, oriC, and the dnaA protein are dispensable in stable DNA replication (sdrA) mutants of Escherichia coli K-12. EMBO J. 1983;2: 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ilina T, LaBarge K, Sarafianos SG, Ishima R, Parniak MA. Inhibitors of HIV-1 Reverse Transcriptase—Associated Ribonuclease H Activity. Biology. 2012;1: 521–541. 10.3390/biology1030521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Davis CA, Parniak MA, Hughes SH. The effects of RNase H inhibitors and nevirapine on the susceptibility of HIV-1 to AZT and 3TC. Virology. 2011;419: 64–71. 10.1016/j.virol.2011.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Bacterial cultures were grown on 7H9 media supplemented with OADC and Tween80. At determined intervals of time optical density of the cultures was measured by spectrophotometer (wave length 600 nm). Results were transformed into growth curves.
(XLSX)
Bacterial cultures were grown on 7H9 media supplemented with OADC, Tween80 and vitamin B12. At determined intervals of time optical density of the cultures was measured by spectrophotometer (wave length 600 nm). Results were transformed into growth curves.
(XLSX)
Bacteria were grown for 24 hours in 7H9 medium supplemented with OADC and Tween80. Cell lengths were measured on Nikon Eclipse TE2000 microscope. Lengths are presented in (nm).
(XLSX)
Bacteria were grown for 24 hours in 7H9 medium supplemented with OADC, Tween80 and vitamin B12. Cell lengths were measured on Nikon Eclipse TE2000 microscope. Lengths are presented in (nm).
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.