

Abstract
Protein synthesis by the ribosome is highly dependent on the ionic conditions in the cellular environment, but the roles of ribosome solvation remain poorly understood. Moreover, the function of modifications to ribosomal RNA and ribosomal proteins are unclear. Here we present the structure of the Escherichia coli 70S ribosome to 2.4 Å resolution. The structure reveals details of the ribosomal subunit interface that are conserved in all domains of life, and suggest how solvation contributes to ribosome integrity and function. The structure also suggests how the conformation of ribosomal protein uS12 likely impacts its contribution to messenger RNA decoding. This structure helps to explain the phylogenetic conservation of key elements of the ribosome, including posttranscriptional and posttranslational modifications and should serve as a basis for future antibiotic development.
Introduction
The structure and function of the ribosome in protein synthesis is exquisitely sensitive to ionic conditions1. Bacterial ribosomes carry a formal charge of nearly −4,000, as ribosomal proteins neutralize very little of the charge in the phosphodiester backbone in ribosomal RNA (rRNA). In the bacterium Escherichia coli, ribosomes harbor large numbers of different cations, including potassium, magnesium, and polyamines. These ions influence ribosome dynamics, including intersubunit rotation2 as well as the mechanisms of mRNA decoding3 and translocation4,5. Furthermore, the ribosome is highly hydrated, due to the relatively open nature of RNA secondary and tertiary structure6.
In addition to high levels of solvation and counterion binding, both rRNA and ribosomal proteins undergo numerous posttranscriptional and posttranslational modifications, respectively. The degree of these modifications increases with the complexity of the organism, suggesting that they have important functional roles7. The most common posttranscriptional modification is isomerization of uridine to pseudouridine, which bears a free imino group that can stabilize RNA structure through base-stacking interactions and the use of its N1 imino group as a hydrogen bond donor8. Further modifications of the E. coli ribosome include methylation of the ribose 2′-hydroxyl group, methylation of nucleobases and reduction of the nucleobase uridine to dihydrouridine8. Modification of RNA nucleotides leads to altered electronic and steric effects that result in altered base pairing potentials, preferred sugar puckers, and different base stacking properties9.
Notably, posttranscriptional and posttranslational modifications cluster in or near key functional sites of the ribosome, such as the decoding center, the peptidyl transferase center (PTC), the exit tunnel, and intersubunit bridges. However, the functional role of individual modifications remains poorly understood. Although some modifications are nearly universal in bacteria10 the inhibition of specific rRNA modifications often has little effect on cell growth, hence, the function of each individual rRNA modification is not clear11–13. Notably, ribosomes lacking all pseudouridines cause severe growth defects in yeast14. Furthermore, even if individual rRNA modifications are dispensable, their presence leads to a competitive advantage for growth15,16. In addition, some ribosomal modifications are incorporated as an environmental response, and can have widespread impacts on the translation capacity of the ribosome17–19.
To date, it has not been possible to accurately model solvation characteristics of the intact ribosome due to the generally low resolution of ribosome X-ray crystal structures20, most at a resolution lower than ~2.8 Å. Recent high-resolution structures of the Thermus thermophiluss 70S ribosome used crystallographic data to 2.55 Å21 and 2.4 Å22 resolution, but the resulting structural models do not yet include solvation beyond inclusion of inner-sphere and outer-sphere coordinated Mg2+ ions, and do not include posttranscriptional and posttranslational modifications. We therefore set out to solve the structure of the bacterial ribosome in order to elucidate its solvation characteristics and the structural impacts of ribosomal modifications. Here, we present a high-resolution structure of the E. coli ribosome, which enabled us to greatly improve the model of the bacterial ribosome by removing many previous modeling errors. The structure reveals unprecedented views of solvation as well as posttranscriptional and posttranslational modifications of the ribosome in key functional sites and antibiotic binding sites. Comparisons to biochemical, genetic and phylogenetic data allows us to propose functional roles for a number of the structural features seen for the first time at high resolution.
Results
We significantly improved the diffraction power of E. coli 70S ribosome crystals23 by extensively optimizing the cryo-stabilization procedure prior to crystal freezing, as well as detector geometry and crystal orientation to reduce diffraction overlap (Online Methods). Due to radiation damage caused by the X-ray beam, we merged diffraction data from more than one hundred high-quality crystals for the structure presented here. The 70S ribosome crystallized with two 70S ribosomes in the crystallographic asymmetric unit (Table 1), with the small (30S) subunit of ribosome I rotated to an intermediate position relative to the large (50S) subunit and with ribosome II in an unrotated state23. The electron density maps for most parts of ribosome I were of high quality, except for peripheral elements including protein L9, the dynamic L1 arm, the stalk proteins L10, L11 and L7–L12, sections of the GTPase center and A-site finger in the large (50S) subunit, and regions of the head domain of the small (30S) subunit (Fig. 1a). Ribosome II was more dynamic in this crystal form, resulting in electron density maps of lower quality in most regions (Supplementary Fig. 1a).
Table 1.
Data collection and refinement statistics
| Merged data | |
|---|---|
| Data collection | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 212.1, 433.9, 624.3 |
| α, β, γ (°) | 90.0, 90.0, 90.0 |
| Resolution (Å) | 69.39 – 2.11 (2.17 – 2.11)* |
| Rmerge | 16.9 (205.2) |
| I/σI | 5.1 (0.28) |
| CC(1/2) (%) | 99.6 (13.7)* |
| Completeness (%) | 93.2 (80.9) |
| Redundancy | 4.15 (2.09) |
| Refinement | |
| Resolution (Å) | 69.39 – 2.1 |
| No. reflections | 3,003,826 |
| Rwork/Rfree | 21.85/23.38 |
| No. atoms (without H) | 295,744 |
| Protein/RNA | 286,907 |
| Ligand/ion | 1,083 |
| Water | 7,754 |
| B-factors (Å2) | |
| Protein/RNA, ribosome I | 69.89 |
| Protein/RNA, ribosome II | 122.98 |
| Ligand/ion | 64.13 |
| Water | 53.25 |
| r.m.s. deviations | |
| Bond lengths (Å) | 0.008 |
| Bond angles (°) | 1.270 |
Values in parentheses are for highest-resolution shell. 109 crystals were used to measure the data.
Figure 1.

E. coli 70S ribosome I in an intermediate rotated state. (a) Ribosomal subunits of ribosome I colored by atomic displacement factor (ADP) from 20 to 150 Å2. The views are from the perspective of the subunit interface. Features in the 50S subunit include the central protuberance (CP), L1 arm (L1), protein L9 (L9), L7–L12 region (L12), A-site finger (ASF) and the GTPase center (G). In the 30S subunit, these include the head (H), body (B), and platform (PL). (b) Single-molecule imaging of the modulation of ribosome dynamics by magnesium ions. Occupancy of unrotated (red) and rotated (black) states of the ribosome as measured by smFRET between fluorophores on uS13 and uL1, shown with standard deviations of three technical replicates (Online methods).
Ribosome modeling in high-resolution electron density maps
We determined the 70S ribosome crystal structures at a resolution of approximately 2.4 – 2.5 Å (I/_I=1). Using the recently introduced cross-correlation parameter24 CC1/2, the scaled and merged X-ray diffraction data measured from the above ribosome crystals extended to 2.11 Å, where the value of CC1/2 was still significant (13.7%, Supplementary Table 1), and were therefore included in the structure refinement. Due to the inherent dynamics of the ribosome and regions of disorder, the quality of the model varied by region, as reflected by the range of atomic displacement parameters (ADPs) (Fig. 1a, Supplementary Fig. 1a). In the following description, we therefore focused our modeling and interpretation primarily on the regions that are well-ordered in ribosome I (Online Methods).
Consistent with the CC1/2 resolution, the electron density maps were highly detailed in many regions of the ribosome (Online Methods) (Supplementary Fig. 2). The directionality of non-bridging phosphate oxygen atoms in the rRNA was resolved and ribose 2′-hydroxyl groups were clearly visible, as were nucleotide exocyclic amines and carbonyl groups (Supplementary Fig. 2 and Supplementary Fig. 3). In well-ordered ribosomal proteins, the peptide backbone and methyl groups of valine, leucine and isoleucine side chains, as well as guanidino groups of arginines were clearly resolved (Supplementary Fig. 3). This level of resolution allowed us to greatly improve the ribosomal protein models compared to a prior 2.8 Å E. coli ribosome X-ray structure as judged by MolProbity25 (Supplementary Table 2–Supplementary Table 5). The data also allowed placement of more than 8,800 solvent molecules and ions. At this resolution, magnesium ions were readily distinguished from water and ammonium ions, and we could see specific polyamines. However, water and ammonium ions were difficult to tell apart, and were therefore modeled as waters throughout the structure.
The quality of the electron density in many regions revealed the contribution of solvent molecules to key functional sites of the ribosome, including the subunit interface, the peptidyl transferase center (PTC) and the exit tunnel. Furthermore, the electron density allowed modeling of many posttranslational and posttranscriptional modifications, including those in three ribosomal proteins26,27 and 35 of 36 posttranscriptional modifications except for the substoichiometric s2C2501 modification in 23S rRNA8,28 (Supplementary Fig. 4, Supplementary Table 6). We also determined the absolute configuration of the posttranslational modification of β-methylthioaspartate at position 89 of protein uS12 in the small ribosomal subunit, the orientation of six out of seven nucleobase methylations in m2G and m6A (Supplementary Fig. 4, Supplementary Table 7) and of the four 2′-hydroxymethylations.
Solvation of the ribosome at the subunit interface
Single-molecule Förster Resonance Energy Transfer (smFRET) experiments demonstrated that lower Mg2+ concentrations promote rotated ribosome configurations in which tRNAs adopt hybrid site binding (Fig. 1b, Supplementary Fig. 1b). In line with the smFRET data, lower Mg2+ concentrations favored crystallization of one ribosome in the crystallographic asymmetric unit in an intermediate rotated state23 (ribosome I, Fig. 1a). Putrescine and spermidine were included in the crystallization and cryoprotection solutions due to their essential role in cellular physiology and the fidelity of translation1,29. These cellular polyamines are also required for 70S complex stability in smFRET experiments30,31, although their contribution to ribosome dynamics are less clear.
In the new high-resolution structure of the ribosome, many of the RNA-rich intersubunit bridges in ribosome I showed clear evidence for water-mediated interactions between the two subunits, but there were few if any examples of clear magnesium ions or polyamines at the subunit interface (Supplementary Table 8). We observed electron density consistent with bound polyamines in the large subunit near the subunit interface in bridge B3 and near the base of helix H69 in 23S rRNA (Supplementary Fig. 3), but few other polyamines were clearly visible in the electron density. These results suggested that charge neutralization occurs primarily through diffusely bound ions32, or possibly through ions bound in the RNA major groove33, which may help to promote ribosome subunit association while also allowing for intersubunit rotation.
Bridge B3, considered the “pivot point” of intersubunit rotation23,34, involves a type I A-minor motif interaction involving the minor-groove faces of two adenines in tandem sheared GA base pairs in 16S rRNA helix h44 of the 30S subunit that contact the minor-groove of two G-C base pairs in 23S rRNA helix H71 of the 50S subunit33,35. We identified three water molecules that buttress this A-minor motif, increasing the number of base pairing planes that interact between the small and the large subunit in bridge B3 from three to four (Fig. 2a–c).
Figure 2.

Solvation at the ribosomal subunit interface and in the nascent peptide exit tunnel. (a–c) Water molecules that bridge the 16S (blue) and 23S (grey) rRNA in bridge B3 of ribosome I. 2′-hydroxyls involved in the water interactions are marked with asterisks. The location of bridge B3 is indicated in the inset. The feature enhanced maps are contoured at 2.5 standard deviations from the mean. (d) Solvation at the entrance of the nascent peptide exit tunnel. Labeled residues of 23S rRNA are shown in orange. The feature enhanced map is contoured at 2.0 standard deviations from the mean.
Whereas bridge B3 likely adopts a fairly constant conformation during intersubunit rotation, the remaining bridges change position23,34,36. Although we observed ordered solvent molecules in other bridges (Supplementary Table 8), bridge B6 is nearly devoid of ordered solvent, although the space between the two RNA helices that make up the bridge leaves room for several water molecules33. This is true in both copies of the 70S ribosome in the crystallographic asymmetric unit, in which bridge B6 is well ordered in terms of the RNA, and for which we could model solvent in the vicinity of the bridge.
Solvation of antibiotic binding sites in the exit tunnel
Macrolides, ketolides and streptogramin B are clinically important antibiotic classes that partially block the nascent peptide exit tunnel, leading to bacterial cell death37,38. In the exit tunnel, nucleotide A2058 of 23S rRNA is critical for antibiotic binding, and antibiotic resistance results from mutation or methylation of this nucleotide39. In the high-resolution ribosome structure, a number of solvent molecules in the proximity of A2058 overlap with the binding site of macrolides, ketolides and streptogramin B antibiotics (Fig. 2d, Supplementary Fig. 5a). These solvent molecules (here interpreted as water) form a layer between nucleotides A2058 and ψ746, and extend towards the base pair formed by A752 and U609, which stacks on the alkyl-aryl arm of the ketolide telithromycin40,41 (Fig. 2d). These solvent molecules would have to be displaced upon antibiotic binding, the effects of which can be included in future computational docking methods.
The narrow “constriction site” of the nascent peptide exit tunnel is only about 10 Å wide and is confined by residues of ribosomal proteins uL4, uL22 and A751 of 23S rRNA42,43. Notably, clear electron density for 2-methyl-2,4-pentanediol (MPD), a moderately hydrophobic molecule used in the crystallization solution, stacks on A751 and partly blocks the exit tunnel (Supplementary Fig. 5b). Cryo-electron microscopy studies44 and molecular dynamics simulations45 of the SecM stalled ribosome had identified that stacking of the hydrophobic side chain of amino acid Trp155 of SecM on nucleotide A751 is crucial for stalling (Supplementary Fig. 5c). Mutations of either the SecM nascent chain or A751 abolish stalling, indicating that this hydrophobic interaction is involved in regulation of gene expression of the protein translocase SecA, and may possibly contribute to the regulation of other nascent chains46–48.
Ribosomal modifications near the PTC
A number of posttranscriptional and posttranslational modifications are clustered near the peptidyl transferase center (Fig. 3a). In the classical state, nucleotides C74 and C75 in the 3′ CCA acceptor arm of the P-site tRNA base pair with G2252 and Gm2251 of the P loop of 23S rRNA, respectively, which help orient the peptidyl-tRNA for the peptidyl transfer reaction21,49. In the E. coli ribosome structure, Gm2251 assumes a C3′-endo sugar pucker, which may be stabilized by the 2′-O-methylation, since a C2′-endo pucker could lead to altered positioning of the nucleobase50. Notably, the remaining nucleotides of the E. coli ribosome carrying 2′-O-methylations (Supplementary Table 6) also adopt a C3′-endo pucker. The non-bridging phosphate oxygen of Gm2251 contacts the guanidinium group of Arg81 of uL16, which itself is modified with a hydroxyl group at the β-carbon (Fig. 3a,b). In the present structure, the hydroxyarginine 81 is present in two alternate conformations and its absolute configuration at the β-carbon is R, consistent with the hydroxylation catalyzed by ribosomal oxygenase ycfD27. On the opposite side of Arg81, Gm2251 is positioned next to the only dihydroudirine in the E. coli ribosome, D2449. A dihydrouridine nucleotide disrupts base stacking interactions due to the non-planar nature of the dihydrouridine nucleobase, and can stabilize its ribose and that of the 5′ neighboring nucleotide in C2′-endo puckers51, as observed in the E. coli ribosome structure (Fig. 3c). In close proximity to the dihydrouridine, the methyl group on the exocyclic amine of m2G2445 adopts an s-trans position and occupies a cavity created by neighboring nucleobases including D2449 (Fig. 3a and Supplementary Table 7).
Figure 3.

Posttranscriptional and posttranslational modifications in functional centers of the ribosome. (a) View of the peptidyl transferase center (PTC) with C75 of the P-site tRNA (orange) modeled from PDB ID 3I8H and 3I8I60. Hydroxyarginine 81 (magenta) is in protein uL16 and Gm2251, dihydrouridine 2449 and m2G2445 are in 23S rRNA (grey). (b) Hydroxylation of arginine 81 of uL16 at the Cβ position is in the R configuration. The feature enhanced map is contoured at 2.5 standard deviations from the mean. (c) Dihydrouridine 2449 and A2448 assume a C2′-endo sugar pucker. Feature-enhanced electron density map shown as in (b). (d) Geometry of protein uS12 (cyan) near the mRNA decoding center. Proline 45 of uS12 in the cis-peptide conformation would be positioned less than 4 Å away from the 3rd nucleotide in the mRNA A-site codon (green). mRNA is modeled from PDB ID 4QCQ22. (e) Pro45 in uS12 with a cis-peptide bond and β-methylthioaspartate in the R configuration at position 89 shown with feature enhanced maps contoured at 2.5 and 2.2 standard deviations from the mean, respectively. (f) Model of mRNA (PDB ID: 4QCQ)22 superimposed on the E. coli high-resolution structure, in which protein uS12 in the decoding site is modeled with a trans-3-hydroxy-proline at position 45. The 3-hydroxy group of Pro45 would be in hydrogen bonding distance of the O3′ of the 3rd position of the A-site codon.
Conformation of uS12 near the mRNA decoding center
Protein uS12 forms key parts of the mRNA decoding center of the small ribosomal subunit, with proline 45 of uS12 positioned less than 4 Å away from the third nucleotide of the mRNA A-site codon21,22 (Fig. 3d,e). Interestingly, Pro45 clearly adopted a cis-peptide conformation in the present high-resolution structure (Fig. 3d,e), a conformation that persists through mRNA decoding22, even though cis-prolines occur at a low frequency in proteins52. This stable conformation could have functional implications in mRNA decoding and mRNA and tRNA translocation. For example, in the eukaryotic homolog of uS12, rpS23, the corresponding proline residue (Pro62 in human, Pro64 in yeast), is mono- or dihydroxylated, respectively, depending on environmental or stress conditions17–19. Interestingly, the hydroxylation state of uS12’s Pro64 in yeast has been shown to affect stop-codon readthrough in a sequence- and context dependent manner18. Hydroxylation or dihydroxylation of this proline would change its interaction from a van der Waals contact to the mRNA to one mediated by a hydrogen bond with the O3′ of the third position of the A-site codon (Fig. 3f)22, which may increase the binding energy of the ribosome-mRNA complex by about 1–2 kcal/mol, corresponding to the energy of a hydrogen bond.
In E. coli uS12, aspartate 89 is posttranslationally modified to β-methylthioaspartate26, with an absolute configuration of 3R in the present structure (Fig. 3d–e). In this configuration, the side chain carboxylate is in hydrogen bonding distance with the guanidinium group of Arg50 of uS12, neutralizing its charge, and the methylthio group points towards the positively charged m7G527. The β-methylthio modification on Asp89, specific to bacteria, is introduced by RimO and YcaO53. Although mutations of Asp89 are lethal, E. coli strains with a rimO knockout have only a slightly slower cell growth phenotype54. However, in light of the role of Pro64 hydroxylations in eukaryotes (Pro45 in E. coli), the β-methylthio modification of Asp89 in bacteria may serve important functions under stress or variable growth conditions.
Pseudouridines in the ribosome
Pseudouridine has been proposed to stabilize RNA structures by presenting an extra hydrogen bond donor in the major groove, relative to uridine8. For six of the seven well-ordered pseudouridines in the present high-resolution structure (Supplementary Table 9), we observed a water-mediated contact between the pseudouridine N1 imino group in the major groove and the rRNA phosphate backbone. This is consistent with the model that these pseudouridines stabilize the local ribosomal structure by locking the nucleobase in place with respect to the rRNA backbone. One example in the 50S subunit tRNA P-site involves a network of water-mediated interactions including G954 and ψ955 of 23S rRNA helix H39 (Fig. 4a).
Figure 4.
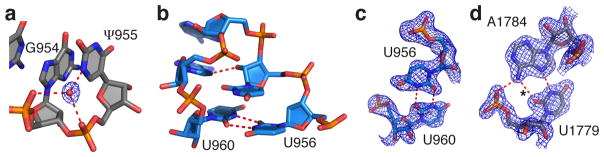
Pseudouridines and syn-pyrimidines in the ribosome. (a) Pseudouridine with a major groove water molecule. Shown is the N1 imino group of ψ955 in 23S rRNA hydrogen bonding to a water molecule that in turn hydrogen bonds to the non-bridging phosphate oxygen of ψ955 and G954. The feature enhanced map is contoured at 2.5 standard deviations from the mean. (b–d) Examples of syn-pyrimidines in the ribosome. Nucleotide U960 in a syn conformation stabilizes a triloop capping helix h31 in 16S rRNA by forming a reverse U-U base pair with U956. (c) The feature enhanced map for the U960–U956 base pair is contoured at 2.5 standard deviations from the mean. (d) Nucleotide U1779 in 23S rRNA adopts a syn conformation to form an unusual reverse Hoogsteen A–U base pair with A1784, allowing the N6 exocyclic amino group of A1784 to hydrogen bond with a non-bridging phosphate oxygen of U1779. The feature-enhanced electron density map is contoured at 2.5 standard deviations from the mean. Hydrogen bonds are indicated by red dashed lines. The hydrogen bond marked with an asterisk is 3.3 Å long.
Syn pyrimidines in the ribosome
Although the favorable conformation of the nucleobase is anti with respect to the ribose, purines can assume sterically less favorable syn conformations. We observed 120 examples of syn purines with good electron density in the E. coli ribosome (defined with a Chi angle in the range of −90° to +90°; Supplementary Table 10). However, syn pyrimidines are rare, due to a larger energetic penalty resulting from the steric clash of the O2 of pyrimidines with the ribose compared to the N3 in purines. Syn nucleotides seem to occur more frequently in functional sites of RNA structures, where the energetic cost for adopting the syn conformation may be compensated for by its favorable effects on RNA structure and function55. In highly ordered regions of the E. coli ribosome structure, we observed 15 pyrimidines in the syn conformation (Supplementary Table 11 and Supplementary Fig. 6). Intriguingly, many of these are supported by phylogenetic analysis as being nearly universal in bacteria or across all domains of life (Supplementary Table 11). For example, U960 of 16S rRNA adopts a syn conformation to form a reverse U-U base pair with U956 that stabilizes a tight triloop located at the base of helix h31 (Fig. 4b,c), which plays an active role in movement of A-site tRNA into the P-site on the small ribosomal subunit during translocation56. In the 50S subunit, U1779 assumes a syn conformation to form an unusual reverse Hoogsteen AU type base pair with A1784 in the loop capping helix H65 of 23S rRNA that forms part of the peptide exit tunnel in close proximity to nucleotides U1781–U1782, which line the binding pocket of several antibiotics (Fig. 4d).
Discussion
The new high-resolution structure of the E. coli 70S ribosome presented here provides a foundation for unraveling key mechanisms shared by ribosomes in all domains of life. For example, the geometry of ribosomal protein uS12 near the mRNA decoding site suggests a model for how hydroxylation of Pro45 could influence the energetics of mRNA decoding and mRNA and tRNA translocation. Sites of syn-pyrimidines, rare in most RNA contexts, reveal conserved base-pair patterns that could contribute to ribosomal RNA tertiary folding and key ribosomal functions, such as tRNA translocation56. Structural knowledge of clustered rRNA and protein modifications near functional centers can now be used to test the mechanisms of fine-tuning translation that likely confer a selective advantage to microbes in their natural habitats.
The present E. coli ribosome structure can also be used for comparisons to other structures of the 50S subunit and 70S ribosome. For example, the 2.2–2.4 Å structures of the archaeal H. marismortui 50S subunit42,57,58, which serve as a model for the bacterial A2058G mutation that confers antibiotic resistance, reveal that the A2058G mutation would substantially alter the architecture and solvation of the macrolide, ketolide, and streptogramin B binding sites. Additionally, Um2552 in the A loop of the PTC adopts the same conformation in the E. coli 70S ribosome and H. marismortui 50S subunit, with its 2′-O-methyl group wedged between the nucleobases of U2554 and G2553, which pairs with C75 in A-site tRNA21. This positioning suggests that this modification could have functional importance in modulating the base pairing between 23S rRNA and A-site tRNA. It will be interesting in the future to compare the present structure to the recent 2.4 – 2.55 Å crystal structures of the Thermus thermophilus 70S ribosome21,22, which are likely at sufficient resolution to allow modeling of posttranscriptional and posttranslational modifications and solvation.
Finally, the new high-resolution structure of the E. coli 70S ribosome can be used as a resource and reference for comparisons to other RNA structures, future ribosome structures in other conformational states, analyses of ribosome phylogenetics, and biochemical studies. For example, it should now be possible to probe the functional roles of pseudouridines and conserved syn-pyrimidines more directly. It is striking that, although some ordered solvent molecules could be identified in key locations at the subunit interface, i.e. in universally conserved bridge B3, most of the solvation at the subunit interface is seemingly diffuse in nature32,59, which could be important for ribosome dynamics. Future studies of the ribosome by smFRET experiments and molecular dynamics simulations should be helpful in deciphering the roles of ordered and diffuse water and ions in the structure and dynamics of the ribosome.
Online Methods
Preparation of ribosome initiation complexes for smFRET experiments
Initiation complexes for the smFRET intersubunit rotation assay were formed using E. coli Cy3-S13 (N-Sfp) 30S and Cy5-L1 (T202C) 50S subunits and initiated with unlabeled fMet-tRNAfMet, as previously described61. Initiation complexes for the tRNA hybrid state fluctuations assay were formed using unlabeled E. coli subunits and initiated with fMet-tRNAfMet (Cy3-s4U8), as previously described30,62.
Single-molecule intersubunit rotation and tRNA hybrid state assays
smFRET experiments were performed using a prism-based total internal reflection (TIR) microscope30,62 at 25 °C in Tris-polymix buffer61 with an oxygen scavenging system63, triplet-state quenching compounds64 (1 mM cyclooctatetraene, 1 mM nitrobenzyl alcohol, 1 mM Trolox) and 2.5 to 30 mM Mg2+. After surface immobilization of initiation complexes programmed with biotinylated mRNA to PEG-passivated, streptavidin-coated quartz microfluidic chambers30,62, pre-translocation complexes were prepared by incubating with either unlabeled ternary complex of EF-Tu(GTP)Phe-tRNAPhe (for the intersubunit rotation assay) or EF-Tu(GTP)Phe-tRNAPhe(Cy5-acp3U47) (for the tRNA hybrid state assay) for 2 min in Tris-polymix buffer, as previously described4. Cy3 fluorophores on either uS13 or P-site tRNA were illuminated with a 532 nm diode-pumped solid-state laser (Opus, LaserQuantum). Fluorescence emission from Cy3 and Cy5 fluorophores was collected using a 1.27 N.A. PlanApo water immersion objective (Nikon) and spectrally separated using a DualCam device (Photometrics) using a 640dcxr dichroic mirror (Chroma). smFRET data were acquired at 25 ms integration time and analyzed using analytical software implemented in MATLAB (MathWorks). FRET trajectories were idealized using the segmental k-means algorithm65, as previously described4,61. Experiments were performed using three technical replicates, i.e. with frozen aliquots of the same ribosome complex preparations used on three separate days.
Ribosome Crystallization and Cryo-protection
Ribosomes purified from E. coli strain MRE60033 were crystallized as described previously23. A highly optimized seven-step cryo-stabilization procedure was established to prepare ribosome crystals for cryo-cooling. This procedure ensured robust crystal-to-crystal reproducibility, allowing diffraction data to be measured and merged from multiple crystals. Crystals were soaked in crystallization buffer containing 6–7% PEG 8,000, 7% MPD, 0–24% PEG 400, 3.75–4.5 mM MgCl2, 5–5.75 mM putrescine, 4.35–5 mM spermidine, pH 4.8–5.7. More specifically the concentrations of the variable parameters for the individual cryostabilizers were as follows. Stabilizer #1: 7% PEG 8,000, 7% MPD, 3.75 mM MgCl2, 5 mM putrescine, 4.35 mM spermidine, 380 mM NH4Cl, 0% PEG 400, pH 5.7; stabilizer #2 to #6: 7% PEG 8,000, 7% MPD, 3.75 mM MgCl2, 5.75 mM putrescine, 5 mM spermidine, 380 mM NH4Cl, PEG 400 6% (#2), 9% (#3), 12% (#4), 15% (#5), 18% (#6), pH 4.8; stabilizer #7: 6% PEG 8,000, 7% MPD, 4.5 mM MgCl2, 5.75 mM putrescine, 5 mM spermidine, 380 mM NH4Cl,, 24% PEG 400, pH 5.0. Crystals were soaked in stabilizer #1 for 100 min., then soaked in each of stabilizers #2 through #6 for 5–10 min., and stored overnight in stabilizer #7.
X-ray diffraction data measurement
Diffraction data were measured from crystals cooled to 100 K using 0.1° oscillations at beamline 8.3.1 at the Advanced Light Source (ALS) at Lawrence Berkeley National Laboratory (LBNL), which is equipped with an ADSC Q315r detector. In order to reduce diffraction spot overlap due to the large unit cell size of the ribosome crystals, crystals were adjusted to a position in which the c* axis ran across the detector at a ~45° angle to the horizontal plane, when c* was oriented parallel to the detector, i.e. normal to the x-ray beam. Furthermore, the crystal-to-detector distance was increased to 650 mm and the slit width was reduced to ~0.35 mradians during x-ray diffraction data collection. With these settings and exposure times of 3 minutes, diffraction spots to at least 2.1 Å could be observed. These were captured by a face-normal offset of the detector by about 17.5°. Diffraction data frames usually spanning only a total of 0.6° were indexed, integrated, and scaled using XDS from the XDS Program Package66. Over 470 data sets collected from 109 crystals were used.
Structure modeling and refinement
Structure factor phases were obtained by molecular replacement using the phenix.refine component of PHENIX67 and the following PDB entries as model input with mRNA, tRNAs, and ribosome recycling factor removed: 3R8S, 3R8T, 4GD1, and 4GD2. The resulting model was manually improved using Coot68 followed by multiple rounds of positional, atomic displacement parameter (ADP), and TLS refinement using the phenix.refine component of PHENIX. In order to improve the model of the second ribosome in the asymmetric unit, coordinate information of the first ribosome were used as a reference model during refinement.
For initial model building, electron density maps were calculated using structure factors from PHENIX refinements sharpened by applying a negative B-factor of −50 Å2, and with the figure of merit was dampened by applying a positive B-factor of 50 Å2. The resulting structure factors and figures of merit were subjected to density modification in DM69, using a resolution range of 30 – 2.1 Å and Sayre’s equation70,71. Finally, the DM-modified structure factors and figures of merit were used to calculate sharpened (2FObs–FCalc) and (FObs–FCalc) electron density maps. Homology models for various parts of the ribosome were used to guide manual rebuilding72–77 (including PDB 3A1P). Subsequent refinement and model building used feature-enhanced maps generated by phenix.fem in the PHENIX-1.9–1692 release, with default parameters plus the combined-omit map algorithm78. All maps presented in the figures use feature enhanced maps.
Molprobity25 analyses were carried out as follows. Prior to running the MolProbity web server, nucleotides 77–93, 841–847, 999–1012, and 1016–1042 were removed from 16S rRNA in the 30S subunit due to disorder. From the 50S subunit, E. coli ribosomal proteins L1, L9, L10, L11, and nucleotides 883–897, 1057–1090 and 2099–2188 of 23S rRNA were removed due to their high degree of disorder. High-resolution ribosome II in the present structure and PDB ID: 4U26 for the 2.8 Å structure79 were used for the analysis.
Phylogenetic comparisons of nucleotides in the ribosome were carried out using “The Comparative RNA Web Site and Project”80.
Supplementary Material
Acknowledgments
The authors wish to thank J. Doudna, A. S.-Y. Lee, A. Pulk, and P. Kranzusch for helpful discussions; P. Afonine and P. Adams for advice on the use of feature enhanced maps; G. Meigs and J. Holton for assistance at beam line 8.3.1 at the Advanced Light Source (ALS); and K. Diederichs for help with XDS. This work was supported by US National Institutes of Health (NIH) grant R01-GM65050 to J.H.D.C., by NIH grant 2R01GM079238 to S.C.B., D.S.T., M.R.W., and R.B.A., by the NIH project MINOS (Macromolecular Insights on Nucleic acids Optimized by Scattering)–grant R01GM105404–for beam line 8.3.1 at the ALS, and by the US Department of Energy–grant DEAC02-05CH11231–for beam line 8.3.1 at the ALS. J.N. was funded by a Human Frontiers in Science Program Long-Term Postdoctoral Fellowship.
Footnotes
Accession Codes
Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes ____, ____, ____, and _____.
References
- 1.Jelenc PC, Kurland CG. Nucleoside triphosphate regeneration decreases the frequency of translation errors. Proc Natl Acad Sci USA. 1979;76:3174–3178. doi: 10.1073/pnas.76.7.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim HD, Puglisi JD, Chu S. Fluctuations of transfer RNAs between classical and hybrid states. Biophysical Journal. 2007;93:3575–3582. doi: 10.1529/biophysj.107.109884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wohlgemuth I, Pohl C, Rodnina MV. Optimization of speed and accuracy of decoding in translation. EMBO J. 2010;29:3701–3709. doi: 10.1038/emboj.2010.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feldman MB, Terry DS, Altman RB, Blanchard SC. Aminoglycoside activity observed on single pre-translocation ribosome complexes. Nature Chemical Biology. 2010;6:244. doi: 10.1038/nchembio0310-244c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munro JB, Wasserman MR, Altman RB, Wang L, Blanchard SC. Correlated conformational events in EF-G and the ribosome regulate translocation. Nature Structural & Molecular Biology. 2010;17:1470–1477. doi: 10.1038/nsmb.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alden CJ, Kim SH. Solvent-accessible surfaces of nucleic acids. J Mol Biol. 1979;132:411–434. doi: 10.1016/0022-2836(79)90268-7. [DOI] [PubMed] [Google Scholar]
- 7.Rozenski J, Crain PF, McCloskey JA. The RNA Modification Database: 1999 update. Nucleic Acids Res. 1999;27:196–197. doi: 10.1093/nar/27.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chow CS, Lamichhane TN, Mahto SK. Expanding the nucleotide repertoire of the ribosome with post-transcriptional modifications. ACS Chemical Biology. 2007;2:610–619. doi: 10.1021/cb7001494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agris PF. The importance of being modified: roles of modified nucleosides and Mg2+ in RNA structure and function. Progress in Nucleic Acid Research and Molecular Biology. 1996;53:79–129. doi: 10.1016/s0079-6603(08)60143-9. [DOI] [PubMed] [Google Scholar]
- 10.Grosjean H, et al. Predicting the minimal translation apparatus: lessons from the reductive evolution of mollicutes. PLoS Genet. 2014;10:e1004363. doi: 10.1371/journal.pgen.1004363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Connor M, Gregory ST. Inactivation of the RluD pseudouridine synthase has minimal effects on growth and ribosome function in wild-type Escherichia coli and Salmonella enterica. Journal of Bacteriology. 2011;193:154–162. doi: 10.1128/JB.00970-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutgsell NS, Deutscher MP, Ofengand J. The pseudouridine synthase RluD is required for normal ribosome assembly and function in Escherichia coli. RNA. 2005;11:1141–1152. doi: 10.1261/rna.2550105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ejby M, Sorensen MA, Pedersen S. Pseudouridylation of helix 69 of 23S rRNA is necessary for an effective translation termination. Proc Natl Acad Sci USA. 2007;104:19410–19415. doi: 10.1073/pnas.0706558104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tollervey D, Lehtonen H, Jansen R, Kern H, Hurt EC. Temperature-sensitive mutations demonstrate roles for yeast fibrillarin in pre-rRNA processing, pre-rRNA methylation, and ribosome assembly. Cell. 1993;72:443–457. doi: 10.1016/0092-8674(93)90120-f. [DOI] [PubMed] [Google Scholar]
- 15.Basturea GN, Rudd KE, Deutscher MP. Identification and characterization of RsmE, the founding member of a new RNA base methyltransferase family. RNA. 2006;12:426–434. doi: 10.1261/rna.2283106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Purta E, O’Connor M, Bujnicki JM, Douthwaite S. YgdE is the 2′-O-ribose methyltransferase RlmM specific for nucleotide C2498 in bacterial 23S rRNA. Molecular microbiology. 2009;72:1147–1158. doi: 10.1111/j.1365-2958.2009.06709.x. [DOI] [PubMed] [Google Scholar]
- 17.Katz MJ, et al. Sudestada1, a Drosophila ribosomal prolyl-hydroxylase required for mRNA translation, cell homeostasis, and organ growth. Proc Natl Acad Sci USA. 2014;111:4025–4030. doi: 10.1073/pnas.1314485111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loenarz C, et al. Hydroxylation of the eukaryotic ribosomal decoding center affects translational accuracy. Proc Natl Acad Sci USA. 2014;111:4019–4024. doi: 10.1073/pnas.1311750111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singleton RS, et al. OGFOD1 catalyzes prolyl hydroxylation of RPS23 and is involved in translation control and stress granule formation. Proc Natl Acad Sci USA. 2014;111:4031–4036. doi: 10.1073/pnas.1314482111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore PB. How should we think about the ribosome? Annual Review of Biophysics. 2012;41:1–19. doi: 10.1146/annurev-biophys-050511-102314. [DOI] [PubMed] [Google Scholar]
- 21.Polikanov YS, Steitz TA, Innis CA. A proton wire to couple aminoacyl-tRNA accommodation and peptide-bond formation on the ribosome. Nature Structural & Molecular Biology. 2014;21:787–793. doi: 10.1038/nsmb.2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Polikanov YS, et al. Amicoumacin A Inhibits Translation by Stabilizing mRNA Interaction with the Ribosome. Molecular Cell. 2014;56:531–540. doi: 10.1016/j.molcel.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang W, Dunkle JA, Cate JHD. Structures of the ribosome in intermediate states of ratcheting. Science. 2009;325:1014–1017. doi: 10.1126/science.1175275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta crystallographica Section D, Biological Crystallography. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nesterchuk MV, Sergiev PV, Dontsova OA. Posttranslational Modifications of Ribosomal Proteins in Escherichia coli. Acta Naturae. 2011;3:22–33. [PMC free article] [PubMed] [Google Scholar]
- 27.Ge W, et al. Oxygenase-catalyzed ribosome hydroxylation occurs in prokaryotes and humans. Nature Chemical Biology. 2012;8:960–962. doi: 10.1038/nchembio.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersen TE, Porse BT, Kirpekar F. A novel partial modification at C2501 in Escherichia coli 23S ribosomal RNA. RNA. 2004;10:907–913. doi: 10.1261/rna.5259404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMurry LM, Algranati ID. Effect of polyamines on translation fidelity in vivo. European Journal of Biochemistry/FEBS. 1986;155:383–390. doi: 10.1111/j.1432-1033.1986.tb09502.x. [DOI] [PubMed] [Google Scholar]
- 30.Blanchard SC, Kim HD, Gonzalez RLJ, Puglisi JD, Chu S. tRNA dynamics on the ribosome during translation. Proc Natl Acad Sci USA. 2004;101:12893–12898. doi: 10.1073/pnas.0403884101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blanchard SC, Gonzalez RL, Kim HD, Chu S, Puglisi JD. tRNA selection and kinetic proofreading in translation. Nature structural & molecular biology. 2004;11:1008–1014. doi: 10.1038/nsmb831. [DOI] [PubMed] [Google Scholar]
- 32.Draper DE. A guide to ions and RNA structure. RNA. 2004;10:335–343. doi: 10.1261/rna.5205404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schuwirth BS, et al. Structures of the bacterial ribosome at 3.5 Å resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 34.Dunkle JA, et al. Structures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science. 2011;332:981–984. doi: 10.1126/science.1202692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nissen P, Ippolito JA, Ban N, Moore PB, Steitz TA. RNA tertiary interactions in the large ribosomal subunit: the A-minor motif. Proc Natl Acad Sci USA. 2001;98:4899–4903. doi: 10.1073/pnas.081082398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ben-Shem A, et al. The structure of the eukaryotic ribosome at 3.0 Å resolution. Science. 2011;334:1524–1529. doi: 10.1126/science.1212642. [DOI] [PubMed] [Google Scholar]
- 37.Kannan K, Vazquez-Laslop N, Mankin AS. Selective protein synthesis by ribosomes with a drug-obstructed exit tunnel. Cell. 2012;151:508–520. doi: 10.1016/j.cell.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 38.Sothiselvam S, et al. Macrolide antibiotics allosterically predispose the ribosome for translation arrest. Proc Natl Acad Sci USA. 2014;111:9804–9809. doi: 10.1073/pnas.1403586111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts MC. Resistance to macrolide, lincosamide, streptogramin, ketolide, and oxazolidinone antibiotics. Molecular Biotechnology. 2004;28:47–62. doi: 10.1385/MB:28:1:47. [DOI] [PubMed] [Google Scholar]
- 40.Bulkley D, Innis CA, Blaha G, Steitz TA. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc Natl Acad Sci USA. 2010;107:17158–17163. doi: 10.1073/pnas.1008685107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dunkle JA, Xiong L, Mankin AS, Cate JH. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc Natl Acad Sci USA. 2010;107:17152–17157. doi: 10.1073/pnas.1007988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 43.Harms J, et al. High resolution structure of the large ribosomal subunit from a mesophilic eubacterium. Cell. 2001;107:679–688. doi: 10.1016/s0092-8674(01)00546-3. [DOI] [PubMed] [Google Scholar]
- 44.Bhushan S, et al. SecM-stalled ribosomes adopt an altered geometry at the peptidyl transferase center. PLoS Biology. 2011;9:e1000581. doi: 10.1371/journal.pbio.1000581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gumbart J, Schreiner E, Wilson DN, Beckmann R, Schulten K. Mechanisms of SecM-mediated stalling in the ribosome. Biophysical Journal. 2012;103:331–341. doi: 10.1016/j.bpj.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bischoff L, Berninghausen O, Beckmann R. Molecular Basis for the Ribosome Functioning as an L-Tryptophan Sensor. Cell Reports. 2014;9:469–475. doi: 10.1016/j.celrep.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 47.Bhushan S, et al. Structural basis for translational stalling by human cytomegalovirus and fungal arginine attenuator peptide. Molecular Cell. 2010;40:138–146. doi: 10.1016/j.molcel.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 48.Martinez AK, et al. Crucial elements that maintain the interactions between the regulatory TnaC peptide and the ribosome exit tunnel responsible for Trp inhibition of ribosome function. Nucleic Acids Res. 2012;40:2247–2257. doi: 10.1093/nar/gkr1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Selmer M, et al. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 50.Kawai G, et al. Conformational rigidity of specific pyrimidine residues in tRNA arises from posttranscriptional modifications that enhance steric interaction between the base and the 2′-hydroxyl group. Biochemistry. 1992;31:1040–1046. doi: 10.1021/bi00119a012. [DOI] [PubMed] [Google Scholar]
- 51.Dalluge JJ, Hashizume T, Sopchik AE, McCloskey JA, Davis DR. Conformational flexibility in RNA: the role of dihydrouridine. Nucleic Acids Res. 1996;24:1073–1079. doi: 10.1093/nar/24.6.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song J, Burrage K, Yuan Z, Huber T. Prediction of cis/trans isomerization in proteins using PSI-BLAST profiles and secondary structure information. BMC Bioinformatics. 2006;7:124. doi: 10.1186/1471-2105-7-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Strader MB, et al. A proteomic and transcriptomic approach reveals new insight into beta-methylthiolation of Escherichia coli ribosomal protein S12. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M110.005199. M110 005199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anton BP, et al. RimO, a MiaB-like enzyme, methylthiolates the universally conserved Asp88 residue of ribosomal protein S12 in Escherichia coli. Proc Natl Acad Sci USA. 2008;105:1826–1831. doi: 10.1073/pnas.0708608105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sokoloski JE, Godfrey SA, Dombrowski SE, Bevilacqua PC. Prevalence of syn nucleobases in the active sites of functional RNAs. RNA. 2011;17:1775–1787. doi: 10.1261/rna.2759911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou J, Lancaster L, Donohue JP, Noller HF. How the ribosome hands the A-site tRNA to the P site during EF-G-catalyzed translocation. Science. 2014;345:1188–1191. doi: 10.1126/science.1255030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmeing TM, Huang KS, Kitchen DE, Strobel SA, Steitz TA. Structural insights into the roles of water and the 2′ hydroxyl of the P site tRNA in the peptidyl transferase reaction. Molecular Cell. 2005;20:437–448. doi: 10.1016/j.molcel.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 58.Gabdulkhakov A, Nikonov S, Garber M. Revisiting the Haloarcula marismortui 50S ribosomal subunit model. Acta Crystallographica Section D, Biological Crystallography. 2013;69:997–1004. doi: 10.1107/S0907444913004745. [DOI] [PubMed] [Google Scholar]
- 59.Karplus PA, Faerman C. Ordered water in macromolecular structure. Current Opinion in Structural Biology. 1994;4:770–776. [Google Scholar]
- 60.Jenner LB, Demeshkina N, Yusupova G, Yusupov M. Structural aspects of messenger RNA reading frame maintenance by the ribosome. Nature Structural & Molecular Biology. 2010;17:555–560. doi: 10.1038/nsmb.1790. [DOI] [PubMed] [Google Scholar]
- 61.Wang L, et al. Allosteric control of the ribosome by small-molecule antibiotics. Nature Structural & Molecular Biology. 2012;19:957–963. doi: 10.1038/nsmb.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Munro JB, Altman RB, O’Connor N, Blanchard SC. Identification of two distinct hybrid state intermediates on the ribosome. Molecular Cell. 2007;25:505–517. doi: 10.1016/j.molcel.2007.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aitken CE, Marshall RA, Puglisi JD. An oxygen scavenging system for improvement of dye stability in single-molecule fluorescence experiments. Biophysical Journal. 2008;94:1826–1835. doi: 10.1529/biophysj.107.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dave R, Terry DS, Munro JB, Blanchard SC. Mitigating unwanted photophysical processes for improved single-molecule fluorescence imaging. Biophysical Journal. 2009;96:2371–2381. doi: 10.1016/j.bpj.2008.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Qin F. Restoration of single-channel currents using the segmental k-means method based on hidden Markov modeling. Biophysical Journal. 2004;86:1488–1501. doi: 10.1016/S0006-3495(04)74217-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kabsch W. Xds. Acta crystallographica Section D, Biological Crystallography. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D, Biological Crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta crystallographica Section D, Biological Crystallography. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 69.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta crystallographica Section D, Biological Crystallography. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sayre D. Least-squares phase refinement. II High-resolution phasing of a small protein. Acta Crystallographica Section A. 1974;30:180–184. [Google Scholar]
- 71.Zhang KYJ, Main P. The use of Sayre’s equation with solvent flattening and histogram matching for phase extension and refinement of protein structures. Acta Crystallographica Section A. 1990;46:377–381. [Google Scholar]
- 72.Wimberly BT, Guymon R, McCutcheon JP, White SW, Ramakrishnan V. A detailed view of a ribosomal active site: the structure of the L11-RNA complex. Cell. 1999;97:491–502. doi: 10.1016/s0092-8674(00)80759-x. [DOI] [PubMed] [Google Scholar]
- 73.Cho JH, et al. Energetically significant networks of coupled interactions within an unfolded protein. Proc Natl Acad Sci USA. 2014;111:12079–12084. doi: 10.1073/pnas.1402054111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Byrgazov K, et al. Structural basis for the interaction of protein S1 with the Escherichia coli ribosome. Nucleic Acids Res. 2015;43:661–673. doi: 10.1093/nar/gku1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lombardi C, et al. A compact viral processing proteinase/ubiquitin hydrolase from the OTU family. PLoS Pathog. 2013;9:e1003560. doi: 10.1371/journal.ppat.1003560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wallgren M, et al. Extreme temperature tolerance of a hyperthermophilic protein coupled to residual structure in the unfolded state. J Mol Biol. 2008;379:845–858. doi: 10.1016/j.jmb.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 77.Luo X, et al. Structural and functional analysis of the E. coli NusB-S10 transcription antitermination complex. Molecular Cell. 2008;32:791–802. doi: 10.1016/j.molcel.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Afonine PV, et al. FEM: Feature Enhanced Map. Acta Crystallographica. Section D, Biological Crystallography. doi: 10.1107/S1399004714028132. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Noeske J, et al. Synergy of streptogramin antibiotics occurs independently of their effects on translation. Antimicrobial Agents and Chemotherapy. 2014;58:5269–5279. doi: 10.1128/AAC.03389-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cannone JJ, et al. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3:2. doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.