

Abstract
Purpose
The outcome for patients with metastatic or recurrent sarcoma remains poor. Adoptive therapy with tumor-directed T cells is an attractive therapeutic option but has never been evaluated in sarcoma.
Patients and Methods
We conducted a phase I/II clinical study in which patients with recurrent/refractory human epidermal growth factor receptor 2 (HER2) –positive sarcoma received escalating doses (1 × 104/m2 to 1 × 108/m2) of T cells expressing an HER2-specific chimeric antigen receptor with a CD28.ζ signaling domain (HER2-CAR T cells).
Results
We enrolled 19 patients with HER2-positive tumors (16 osteosarcomas, one Ewing sarcoma, one primitive neuroectodermal tumor, and one desmoplastic small round cell tumor). HER2-CAR T-cell infusions were well tolerated with no dose-limiting toxicity. At dose level 3 (1 × 105/m2) and above, we detected HER2-CAR T cells 3 hours after infusion by quantitative polymerase chain reaction in 14 of 16 patients. HER2-CAR T cells persisted for at least 6 weeks in seven of the nine evaluable patients who received greater than 1 × 106/m2 HER2-CAR T cells (P = .005). HER2-CAR T cells were detected at tumor sites of two of two patients examined. Of 17 evaluable patients, four had stable disease for 12 weeks to 14 months. Three of these patients had their tumor removed, with one showing ≥ 90% necrosis. The median overall survival of all 19 infused patients was 10.3 months (range, 5.1 to 29.1 months).
Conclusion
This first evaluation of the safety and efficacy of HER2-CAR T cells in patients with cancer shows the cells can persist for 6 weeks without evident toxicities, setting the stage for studies that combine HER2-CAR T cells with other immunomodulatory approaches to enhance their expansion and persistence.
INTRODUCTION
Sarcomas are a diverse group of malignancies that include osteosarcoma, Ewing sarcoma, rhabdomyosarcoma, and nonrhabdomyosarcoma soft tissue sarcomas, such as synovial sarcoma or desmoplastic small round cell tumors. Although patients with local disease have an excellent outcome, the prognosis of patients with advanced-stage disease remains poor.1,2 Cell therapy in the form of high-dose chemotherapy with autologous stem-cell rescue has been extensively explored for sarcomas. However, most studies have not shown a significant survival benefit compared with standard chemotherapy, indicating that more specific cell therapies are needed to improve outcomes.3,4
Immunotherapy with antigen-specific T cells may benefit patients with sarcoma because immune-mediated killing does not rely on pathways used by conventional therapies to which such tumors are often resistant.5,6 Adoptive transfer of T cells, genetically modified to express chimeric antigen receptors (CARs), has shown great promise in early-phase clinical studies for the therapy of CD19-positive malignancies.7–10 Clinical experience using this approach for solid tumors, however, is much more limited.11,12 CARs recognize antigens expressed on the cell surface of tumor cells,13 and several potential CAR target antigens have been identified for sarcoma, including human epidermal growth factor receptor 2 (HER2), GD2, interleukin (IL) -11Rα, and B7H3.14–17 Although sarcoma cells are often HER2-positive, the HER2 gene locus is not amplified in this disease.18,19 Thus, sarcomas belong to a large group of malignancies, including cancers of the lung, ovary, prostate, and brain, that express HER2 at levels too low for HER2 monoclonal antibodies (MAbs) to be effective.14,20
We and others have previously shown that even malignancies that express HER2 at low levels can be targeted with T cells that express HER2-specific CARs.14 These HER2-CAR T cells kill both bulk tumor cells and tumor-initiating cells6 and have potent antitumor activity in preclinical animal models.
Despite the potential value of HER2-specific CARs, significant safety concerns about the use of these receptors arose after the rapid onset of fatal respiratory failure in a patient who received 1 × 1010 T cells expressing a CAR containing HER2-specific ectodomain derived from the HER2-specific MAb trastuzumab and a CD28.CD137.ζ endodomain and IL-2 after lymphodepleting chemotherapy.21 Therefore, we developed a dose-escalation study of a second-generation HER2-specific CAR containing an ectodomain derived from the HER2-specific MAb FRP5 and a CD28.ζ endodomain in patients with recurrent/refractory HER2-positive sarcoma. We began with an ultra-low dose of HER2-CAR T cells (1 × 104/m2) as a single agent without the administration of IL-2 or lymphodepleting chemotherapy and escalated the cell dose to 1 × 108/m2. We now report the safety, persistence, and antitumor activity of the infused cells.
PATIENTS AND METHODS
Patients
This study (ClinicalTrials.gov identifier: NCT00902044) was approved by the institutional review board at Baylor College of Medicine (Houston, TX) and by the US Food and Drug Administration. Patients were eligible for the study if they had a diagnosis of refractory or metastatic HER2-positive osteosarcoma (later modified to sarcoma) not treatable by surgical resection and with disease progression after receiving at least one prior systemic therapy. HER2 positivity was determined by immunohistochemistry.14 Patients had to have completed (and recovered from) experimental or cytotoxic therapies at least 4 weeks before study entry. Patients were excluded if they had abnormal left ventricular function (LVEF). In addition, patients with a serum bilirubin of more than 3× the upper limit of normal, ALT or AST more than 5× upper limit of normal, hemoglobin less than 9 g/dL, WBC less than 2,000/μL, absolute neutrophil count less than 1,000/μL, or platelets less than 100,000/μL were excluded, as were patients with a Karnofsky/Lansky score of less than 50 or positive serology for HIV.
Study Description
All patients had imaging with computed tomography, magnetic resonance imaging, and/or positron emission tomography to assess overall disease burden before T-cell infusion. Patients received escalating doses of HER2-CAR T cells (1 × 104 to 1 × 108/m2) with the following eight dose levels: 1 × 104/m2, 3 × 104/m2, 1 × 105/m2, 1 × 106/m2, 3 × 106/m2, 1 × 107/m2, 3 × 107/m2, and 1 × 108/m2. Peripheral-blood samples were obtained before T-cell infusion and at predetermined time points after infusion to evaluate for toxicity and T-cell persistence and expansion. Clinical response to HER2-CAR T cells was assessed by radiographic imaging 6 weeks after the T-cell infusion. Patients were eligible to receive additional T-cell infusions if they had clinical benefit, defined as a complete response, partial response, or stable disease. All patients were infused between June 2010 and March 2013. Follow-up continued until September 1, 2013.
Generation and Transduction of HER2-CAR T Cells
HER2-CAR T cells were generated according to current Good Manufacturing Practice guidelines. Briefly, peripheral-blood mononuclear cells were activated with immobilized CD3 antibody (Ortho Biotech, Toronto, Ontario, Canada) or immobilized CD3 and CD28 antibodies (P 6, 12, 13, 16, 17, 18; Miltenyi, Cologne, Germany) and recombinant IL-2 (100 U/mL; Proleukin, Chiron, Emeryville, CA) and then transduced on day 3 with retroviral particles encoding an HER2.CD28.ζ-CAR (Appendix, online only) in 24-well plates precoated with Retronectin (FN CH-296; Takara, Shiga, Japan). After transduction, T-cell lines were expanded in the presence of IL-2 (50 to 100 U/mL) added twice weekly until the specified cell dose was achieved. After expansion, HER2-CAR T cells were tested for sterility, HLA identity, immunophenotype, and HER2 specificity at the time of cryopreservation. Specificity was tested in a 4-hour chromium-51 release cytotoxicity assay.
Clinical Response Criteria
Clinical response to T-cell infusion was evaluated by comparing disease identified by computed tomography, magnetic resonance imaging, and/or positron emission tomography images obtained before infusion to images obtained 6 weeks after infusion or as clinically indicated. Rebiopsy of residual masses was not mandatory for study participation. All responses were determined using RECIST.22
Statistical Analysis
This phase I/II trial used the modified continual reassessment method to determine the maximum-tolerated dose. Three patients were enrolled onto dose level 1, two patients each onto dose levels 2 through 7, and four patients onto dose level 8. Transgene expression and multiplex analyses were summarized over time using descriptive statistics. The significance between groups was determined using the t test or Fisher's exact test. P < 0.05 was considered statistically significant. The survival curve was constructed using the Kaplan-Meier method.
RESULTS
Patient Characteristics
The clinical and disease-specific characteristics of the 19 patients who received HER2-CAR T cells are listed in Table 1. Their median age at the time of T-cell infusion was 17 years (range, 7.7 to 29.6 years). Sixteen patients had osteosarcoma, one had Ewing sarcoma, one had a primitive neuroectodermal tumor, and one had a desmoplastic small round cell tumor. HER2 positivity was confirmed by immunohistochemistry (Appendix Table A1, online only). All patients had refractory or recurrent metastatic disease at the time of T-cell infusion and had experienced treatment failure with one or more conventional chemotherapy regimens. Seventeen of 19 patients had undergone metastasectomies (median, four metastasectomies; range, one to six metastatectomies), 11 of 19 patients had received radiation therapy, and 18 of 19 patients had received one or more salvage regimens (median, three regimens; range, one to five regimens) before T-cell infusion. All enrolled patients had a Karnofsky/Lansky performance score of ≥ 60 and a normal LVEF.
Table 1.
Patient Characteristics
| Patient No. | Age (years) | Sex | Diagnosis and Stage | Prior Treatment |
|||
|---|---|---|---|---|---|---|---|
| Chemotherapy | Surgery | XRT | Other and/or Investigational Agents | ||||
| 1 | 21.2 | Female | OS, M | (1) MAPIE; (2) Ifos, VP16, HDMTX | LS | Y | (1) Bevacizumab; (2) sunitinib |
| 2 | 17.4 | Female | OS, L | (1) MAP; (2) Ifos | LS; M (3)* | N | (1) L-MTP-PE |
| 3 | 14.0 | Female | OS, M | (1) MAP; (2) Ifos, VP16, HDMTX | LS; M (5) | Y | (1) GCB; (2) L-MTP-PE, oral CPM; (3) sunitinib |
| 4 | 17.1 | Male | OS, L | (1) MAPIE | LS; M (4) | Y | (1) SCH717454; (2) L-MTP-PE; (3) sunitinib; (4) GCB, docetaxel, bevacizumab; (5) Doxo |
| 5 | 7.7 | Female | OS, M | (1) MAP; (2) Ifos, Carbo, VP16 (3) HD Ifos, HDMTX; (4) HDMTX | LS; M (3) | N | (1) L-MTP-PE, GCB; (2) L-MTP-PE, oral CPM; (3) denosumab, Doxo |
| 6 | 25.3 | Female | OS, L | (1) MAP; (2) Doxo, Ifos (3) HDMTX; (4) Ifos | Primary en bloc; M (1) | N | (1) L-MTP-PE, docetaxel; (2) GCB, bevacizumab, L-MTP-PE |
| 7 | 29.6 | Male | OS, L | (1) MAPIE | LS; M (3) | N | (1) L-MTP-PE; (2) L-MTP-PE, GCB |
| 8 | 15.4 | Female | OS, L | (1) MAP; (2) HD Ifos; (3) HDMTX, Cis; (4) oral CPM | Amp for quarter; M (4) | Y | (1) GCB; (2) Doxo, bevacizumab; (3) L-MTP-PE; (4) sorafenib |
| 9 | 21.1 | Female | OS, M | (1) MAP; (2) HD Ifos | LS; M (2) | Y | (1) Doxo; (2) L-MTP-PE |
| 10 | 14.0 | Female | PNET, L | (1) VDCIE; (2) Carbo, VP16, Mel + auto; (3) metronomic VCR; (4) TMZ, irinotecan | Rt kidney; M (1) | Y | (1) Sorafenib |
| 11 | 16.6 | Male | OS, L | (1) MAP; (2) Ifos, VP16 | LS; M (3) | N | (1) L-MTP-PE; (2) sunitinib; (3) Doxo |
| 12 | 11.3 | Male | DSCRT, L | (1) VDC × 2; (2) topotecan, CPM (3) TMZ, irinotecan; (4) vinorelbine, CPM | Primary en bloc; Hep emb | Y | (1) Sorafenib; (2) PEG-IFN |
| 13 | 20.6 | Male | OS, M | (1) MAPIE; (2) TMZ | LS; M (4) | Y | (1) L-MTP-PE; (2) bevacizumab, GCB, docetaxel; (3) sorafenib |
| 14 | 21.8 | Female | OS, L | (1) Intra-arterial Cis, Doxo, HDMTX, Ifos; (2) intrapleural Cis × 2; (3) HDMTX | LS; M (3) | N | (1) L-MTP-PE, oral CPM (2) Doxo, bevacizumab, HDMTX |
| 15 | 11.0 | Male | OS, M | (1) MAP | Amp; M (6) | Y | (1) L-MTP-PE; (2) GCB; (3) Doxo, bevacizumab; (4) sorafenib; (5) pazopanib, lapatinib |
| 16 | 19.3 | Male | OS, L | (1) Doxo, intra-arterial Cis, HDMTX (2) HD Ifos; (3) oral CPM; (4) Doxo, Ifos | LS; M (2) | N | (1) L-MTP-PE; (2) SCH717454 (IGF-1R MAb); (3) IFN; (4) sunitinib |
| 17 | 16.8 | Male | ES, M | (1) VDCIE; (2) VCR, TMZ, irinotecan; (3) vinoralbine, CPM | M (1) | Y | (1) Temsirolimus, IMC-A12; (2) Doxo; (3) vorinostat, pazopanib; (4) pazopanib, lapatinib |
| 18 | 14.5 | Female | OS, L | (1) MAPIE | LS; M (3) | N | None |
| 19 | 16.5 | Male | OS, M | (1) MAP; (2) HD Ifos, VP16 × 2 | LS; M (5) | Y | (1) Imetelstat |
Abbreviations: Amp, amputation; auto, autologous transplantation; Carbo, carboplatin; Cis, cisplatin; CPM, cyclophosphamide; Doxo, doxorubicin; DSRCT, desmoplastic small round cell tumor; ES, Ewing sarcoma; GCB, gemcitabine; HD, high dose; HDMTX, high-dose methotrexate; Hep emb, hepatic embolization; IFN, interferon; Ifos, ifosfamide; IGF-1R, insulin-like growth factor 1 receptor; L, localized; L-MTP-PE, liposome-encapsulated muramyl tripeptide phosphatidylethanolamine; LS, limb salvage; M, metastatic; M, metastasectomy; MAP, methotrexate, doxorubicin, and cisplatin; MAPIE, methotrexate, doxorubicin, cisplatin, ifosfamide, and etoposide; Mel, melphalan; N, no; OS, osteosarcoma; PEG-IFN, pegylated interferon; PNET, primitive neuroectodermal tumor; Rt, right; TMZ, temozolomide; VCR, vincristine; VDC, vincristine, doxorubicin, and cyclophosphamide; VDCIE, vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide; VP16, etoposide; XRT, radiation therapy; Y, yes.
No. of procedures.
Generation and Characterization of HER2-CAR T Cells
HER2-CAR T cells were successfully generated for all patients. The median time to manufacture the cell product for clinical use was 13.5 days (range, 10 to 21 days). Greater than 97.8% of the transduced cells were CD3+ (mean, 99.2%; range, 97.8% to 99.6%), and both CD3+/CD8+ (mean, 62.7%; range, 37.8% to 80.1%) and CD3+/CD4+ (mean, 31.5%; range, 17.2% to 55.3%) subsets were present in all products (Appendix Fig A1A, online only). CAR T-cell products also contained naive (CD3+/CD45RA+; mean, 22.6%; range, 6.0% to 37.2%), effector memory (CD3+/CD45RO+/CCR7–/CD62L–; mean, 32.2%; range, 7.7% to 57.9%), and central memory T cells (CD3+/CD45RO+/CD62L+; mean, 45.8%; range, 17.9% to 88.0%). A median of 65.2% (range, 36.2% to 88%) of T cells were positive for HER2-CAR expression as judged by fluorescence-activated cell sorting analysis (Appendix Fig A1B). In a standard chromium-51 release cytotoxicity assay, HER2-CAR T cells had significant cytotoxic activity against HER2-positive (NCI-H1299, LM7) target cells, whereas nontransduced T cells did not (P < .001; Appendix Fig A1C). Only background killing was present when HER2-negative (K562, MDA-MB468) target cells were cultured with HER2-CAR and nontransduced T cells.
Administration and Safety of HER2-CAR T Cells
Patients received between 1 × 104/m2 and 1 × 108/m2 HER2-CAR T cells on eight dose levels. Thirteen patients received one infusion, four patients received two infusions, one patient received four infusions, and one patient received nine infusions. None of the patients had adverse events related to the T-cell infusion except for one patient (patient 16) on the highest dose levels, who developed fever within 12 hours after T-cell infusion, which resolved with ibuprofen (Appendix Tables A2 and A3, online only). Concentrations of plasma cytokines (granulocyte-macrophage colony-stimulating factor, interferon gamma, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12p70, IL-13, and tumor necrosis factor α) were determined after infusion by multiplex analysis at 3 hours and again at 1, 2, 4, and 6 weeks after infusion (Fig 1; Appendix Fig A2, online only). There was a significant increase (P < .05) in the plasma concentration of IL-8 as early as 1 week after infusion, and this persisted for up to 4 weeks. No significant change in any other cytokine was observed. At 6 weeks after infusion, repeat cardiac function studies showed LVEFs unchanged from baseline.
Fig 1.
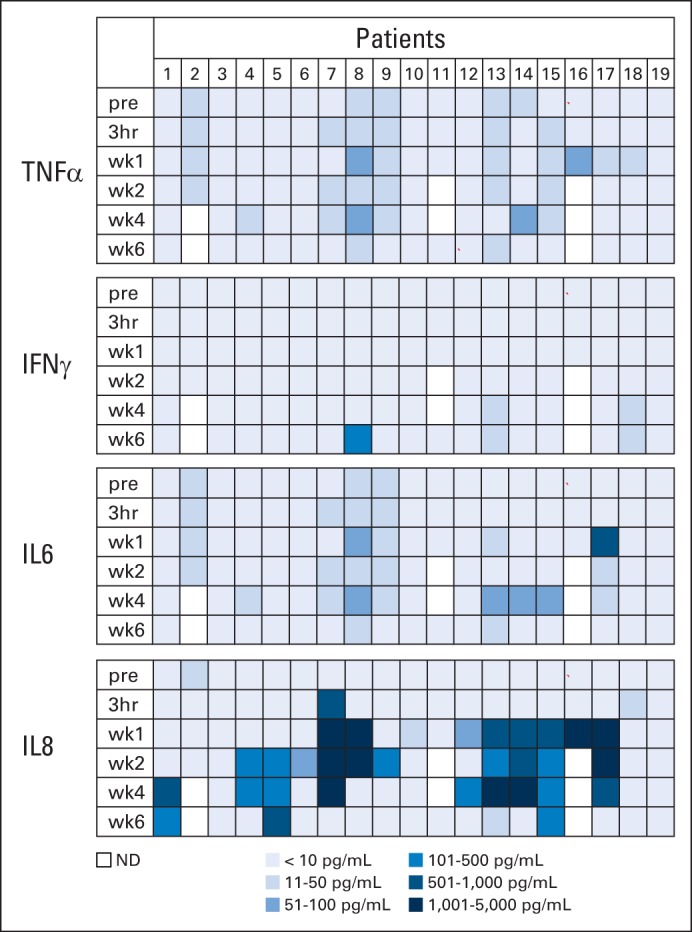
Plasma cytokine levels after human epidermal growth factor receptor 2 (HER2) chimeric antigen receptor (CAR) T-cell infusion. Plasma cytokine levels after HER2-CAR T-cell infusion were measured by multiplex analysis. Results for interferon gamma (IFNγ), tumor necrosis factor α (TNF-α), interleukin (IL) -6, and IL-8 are shown. There was a significant increase of plasma IL-8 levels at 1 (P = .028), 2 (P = .006), and 4 (P = .001) weeks after T-cell infusion. Results for granulocyte-macrophage colony-stimulating factor, IL-1β, IL-2, IL-4, IL-5, IL-7, IL-10, IL-12p70, and IL-13 are shown in Appendix Figure A2. pre, 3 hours (hr) and week (wk) 1, 2, 4, and 6 after infusion; ND, not detected.
In Vivo Detection and Persistence of HER2-CAR T Cells
We detected HER2-CAR T cells in vivo by quantitative polymerase chain reaction (qPCR) analysis of peripheral-blood mononuclear cells. From dose level 3 (1 × 105/m2) and higher, we detected HER2-CAR T cells in the peripheral blood of 14 of 16 patients (median, 6.5 copies/μg DNA; range, 0 to 944 copies/μg DNA; Figs 2A to 2C), and the copy number correlated with the infused T-cell dose (Fig 2D). After the 3-hour time point, there was a rapid decline in the frequency of HER2-CAR T cells, but low-level signal could be detected 6 weeks after infusion in seven of the nine evaluable patients who had received greater than 1 × 106/m2 HER2-CAR T cells (≤ v > 1 × 106/m2, P = .002; Fig 2E). At 3 months, we could detect HER2-CAR T cells in four of 13 evaluable patients, at 6 months in three of seven patients, at 9 months in one of two patients, at 12 months in none of five patients, at 18 months in one of two patients, and at 24 months in none of one patient (Fig 2F). Thus, although we found no evidence for HER2-CAR T-cell expansion in peripheral blood after infusion, these cells could persist long term. Five patients received at least two doses of HER2-CAR T cells, and we observed a similar pattern of HER2-CAR T-cell persistence after both infusions (Appendix Fig A3, online only).
Fig 2.

In vivo persistence of human epidermal growth factor receptor 2 (HER2) chimeric antigen receptor (CAR) T cells. (A to C) In vivo persistence of T cells at each dose level (DL). (D) Correlation of cell dose and level of transgene detection 3 hours after HER2-CAR T-cell infusion. (E) Detection of HER2-CAR T cells 6 weeks after infusion was dependent on the infused T-cell dose (≤ v > 1 × 106/m2, P = .002). (F) HER2-CAR T cells were detected for up to 18 months after infusion.
HER2-CAR T-Cell Traffic to Tumor Sites
Five patients had tumors removed 9 to 15 weeks after HER2-CAR T-cell infusion. For two of five patients (patient 10, soft tissue metastasis; patient 18, metastatic lesion left femur), we received formalin-fixed slides and fresh frozen tissue. In both tumors, CD3+ T cells were present by immunohistochemistry (Fig 3A) and HER2-CAR T cells were present on qPCR analysis (Fig 3B) even though no qPCR signal from HER2-CAR T cells was detected in the peripheral blood obtained at the same time as the resected tumor (Fig 3B), indicating that HER2-CAR T cells preferentially home to, persist in, or expand at tumor sites. We detected T cells within the other three tumor samples by using a CD3-specific antibody (data not shown) but lacked sufficient material for qPCR analysis.
Fig 3.

Human epidermal growth factor receptor 2 (HER2) chimeric antigen receptor (CAR) T-cell homing to tumor sites. (A, C) patient 10; (B, D) patient 18. (A and B) Immunohistochemistry for CD3 expression in tumor biopsy. (C and D) Transgene detection in tumor biopsy and corresponding peripheral-blood sample.
Clinical Responses After HER2-CAR T-Cell Infusion
We measured clinical responses by pre– and post–T-cell infusion imaging as detailed in Patients and Methods. These data are listed in Table 2. Of 17 evaluable patients, four had stable disease for 12 weeks to 14 months. One of the patients with progressive disease (patient 4) received salvage chemotherapy followed by a second dose of T cells for metastatic lymph node disease. After this second infusion, he had a partial response that lasted for 9 months (Fig 4C). Three patients with stable disease (patients 11, 14, and 18) received no additional therapy and had their residual tumor removed. The sample from patient 14 showed ≥ 90% necrosis, demonstrating antitumor activity of infused HER2-CAR T cells (Fig 4B). All three of these patients remain in remission at 6, 12, and 16 months with no further treatment. With a median follow-up time of 10.1 months (range, 1.1 to 37 months), the median overall survival (OS) time is 10.3 months (range, 5.1 to 29.1 months; Fig 4A).
Table 2.
Patient Outcome
| Patient No. | Diagnosis | Disease at T-Cell Infusion | T-Cell Dose | Outcome | Overall Survival (days) |
|---|---|---|---|---|---|
| 1 | OS | Right hip, multiple bones | 1 × 104/m2 | PD | 1,109* |
| 2 | OS | Sacrum | 1 × 104/m2 | NE | 34 |
| 3 | OS | Right hip, lung | 1 × 104/m2 | PD | 584 |
| 4 | OS | Lung | 3 × 104/m2 | PD; surgery/salvage chemotherapy for PD; second infusion (1 × 105/m2); PR for 9 months | 874 |
| 5 | OS | Lung | 3 × 104/m2 | PD; second infusion (1 × 105/m2); PD | 310 |
| 6 | OS | Lung | 1 × 105/m2 | PD | 107 |
| 7 | OS | Lung | 1 × 105/m2 | PD; second infusion (1 × 105/m2); PD | 303 |
| 8 | OS | Pelvis, spine, lung | 1 × 106/m2 | PD | 120 |
| 9 | OS | Lung | 1 × 106/m2 | PD | 151 |
| 10 | PNET | Sacrum | 3 × 106/m2 | PD; tumor removed (no necrosis) | 451 |
| 11 | OS | Lung/pleura | 3 × 106/m2 | SD for 15 weeks; tumor removed (no necrosis); remains in remission | 584* |
| 12 | DSCRT | Liver | 1 × 107/m2 | 8 additional infusions; SD for 14 months | 528* |
| 13 | OS | Lung | 1 × 107/m2 | PD | 268 |
| 14 | OS | Lung/pleura | 3 × 107/m2 | Second infusion; SD for 12 weeks; tumor removed (≥ 90% necrosis); 2 additional infusions; remains in remission | 446* |
| 15 | OS | Bone, chest wall, brain, spine, marrow | 3 × 107/m2 | PD | 156 |
| 16 | OS | Pleura, liver, subdiaphragmatic | 1 × 108/m2 | NE | 389* |
| 17 | ES | Lung | 1 × 108/m2 | PD | 153 |
| 18 | OS | Left femur | 1 × 108/m2 | Second infusion; SD for 12 weeks; tumor removed (no necrosis); remains in remission | 290* |
| 19 | OS | Lung and bone | 1 × 108/m2 | PD | 164 |
Abbreviations: DSRCT, desmoplastic small round cell tumor; ES, Ewing sarcoma; NE, not evaluable; OS, osteosarcoma; PD, progressive disease; PR, partial response; PNET, primitive neuroectodermal tumor; SD, stable disease.
Patient still alive.
Fig 4.
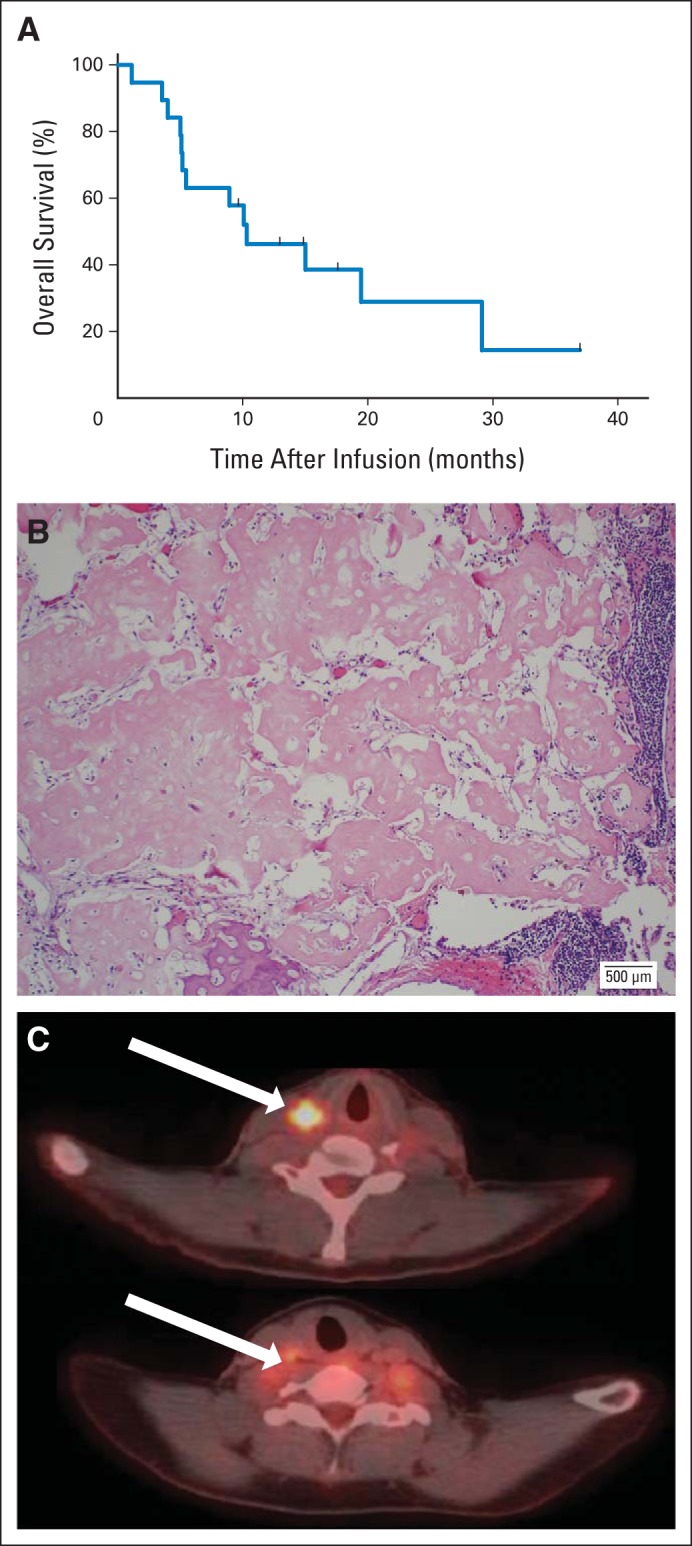
Outcome after human epidermal growth factor receptor 2 (HER2) chimeric antigen receptor (CAR) T-cell infusion. (A) Kaplan-Meier curve of all infused patients (N = 19). (B) Prominent necrosis (patient 14) of tumor after HER2-CAR T-cell infusion. (C) Positron emission tomography images (patient 4) before (top arrow) and 6 weeks after (bottom arrow) HER2-CAR T-cell infusion.
DISCUSSION
In this study, we evaluated the feasibility and safety of administering escalating doses of HER2-CAR T cells to patients with recurrent or refractory osteosarcoma. We found that infusion of up to 1 × 108/m2 HER2-CAR T cells was well tolerated, and four of 17 evaluable patients had stable disease for 12 weeks to 14 months. After removal of residual metastasis, three patients remain in remission at 6, 12, and 16 months.
Targeting HER2 with antigen-specific T cells is an attractive strategy to expand HER2-targeted immunotherapies to malignancies that are HER2 positive but are insensitive to HER2 antibodies because they are not HER2 gene amplified.23,24 In this study, infusion of up to 1 × 108/m2 of HER2-CAR T cells was safe, so we did not reach a dose-limiting toxicity. Our findings agree with two other studies in which T cells with conventional HER2-specific α/ß T-cell receptors have been safely given to human patients in doses as high as 4.1 × 1010 without significant adverse effects, producing objective tumor responses in three of six patients.25,26 Although there is no intrinsically impassable barrier to the use of HER2-specific T cells, a report of death from acute respiratory failure after receiving lymphodepleting chemotherapy and 1 × 1010 T cells expressing a third-generation HER2-CAR with a CD28.CD137.ζ endodomain remains troubling.21 The difference in the observed toxicity profile between this fatal outcome and our own observations may be a result of several factors. In this study, we did not use lymphodepleting chemotherapy before T-cell transfer, gave a 2-log lower maximum dose of cells, omitted postinfusion IL-2, and did not use a third-generation HER2-CAR with a CD137 signaling domain, which has been implicated in antigen-independent T-cell activation.27 In addition, the HER2-specific scFvs of each CAR was derived from different MAbs (trastuzumab v FRP5), which bind to distinct epitopes.28,29
Although we observed no postinfusion expansion of HER2-CAR T cells in the peripheral blood, these cells did persist for up to 18 months after infusion without adverse effects. T-cell persistence correlated with infused T-cell dose alone, but not with the presence of CD4+ T cells and/or central memory T cells in the T-cell product.30 The lack of CAR T-cell expansion we observed may in part be attributable to the lack of lymphodepletion before HER2-CAR T-cell infusion, although other studies have observed CAR T-cell expansion even without prior lymphodepletion when the CAR T cells are directed to the CD19 antigen.7,31,32 CD19, however, is expressed by normal B cells, which also express a multiplicity of costimulatory ligands that may provide additional activation and expansion signals to CD19-CAR T cells. HER2-positive malignant cells, in contrast, lack such costimulatory properties. Although the expansion of HER2-CAR T cells could also be limited by the development of human antimouse antibodies against murine scFvs,33,34 none of our patients had developed such antibodies by 6 weeks after T-cell infusion (data not shown).
Although levels of transduced cells remained low in peripheral blood, this compartment contains only 2% of the entire lymphocyte pool.35 Thus, the lack of expansion in the peripheral blood does not exclude expansion of HER2-CAR T cells in the presence of antigen at tumor sites. Indeed, we detected HER2-CAR T cells by qPCR in two of two fresh frozen biopsy samples at 6 and 12 weeks after T-cell infusion, even when they were absent in the peripheral blood, suggesting specific homing, persistence, or expansion at sites of disease. Although these results are encouraging, T-cell homing to tumor sites has to be confirmed in a larger cohort of patients. Additional genetic modification of T cells to express chemokine receptors may be needed to enhance T-cell homing to tumor sites.36
Although several patients had stable disease after HER2-CAR T-cell infusion, no radiologic complete responses were observed. As others have reported, conventional RECIST criteria may underestimate the antitumor effects of immunotherapies, leading to apparently limited improvements in progression-free survival even in the presence of significantly longer OS rates.37 Indeed, although we observed a median progression-free survival time of 6 weeks, the median OS time was 10.3 months. Sixteen patients infused with HER2-CAR T cells had recurrent osteosarcoma. Only limited data are available regarding median OS for patients with recurrent osteosarcoma who do not achieve a complete response. In two studies, the Cooperative Osteosarcoma Study Group reported median OS times of 5.8 and 7.6 months,38,39 and in another study, the mean time to death was 11 months for patients who did not achieve a second complete response.40
In this dose-escalation study, the clinical benefit of HER2-CAR T-cell infusion was clearly limited, indicating that further manipulation of the immune system will be essential for worthwhile benefits to be obtained. One possibility will be to combine HER2-CAR T cells with one or more of the checkpoint antibodies now becoming available for cancer immunotherapy and thereby increase T-cell activation and prolong in vivo survival. Additional engineering of the CAR T cells themselves may also be of value, for example by forcing transgenic expression of stimulatory cytokines or by rendering HER2-CAR T cells resistant to the inhibitory tumor microenvironment.13 Because enhancing the potency of HER2-CAR T cells may result in on target/off cancer toxicity, additional genetic modifications to increase safety may also be needed, such as an inducible suicide gene or inhibitory receptors to limit the effector function of T cells to tumor sites.41,42
In summary, our study shows that a safe dose of HER2-CAR T cells can be established for patients with cancer and that these cells can traffic to tumor sites and persist at low levels for more than 6 weeks in a dose-dependent manner. These data will facilitate subsequent clinical trials to further augment the expansion, function, and persistence of HER2-directed T cells.
Acknowledgment
We thank the referring physicians, the staff of the clinical research unit for assisting with patient follow-up, and the staff of the Good Manufacturing Practice facility for assisting in T-cell line production and analysis. We also thank the patients who participated in this study; and the parents, who entrusted the care of their children to us.
Appendix
Human Epidermal Growth Factor Receptor Immunohistochemistry
Human epidermal growth factor receptor 2 (HER2) was detected by phospho-HER2 immunohistochemistry as previously described (Gilbertson RJ, et al: Br J Cancer 71:473-477, 1995).14 HER2 staining was scored by an independent pathologist for percent positive cells (grade 1, 1% to 25%; grade 2, 26% to 50%; grade 3, 51% to 75%; grade 4, 76% to 100%) and intensity (0, +, ++, or +++). For patients to be considered HER2 positive, tumors had to have 1% to 25% positive cells (grade 1) and an intensity score of +.
Generation of Retroviral Construct
The generation of the HER2 chimeric antigen receptor (CAR) has been previously described.14,24 Briefly, the HER2-specific murine scFv FRP5 was cloned into an SFG retroviral vector containing a short hinge, a CD28 transmembrane domain, and a CD28.ζ signaling domain.14 A clinical-grade packaging cell line was generated using PG13 cells (gibbon ape leukemia virus pseudotyping packaging cell line; CRL-10686; ATCC, Manassas, VA) as previously described.32 The highest-titer clone was used to establish a master cell bank, which was used to produce a clinical batch of virus.
Flow Cytometry
A FACSCalibur instrument (Becton Dickinson, San Jose, CA) and CellQuest software (Becton Dickinson) were used for flow cytometric analysis. Monoclonal antibodies were obtained from Becton Dickinson and included anti-CD3, -CD4, -CD8, -CD16, -CD19, -CD56, -CD62L, -CCR7, -TCRα/β, and -TCRγ/δ. HER2-CAR expression was detected with a murine scFV-specific monoclonal antibody (Jackson ImmunoResearch Laboratories, West Grove, PA). Negative controls included isotype antibodies.
Multiplex Analysis
A 13-plex human cytokine/chemokine bead array assay kit (Millipore, Billerica, MA) was used to measure granulocyte-macrophage colony-stimulating hormone, interferon gamma, interleukin (IL) -1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12p70, IL-13, and tumor necrosis factor α. Each undiluted plasma sample was assayed in duplicate according to the protocol provided by the manufacturer.
Real-Time Polymerase Chain Reaction Assay
We used an FRP5-specific primer and TaqMan probe (Applied Biosystems, Waltham, MA) to detect HER2-CAR T cells. DNA was extracted with the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany), and quantitative polymerase chain reaction was performed in triplicates using the ABI RPISM 7900HT Sequence Detection System (Applied Biosystems). The baseline range was set at cycles 6 to 15, with the threshold at 10 standard deviations above the baseline fluorescence. To generate DNA standards, we established serial dilution of DNA plasmids encoding each specific cassette.
Table A1.
Results of HER2 Immunohistochemistry
| Patient No. | Intensity Score* | Grade† |
|---|---|---|
| 1 | ++ | 1 |
| 2 | +++ | 4 |
| 3 | + | 1 |
| 4 | +++ | 3 |
| 5 | +++ | 4 |
| 6 | +++ | 3 |
| 7 | ++ | 3 |
| 8 | +++ | 4 |
| 9 | ++ | 3 |
| 10 | +++ | 4 |
| 11 | + | 2 |
| 12 | +++ | 4 |
| 13 | ++ | 2 |
| 14 | +++ | 4 |
| 15 | ++ | 2 |
| 16 | + | 1 |
| 17 | ++ | 2 |
| 18 | ++ | 3 |
| 19 | + | 1 |
NOTE. For the 19 patients enrolled onto this study, the median grade was 3 (range, 1 to 4), and the median intensity was ++ (range, + to +++).
Abbreviation: HER2, human epidermal growth factor receptor 2.
Immunohistochemistry intensity score: 0, +, ++, or +++.
Grade (percent cells positive): 1 (1% to 25%), 2 (26% to 50%), 3 (51% to 75%), or 4 (76% to 100%).
Table A2.
Unrelated Reported Adverse Events Within the First 6 Weeks of HER2-CAR T-Cell Infusion
| Adverse Event | Grade 1 |
Grade 2 |
Grade 3 |
Grade 4 |
||||
|---|---|---|---|---|---|---|---|---|
| No. of Patients | % | No. of Patients | % | No. of Patients | % | No. of Patients | % | |
| Hematologic toxicities | ||||||||
| Anemia | 8 | 42.1 | 1 | 5.3 | ||||
| Leukopenia | 5 | 26.3 | 1 | 5.3 | ||||
| Neutropenia | 4 | 21.1 | 1 | 5.3 | ||||
| Lymphopenia | 5 | 26.3 | 1 | 5.3 | ||||
| Nonhematologic toxicities | ||||||||
| General | ||||||||
| Anxiety | 1 | 5.3 | ||||||
| Fatigue | 3 | 15.8 | ||||||
| Fever | 1 | 5.3 | ||||||
| Hot flashes | 2 | 10.5 | ||||||
| Flu-like symptoms | 1 | 5.3 | ||||||
| Muscle weakness | 1 | 5.3 | 1 | 5.3 | ||||
| HEENT | ||||||||
| Watery eye | 1 | 5.3 | ||||||
| Rhinorrhea | 2 | 10.5 | ||||||
| GI | ||||||||
| Dry mouth | 1 | 5.3 | ||||||
| Nausea/vomiting | 5 | 26.3 | 1 | 5.3 | ||||
| Diarrhea | 1 | 5.3 | ||||||
| Constipation | 1 | 5.3 | ||||||
| Cardiac | ||||||||
| Hypotension | 1 | 5.3 | ||||||
| Paroxysmal tachycardia | 1 | 5.3 | ||||||
| Respiratory | ||||||||
| Cough | 5 | 26.3 | ||||||
| Upper respiratory tract infection | 1 | 5.3 | ||||||
| Wheezing | 1 | 5.3 | ||||||
| Pain | ||||||||
| Abdominal | 2 | 10.5 | ||||||
| Back | 2 | 10.5 | 1 | 5.3 | ||||
| Bone | 1 | 5.3 | ||||||
| Chest | 1 | 5.3 | ||||||
| Extremity | 1 | 5.3 | ||||||
| Neck | 1 | 5.3 | ||||||
| Pain NOS | 2 | 10.5 | ||||||
| Throat | 1 | 5.3 | ||||||
| Tumor site | 1 | 5.3 | 1 | 5.3 | ||||
| CNS | ||||||||
| Headache | 2 | 10.5 | ||||||
| Intracranial hemorrhage | 1 | 5.3 | ||||||
| Seizure | 2 | 10.5 | ||||||
| Numbness | 1 | 5.3 | ||||||
| Musculoskeletal | ||||||||
| Bruising | 1 | 5.3 | ||||||
| Ankle fracture | 1 | 5.3 | ||||||
| Laboratory | ||||||||
| ALT | 3 | 15.8 | ||||||
| Alkaline phosphatase | 3 | 15.8 | 2 | 10.5 | ||||
| AST | 6 | 31.6 | ||||||
| Hyperglycemia | 2 | 10.5 | ||||||
| Hyperkalemia | 1 | 5.3 | 1 | 5.3 | ||||
| Hypernatremia | 2 | 10.5 | ||||||
| Hypoalbuminemia | 1 | 5.3 | ||||||
| Hypocalcemia | 2 | 10.5 | ||||||
| Hypokalemia | 5 | 26.3 | ||||||
| Hyponatremia | 3 | 15.8 | ||||||
Abbreviations: CAR, chimeric antigen receptor; HEENT, head, eyes, ears, nose, and throat; HER2, human epidermal growth factor receptor 2; NOS, not otherwise specified.
Table A3.
Related Reported Adverse Events Within the First 6 Weeks of HER2-CAR T-Cell Infusion
| Adverse Event | Grade 1 |
Grade 2 |
Grade 3 |
Grade 4 |
||||
|---|---|---|---|---|---|---|---|---|
| No. of Patients | % | No. of Patients | % | No. of Patients | % | No. of Patients | % | |
| Hematologic toxicities | ||||||||
| Anemia | ||||||||
| Leukopenia | ||||||||
| Neutropenia | ||||||||
| Lymphopenia | ||||||||
| Nonhematologic toxicities | ||||||||
| General | ||||||||
| Anxiety | ||||||||
| Fatigue | ||||||||
| Fever | 1 | 5.3 | ||||||
| Hot flashes | ||||||||
| Flu-like symptoms | ||||||||
| Muscle weakness | ||||||||
| HEENT | ||||||||
| Watery eye | ||||||||
| Rhinorrhea | ||||||||
| GI | ||||||||
| Dry mouth | ||||||||
| Nausea/vomiting | ||||||||
| Diarrhea | ||||||||
| Constipation | ||||||||
| Cardiac | ||||||||
| Hypotension | ||||||||
| Paroxysmal tachycardia | ||||||||
| Respiratory | ||||||||
| Cough | ||||||||
| Upper respiratory tract infection | ||||||||
| Wheezing | ||||||||
| Pain | ||||||||
| Abdominal | ||||||||
| Back | ||||||||
| Bone | ||||||||
| Chest | ||||||||
| Extremity | ||||||||
| Neck | ||||||||
| Pain NOS | ||||||||
| Throat | ||||||||
| Tumor site | ||||||||
| CNS | ||||||||
| Headache | ||||||||
| Intracranial hemorrhage | ||||||||
| Seizure | ||||||||
| Numbness | ||||||||
| Musculoskeletal | ||||||||
| Bruising | ||||||||
| Ankle fracture | ||||||||
| Laboratory | ||||||||
| ALT | ||||||||
| Alkaline phosphatase | ||||||||
| AST | ||||||||
| Hyperglycemia | ||||||||
| Hyperkalemia | ||||||||
| Hypernatremia | ||||||||
| Hypoalbuminemia | ||||||||
| Hypocalcemia | ||||||||
| Hypokalemia | ||||||||
| Hyponatremia | ||||||||
Abbreviations: CAR, chimeric antigen receptor; HEENT, head, eyes, ears, nose, and throat; HER2, human epidermal growth factor receptor 2; NOS, not otherwise specified.
Fig A1.

Characterization of human epidermal growth factor receptor 2 (HER2) chimeric antigen receptor (CAR) T-cell product. (A) HER2-CAR expression on nontransduced (NT) and transduced T cells (NT v HER2-CAR T cells, P < .001). Individual data points and mean are shown. (B) Phenotypic analysis of HER2-CAR T-cell product. CM, central memory (CD3+/CD45RO+/CD62L+); EM, effector memory (CD3+/CD45RO+/CCR7–/CD62L–). Box plot with whiskers (Tukey method) is shown. (C) Cytotoxicity assay using NT and HER2-CAR T cells as effectors and HER2-negative (K562 and MDA-MB-468 [MDA]) or HER2-positive (NCI-H1299 and LM7) cell lines as targets. Mean with standard deviation at an effector-to-target ratio of 20:1 is shown. K562: NT v HER2-CAR T cells, P = not significant (NS); MDA: NT v HER2-CAR T cells, P = NS; NCI-H1229: NT v HER2-CAR T cells, P < .001; LM7: NT v HER2-CAR T cells, P < .001.
Fig A2.

Plasma cytokine levels after human epidermal growth factor receptor 2 (HER2) chimeric antigen receptor (CAR) T-cell infusion. Plasma cytokine levels after HER2-CAR T-cell infusion were measured by multiplex analysis. (A) Results for granulocyte-macrophage colony-stimulating factor (GMCSF), interleukin (IL) -1β, IL-2, IL-4, IL-5; and (B) IL-7, IL-10, IL-12p70, and IL-13 are shown. There were no significant changes after T-cell infusion. Results for interferon gamma, tumor necrosis factor α, IL-6, and IL-8 are shown in Figure 1. ND, not detected.
Fig A3.
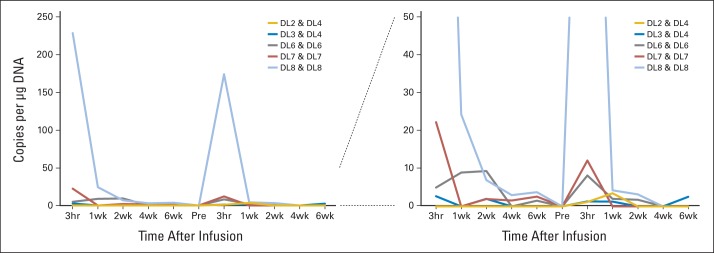
In vivo persistence of T cells. Six patients received at least two doses of human epidermal growth factor receptor 2 (HER2) chimeric antigen receptor (CAR) T cells (shown for patients 5, 7, 12, 14, and 18). HER2-CAR T cells were detected by quantitative polymerase chain reaction after infusion. The pattern of HER2-CAR T-cell persistence was similar after both infusions. Left panel shows the data with a y axis maximum of 250 copies/μg DNA, and right panel shows the data with a y axis maximum of 50 copies/μg DNA.
Footnotes
See accompanying article on page 1703
Supported in part by the V Foundation for Cancer Research, Cancer Prevention and Research Institute of Texas Grant No. RP101335, the Hoag Foundation, the Alliance for Cancer Gene Therapy, Alex's Lemonade Stand Foundation for Childhood Cancer, Stand Up To Cancer St. Baldrick's Pediatric Dream Team Translational Research Grant (SU2CAACR-DT1113; Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer), the Clinical Research Center at Texas Children's Hospital, the Dan L. Duncan Institute for Clinical and Translational Research at Baylor College of Medicine, and by shared resources through Dan L. Duncan Cancer Center Support Grant No. P30CA125123.
Authors' disclosures of potential conflicts of interest are found in the article online at www.jco.org. Author contributions are found at the end of this article.
Clinical trial information: NCT00902044.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Disclosures provided by the authors are available with this article at www.jco.org.
AUTHOR CONTRIBUTIONS
Conception and design: Nabil Ahmed, Meng-Fen Wu, Hao Liu, Gianpietro Dotti, Bambi Grilley, Cliona M. Rooney, Malcolm K. Brenner, Helen E. Heslop, Stephen Gottschalk
Administrative support: Bambi Grilley
Provision of study materials or patients: Nino Rainusso, Winfried S. Wels, Lisa L. Wang, Peter Anderson
Collection and assembly of data: Nabil Ahmed, Vita S. Brawley, Catherine Robertson, Alexia Ghazi, Claudia Gerken, Enli Liu, Olga Dakhova, Aidin Ashoori, Amanda Corder, Tara Gray, John Hicks, Cliona M. Rooney, Malcolm K. Brenner, Helen E. Heslop, Stephen Gottschalk
Data analysis and interpretation: Nabil Ahmed, Vita S. Brawley, Meenakshi Hedge, Catherine Robertson, Enli Liu, Olga Dakhova, Amanda Corder, Meng-Fen Wu, Hao Liu, John Hicks, Nino Rainusso, Zhuyong Mei, Adrian Gee, Cliona M. Rooney, Malcolm K. Brenner, Helen E. Heslop, Winfried S. Wels, Lisa L. Wang, Peter Anderson, Stephen Gottschalk
Manuscript writing: All authors
Final approval of manuscript: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Human Epidermal Growth Factor Receptor 2 (HER2) –Specific Chimeric Antigen Receptor–Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or jco.ascopubs.org/site/ifc.
Nabil Ahmed
Consulting or Advisory Role: Celgene, Bluebird Bio
Patents, Royalties, Other Intellectual Property: I have patent applications in the field of T-cell immunotherapy for cancer
Vita S. Brawley
Patents, Royalties, Other Intellectual Property: I have filed patent disclosures in the field of T-cell therapy for cancer
Meenakshi Hedge
Patents, Royalties, Other Intellectual Property: I have filed patent applications in the field of T-cell therapy for cancer
Catherine Robertson
No relationship to disclose
Alexia Ghazi
No relationship to disclose
Claudia Gerken
No relationship to disclose
Enli Liu
No relationship to disclose
Olga Dakhova
No relationship to disclose
Aidin Ashoori
No relationship to disclose
Amanda Corder
No relationship to disclose
Tara Gray
No relationship to disclose
Meng-Fen Wu
No relationship to disclose
Hao Liu
No relationship to disclose
John Hicks
No relationship to disclose
Nino Rainusso
No relationship to disclose
Gianpietro Dotti
Consulting or Advisory Role: Celgene, Bluebird Bio
Patents, Royalties, Other Intellectual Property: I receive royalties from Bellicum Pharm, and I have patent applications in the field of T-cell therapy for cancer
Zhuyong Mei
No relationship to disclose
Bambi Grilley
Employment: South Texas Nuclear Pharmacy (I)
Adrian Gee
No relationship to disclose
Cliona M. Rooney
Consulting or Advisory Role: Celgene, Cell Medica, Bluebird Bio
Patents, Royalties, Other Intellectual Property: I have patent applications in the field of cellular immunotherapy
Malcolm K. Brenner
Stock or Other Ownership: Bluebird Bio
Consulting or Advisory Role: Celgene, Cell Medica, Jennerex, Bluebird Bio
Patents, Royalties, Other Intellectual Property: I have patent applications in the field of cellular immunotherapy
Helen E. Heslop
Consulting or Advisory Role: Celgene, Cell Medica, Viracyte, Bluebird Bio
Patents, Royalties, Other Intellectual Property: Cell Medica licensing agreement, Celgene collaborative research agreement
Winfried S. Wels
Patents, Royalties, Other Intellectual Property: I have patent applications in the field of cancer immunotherapy (Inst)
Lisa L. Wang
No relationship to disclose
Peter Anderson
No relationship to disclose
Stephen Gottschalk
Consulting or Advisory Role: Celgene, Bluebird Bio
Patents, Royalties, Other Intellectual Property: I have patent applications in the field of T-cell immunotherapy
REFERENCES
- 1.Linch M, Miah AB, Thway K, et al. Systemic treatment of soft-tissue sarcoma-gold standard and novel therapies. Nat Rev Clin Oncol. 2014;11:187–202. doi: 10.1038/nrclinonc.2014.26. [DOI] [PubMed] [Google Scholar]
- 2.Bielack SS, Carrle D, Hardes J, et al. Bone tumors in adolescents and young adults. Curr Treat Options Oncol. 2008;9:67–80. doi: 10.1007/s11864-008-0057-1. [DOI] [PubMed] [Google Scholar]
- 3.Meyers PA. High-dose therapy with autologous stem cell rescue for pediatric sarcomas. Curr Opin Oncol. 2004;16:120–125. doi: 10.1097/00001622-200403000-00006. [DOI] [PubMed] [Google Scholar]
- 4.Finkelstein SE, Fishman M, Conley AP, et al. Cellular immunotherapy for soft tissue sarcomas. Immunotherapy. 2012;4:283–290. doi: 10.2217/imt.12.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buddingh EP, Schilham MW, Ruslan SE, et al. Chemotherapy-resistant osteosarcoma is highly susceptible to IL-15-activated allogeneic and autologous NK cells. Cancer Immunol Immunother. 2011;60:575–586. doi: 10.1007/s00262-010-0965-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rainusso N, Brawley VS, Ghazi A, et al. Immunotherapy targeting HER2 with genetically modified T cells eliminates tumor-initiating cells in osteosarcoma. Cancer Gene Ther. 2012;19:212–217. doi: 10.1038/cgt.2011.83. [DOI] [PubMed] [Google Scholar]
- 7.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pule MA, Savoldo B, Myers GD, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–1270. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park JR, Digiusto DL, Slovak M, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15:825–833. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 13.Dotti G, Gottschalk S, Savoldo B, et al. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257:107–126. doi: 10.1111/imr.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmed N, Salsman VS, Yvon E, et al. Immunotherapy for osteosarcoma: Genetic modification of T cells overcomes low levels of tumor antigen expression. Mol Ther. 2009;17:1779–1787. doi: 10.1038/mt.2009.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roth M, Linkowski M, Tarim J, et al. Ganglioside GD2 as a therapeutic target for antibody-mediated therapy in patients with osteosarcoma. Cancer. 2014;120:548–554. doi: 10.1002/cncr.28461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang G, Yu L, Cooper LJ, et al. Genetically modified T cells targeting interleukin-11 receptor alpha-chain kill human osteosarcoma cells and induce the regression of established osteosarcoma lung metastases. Cancer Res. 2012;72:271–281. doi: 10.1158/0008-5472.CAN-11-2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Modak S, Kramer K, Gultekin SH, et al. Monoclonal antibody 8H9 targets a novel cell surface antigen expressed by a wide spectrum of human solid tumors. Cancer Res. 2001;61:4048–4054. [PubMed] [Google Scholar]
- 18.Gorlick R, Huvos AG, Heller G, et al. Expression of HER2/erbB-2 correlates with survival in osteosarcoma. J Clin Oncol. 1999;17:2781–2788. doi: 10.1200/JCO.1999.17.9.2781. [DOI] [PubMed] [Google Scholar]
- 19.Somers GR, Ho M, Zielenska M, et al. HER2 amplification and overexpression is not present in pediatric osteosarcoma: A tissue microarray study. Pediatr Dev Pathol. 2005;8:525–532. doi: 10.1007/s10024-005-0044-5. [DOI] [PubMed] [Google Scholar]
- 20.Ebb D, Meyers P, Grier H, et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: A report from the children's oncology group. J Clin Oncol. 2012;30:2545–2551. doi: 10.1200/JCO.2011.37.4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morgan RA, Yang JC, Kitano M, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors: European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 23.Ahmed N, Ratnayake M, Savoldo B, et al. Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res. 2007;67:5957–5964. doi: 10.1158/0008-5472.CAN-06-4309. [DOI] [PubMed] [Google Scholar]
- 24.Ahmed N, Salsman VS, Kew Y, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16:474–485. doi: 10.1158/1078-0432.CCR-09-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bernhard H, Neudorfer J, Gebhard K, et al. Adoptive transfer of autologous, HER2-specific, cytotoxic T lymphocytes for the treatment of HER2-overexpressing breast cancer. Cancer Immunol Immunother. 2008;57:271–280. doi: 10.1007/s00262-007-0355-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Disis M, Salazar LG, Coveler A, et al. Phase I study of infusion of HER2/neu (HER2) specific T cells in patients with advanced-stage HER2 overexpressing cancers who have received a HER2 vaccine. J Clin Oncol. 2009;(suppl 15s):27. abstr 3000. [Google Scholar]
- 27.Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cho HS, Mason K, Ramyar KX, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 29.Gerstmayer B, Altenschmidt U, Hoffmann M, et al. Costimulation of T cell proliferation by a chimeric B7-2 antibody fusion protein specifically targeted to cells expressing the erbB2 proto-oncogene. J Immunol. 1997;158:4584–4590. [PubMed] [Google Scholar]
- 30.Louis CU, Savoldo B, Dotti G, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6056. doi: 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Porter DL, Levine BL, Kalos M, et al. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jensen MC, Popplewell L, Cooper LJ, et al. Anti-transgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor re-directed T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–1256. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamers CH, Willemsen R, van Elzakker P, et al. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood. 2011;117:72–82. doi: 10.1182/blood-2010-07-294520. [DOI] [PubMed] [Google Scholar]
- 35.Blum KS, Pabst R. Lymphocyte numbers and subsets in the human blood: Do they mirror the situation in all organs? Immunol Lett. 2007;108:45–51. doi: 10.1016/j.imlet.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 36.Craddock JA, Lu A, Bear A, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 2010;33:780–788. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoos A, Eggermont AM, Janetzki S, et al. Improved endpoints for cancer immunotherapy trials. J Natl Cancer Inst. 2010;102:1388–1397. doi: 10.1093/jnci/djq310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bielack SS, Kempf-Bielack B, Branscheid D, et al. Second and subsequent recurrences of osteosarcoma: Presentation, treatment, and outcomes of 249 consecutive cooperative osteosarcoma study group patients. J Clin Oncol. 2009;27:557–565. doi: 10.1200/JCO.2008.16.2305. [DOI] [PubMed] [Google Scholar]
- 39.Kempf-Bielack B, Bielack SS, Jürgens H, et al. Osteosarcoma relapse after combined modality therapy: An analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS) J Clin Oncol. 2005;23:559–568. doi: 10.1200/JCO.2005.04.063. [DOI] [PubMed] [Google Scholar]
- 40.Bacci G, Briccoli A, Longhi A, et al. Treatment and outcome of recurrent osteosarcoma: Experience at Rizzoli in 235 patients initially treated with neoadjuvant chemotherapy. Acta Oncol. 2005;44:748–755. doi: 10.1080/02841860500327503. [DOI] [PubMed] [Google Scholar]
- 41.Di Stasi A, Tey SK, Dotti G, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5:215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]