

Abstract
Surfactant protein A (SP-A) is a large multimeric protein found in the lungs. In addition to its immunoregulatory function in infectious respiratory diseases, SP-A is also used as a marker of lung adenocarcinoma. Despite the finding that SP-A expression levels in cancer cells has a relationship with patient prognosis, the function of SP-A in lung cancer progression is unknown. We investigated the role of SP-A in lung cancer progression by introducing the SP-A gene into human lung adenocarcinoma cell lines. SP-A gene transduction suppressed the progression of tumor in subcutaneous xenograft or lung metastasis mouse models. Immunohistochemical analysis showed that the number of M1 antitumor tumor-associated macrophages (TAMs) was increased and the number of M2 tumor-promoting TAMs was not changed in the tumor tissue produced by SP-A–expressing cells. In addition, natural killer (NK) cells were also increased and activated in the SP-A–expressing tumor. Moreover, SP-A did not inhibit tumor progression in mice depleted of NK cells. Taking into account that SP-A did not directly activate NK cells, these results suggest that SP-A inhibited lung cancer progression by recruiting and activating NK cells via controlling the polarization of TAMs.
Lung cancer is the major cause of malignancy-related death worldwide. Mortality is 80% to 90%, which makes this disease the leading cause of cancer-related deaths.1 The high mortality rate of this disease is primarily due to the difficulty of early diagnosis, the high metastatic potential, and the poor responses to chemical therapy and radiotherapy. Because there is no established curative therapy for advanced lung cancer to date, clinical management is palliative in many cases. Therefore, it is crucial to investigate and understand the underlying biological and molecular mechanisms of lung cancer progression.
Surfactant protein A (SP-A) is a large multimeric protein found in the airways and alveoli of the lungs. SP-A is a member of the collectin family of proteins, characterized by NH2-terminal collagen-like regions and COOH-terminal lectin domains. Although other SPs, such as SP-B, function to reduce surface tension in the lungs, SP-A (and SP-D) regulates the pulmonary immune response.2 Previous in vivo studies have shown that SP-A regulates responses involved in initiation and potentiation of inflammation by regulating the production of proinflammatory cytokines, such as tumor necrosis factor α (TNF-α), in response to lipopolysaccharide3 or by accelerating the clearance of a variety of pathogens.4–8 Because SP-A has the ability to opsonize and enhance pathogen uptake by phagocytes, the immunoregulatory roles of SP-A have been studied mainly in the field of infectious diseases. Recently, we reported that SP-A has a role in regulating bleomycin-induced acute noninfectious lung injury by inhibiting lung epithelial cell apoptosis.9 Pastva et al10 reported that SP-A regulates TH2 cytokine production in a mouse asthma model. These results suggest that SP-A has diverse functions to control various lung diseases. Considering that SP-A contributes to multiple aspects of pulmonary host defense, we hypothesized that SP-A might have a role in lung cancer progression.
In a lung cancer study, SP-A was expressed in approximately 49% of primary non–small cell lung carcinomas11 and is used as a specific marker of carcinoma that originates in type II pneumocytes. In addition, a previous study demonstrated that deletion of the SFTPA1 (alias, SPA) gene in non–small cell lung cancer cells was associated with tumor progression.12 Tsutsumida et al13 found that patients with lung adenocarcinoma with relatively high MUC1 mucin expression and low SP-A expression in cancer cells had a poor outcome. These clinical studies demonstrate that in addition to use as a diagnostic marker, SP-A expression in lung cancer cells could be a useful biomarker of good prognosis. Although these studies suggested that SP-A might have a role in suppressing lung cancer progression, the role of SP-A in lung cancer has not been extensively studied, and the mechanisms by which SP-A controls lung cancer progression remain unknown.
In this study, we generated SP-A–overexpressing human lung adenocarcinoma cells and evaluated the role of SP-A in lung cancer progression using experimental mouse models.
Materials and Methods
Cell Lines
The human lung adenocarcinoma cell line PC14PE6 was a gift from Dr. Isaiah J. Fidler (The University of Texas MD Anderson Cancer Center, Houston TX). The human lung adenocarcinoma cell line A549 was purchased from ATCC (Manassas, VA). These cell lines were authenticated by BEX Co. Ltd. (Tokyo, Japan) using a multiplex short tandem repeat assay. Both cell lines were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U/mL of penicillin, and 50 μg/mL of streptomycin and were cultured at 37°C in a humidified atmosphere of 5% CO2 in air.
Reagents
An anti-mouse IL-2 receptor β-chain monoclonal antibody, TM-β1 (IgG2b), was a gift from Drs. Masayuki Miyasaka and Toshio Tanaka (Osaka University, Osaka, Japan).
SP-A Purification
SP-A was purified from the lung lavage fluid of patients with alveolar proteinosis as previously described14 and was routinely tested to reduce endotoxin contamination.14 Briefly, SP-A was suspended in 100 mmol/L octylglucoside and 5 mmol/L Tris, pH 7.4, after butanol extraction. Polymyxin-agarose (Sigma-Aldrich, St. Louis, MO) was added 1:5 (v/v) and allowed to incubate at room temperature for 30 minutes. The mixture was then dialyzed (14,000 molecular weight cutoff value) for four changes ≥4 hours each against autoclaved 5 mmol/L Tris, pH 7.4. The mixture was then centrifuged, and the supernatant, containing SP-A, was removed by gentle aspiration. SP-A preparations had final endotoxin concentrations of <0.1 pg/μg of SP-A as determined by the Limulus amoebocyte lysate assay (QCL-1000; BioWhittaker, Walkersville, MD).
SPA Gene Transduction
The human SPA gene–expressed region [SFTPA1 (NM_005411)] (OriGene Technologies, Rockville, MD) was introduced into the pMIG vector (a gift from Dr. Alana L. Welm, University of Utah, Salt Lake City). The Platinum-E packaging cell line (a gift from Dr. Toshio Kitamura, Tokyo University, Tokyo, Japan)15 was transfected with pMIG or derivative vector DNA by using FuGENE 6 transfection reagent (Roche Applied Science, Indianapolis, IN). PC14PE6 or A549 cells were infected using the viral supernatant as described previously.16 The proportion of green fluorescent protein–positive cells was >90% in the entire population.
Animals
Male athymic BALB/c nude mice and SCID mice were obtained from Charles River Laboratories Japan (Yokohama) and CLEA Japan (Tokyo), respectively, and were maintained under specific pathogen-free conditions throughout the study. All the experiments were performed in accordance with the guidelines established by The University of Tokushima Committee on Animal Care and Use, Tokushima, Japan. At the end of each in vivo experiment, the mice were anesthetized with isoflurane and euthanized humanely by cutting the subclavian artery. All the experiment protocols were reviewed and approved by the Animal Research Committee of The University of Tokushima.
In Vivo Subcutaneous Xenograft Model
PC14PE6 cells (1.0 × 106 per mouse) or A549 cells (3.0 × 106 per mouse) suspended in 0.1 mL of PBS were subcutaneously inoculated into the right flank of nude mice. Tumor size was measured using a vernier caliper three times a week (volume = ab2/2, where a indicates long diameter; b, short diameter). The mice were euthanized humanely on day 21, and the tumors were resected for further analyses.
In Vivo Lung Metastasis Model
To establish lung metastasis, nude mice were intravenously inoculated via the tail vein with 1.0 × 106 tumor cells per mouse.17 The mice were euthanized humanely on either day 28 (PC14PE6) or day 42 (A549). The lungs were weighed, and the number of metastatic colonies on the surface of the lungs was determined by visual examination. Because PC14PE6 cells produce large amounts of pleural effusion,18 the volume of the effusion was also evaluated. In some experiments, natural killer (NK) cells were depleted by treating nude mice with TM-β1 5 days after inoculation of PC14PE6 cells.19
Immunofluorescence
The excised tumor tissue was placed into OCT compound (Sakura Finetechnical Co., Tokyo, Japan) and snap frozen. Eight-micrometer-thick frozen tissue sections were fixed with 4% paraformaldehyde solution in PBS and were used for identification of macrophages using 1:150 rat anti-mouse CD68 monoclonal antibody (Serotec, Oxford, UK) and of NK cells using 1:100 goat anti-mouse NKp46/NCR1 monoclonal antibody (R&D Systems, Minneapolis, MN). Alexa Fluor 488–labeled secondary antibodies (dilution 1:250; Invitrogen, Carlsbad, CA) were used for immunofluorescence (IF) detection. To identify M1 or M2 macrophages, the sections were stained with 1:150 fluorescein isothiocyanate–conjugated rat anti-mouse TNF-α antibody (BD Pharmingen, Franklin Lakes, NJ) or 1:150 fluorescein isothiocyanate–conjugated rat anti-mouse CD206 [mannose receptor C type 1 (MRC-1)] antibody (BioLegend, San Diego, CA) after CD68 staining. Alexa Fluor 594–labeled anti-rat secondary antibodies (dilution 1:250; Invitrogen) were used for CD68 IF detection. M1 or M2 macrophages were identified as CD68-positive/TNF-α-–positive or CD68-positive/MRC-1–positive cells, respectively. Nuclei were counterstained with DAPI (blue). In each slide, the number of positive cells was counted in five areas under fluorescent microscopy at ×100 (single staining) or ×200 (double staining) magnification.
RT-qPCR
Total RNA was extracted from the tumors using the RNeasy mini kit (Qiagen, Valencia, CA) and reverse transcribed to cDNA using a high-capacity cDNA Reverse Transcription kit (Applied Biosystems, Carlsbad, CA) according to the manufacturer’s instructions. RT-PCR was performed using the CFX96 real-time PCR system (Bio-Rad Laboratories, Hercules, CA) using SYBR Premix Ex Taq (Takara, Kyoto, Japan). Human RPL2720 and mouse β2m mRNA were used as housekeeping genes, and quantification was determined by using the ΔΔCT method. Specific PCR primer pairs for each studied gene are shown in Table 1.
Table 1.
Primer Sequences Used in Quantitative PCR
| Forward | Reverse | Product size (bp) | |
|---|---|---|---|
| Gene (mouse) | |||
| IL-1β | 5′-TGACGTTCCCATTAGACAAC-3′ | 5′-ATTTTGTCGTTGCTTGGTTC-3′ | 171 |
| IL-6 | 5′-GTACCATAGCTACCTGGAGT-3′ | 5′-GGAAATTGGGGTAGGAAGGA-3, ′ | 154 |
| TNF-α | 5′-CCTATGTCTCAGCCTCTTCT-3′ | 5′-TTGGGAACTTCTCATCCCTT-3′ | 107 |
| IL-12 | 5′-CACACTGGACCAAAGGGACT-3′ | 5′-TGGTTTGATGATGTCCCTGA-3′ | 169 |
| IFN-γ | 5′-TAGCTCTGAGACAATGAACG-3′ | 5′-CACATCTATGCCACTTGAGT-3′ | 145 |
| CCL2 | 5′-TTCACAGTTGCCGGCTGG-3′ | 5′-TGAATGAGTAGCAGCAGGTGAGTG-3′ | 81 |
| CCL5 | 5′-CAGCAGCAAGTGCTCCAATCTT-3′ | 5′-TTCTTGAACCCACTTCTTCTCTGG-3′ | 91 |
| IL-10 | 5′-AAGGACCAGCTGGACAACAT-3′ | 5′-TCTCACCCAGGGAATTCAAA-3′ | 172 |
| MRC-1 | 5′-TGCAAGGATCATACTTCCCT-3′ | 5′-TGATGTTCTCCAGTAGCCAT-3′ | 240 |
| Arg1 | 5′-GAATGGAAGAGTCAGTGTGG-3′ | 5′-AATGACACATAGGTCAGGGT-3′ | 97 |
| CD163 | 5′-GACGACAGATTCAGCGACTT-3′ | 5′-CCGAGGATTTCAGCAAGTCCA-3′ | 114 |
| Prf1 | 5′-GACACAGTAGAGTGTCGCAT-3′ | 5′-TTTTGAAGTCAAGGTGGAGTG-3′ | 70 |
| GzmB | 5′-AGAGAGCAAGGACAACACTC-3′ | 5′-ATCGAAAGTAAGGCCATGTAG-3′ | 176 |
| B2M (β2M) | 5′-GGAAGCCGAACATACTGAACTG-3′ | 5′-TTTCCCGTTCTTCAGCATTTGG-3′ | 80 |
| Gene (human) | |||
| IL-1β | 5′-GACAGGATATGGAGCAACAA-3′ | 5′-GCTGTAGAGTGGGCTTATCA-3′ | 147 |
| IL-6 | 5′-CCTCTTCAGAACGAATTGAC-3′ | 5′-AGTCTCCTCATTGAATCCAG-3′ | 186 |
| TNF-α | 5′-GGCAGTCAGATCATCTTCTCG-3′ | 5′-CAGCTGGTTATCTCTCAGCTC-3′ | 148 |
| CCL2 | 5′-CTCATAGCAGCCACCTTCATT-3′ | 5′-ACAGATCTCCTTGGCCACAA-3′ | 192 |
| CCL3 | 5′-GGCAGTCAGATCATCTTCTCG-3′ | 5′-CAGCTGGTTATCTCTCAGCTC-3 ′ | 81 |
| CCL5 | 5′-CTGTCATCCTCATTGCTACTG-3′ | 5′-GCCACTGGTGTAGAAATACTC-3′ | 140 |
| MRC-1 | 5′-CCATCGAGGAATTGGACTTT-3′ | 5′-TGTCATTTAAGCCGATCCAC-3′ | 78 |
| RPL27 | 5′-ATCGCCAAGAGATCAAAGATAA-3′ | 5′-TCTGAAGACATCCTTATTGACG-3′ | 123 |
SP-A Stimulation of Monocytes, Macrophages, and NK Cells
Human monocytes were separated from the peripheral blood of healthy volunteers as described previously.21 The purity and viability of the monocytes was confirmed to be >98% by staining with Diff-Quik (Baxter Diagnostics, Deerfield, IL) and trypan blue, respectively. Mouse alveolar macrophages (AMs) were collected by using bronchoalveolar lavage as described previously.9 More than 95% of the cells were confirmed to be AMs. For eliciting mouse peritoneal macrophages (PMs), 2 mL of thioglycollate (BD Biosciences, San Jose, CA) was injected into the peritoneal cavity of SCID mice. After 3 days, peritoneal exudative cells were harvested by intraperitoneal lavage with ice-cold PBS. Approximately 80% of isolated cells were macrophages. NK cells from SCID mice were isolated as previously described.22 These immune cells were stimulated with 20 μg/mL of human SP-A for 4 hours in RPMI 1640 medium containing 1% fetal bovine serum. Total RNA was extracted for quantitative RT-PCR.
Cell Migration Assay
The migration assay was performed using 8-μm pore size cell culture inserts (BD Biosciences). After 24 hours of serum starvation, PMs in serum-free media were added to the inner chamber in the presence or absence of 20 μg/mL of SP-A. RPMI 1640 medium containing 10% fetal bovine serum was added to the lower chamber. After 17 hours of incubation, the cells that had migrated to the bottom surface of the filter were counted in six randomly selected fields on each filter under a microscope at ×200 magnification.
Western Blot Analysis
Twenty micrograms of total protein extracted from tumor cell lines was resolved by SDS-PAGE (Invitrogen) and was transferred to polyvinylidene difluoride membrane (Atto Corp., Tokyo, Japan), and Western blot was performed as described previously.9 Immunoreactive bands were visualized using SuperSignal west femto maximum sensitivity substrate (Thermo Scientific, Waltham, MA).
Statistical Analysis
Data are given as means ± SEM. Statistical analysis was performed using the Student’s t-test of unpaired samples or the U-test. Values of P < 0.05 were considered statistically significant.
Results
Effect of SP-A on Lung Cancer Xenografts in Vivo
Human lung adenocarcinoma cell lines (PC14PE6 and A549 cells) were transduced with vectors encoding human SP-A by the retroviral transduction system (termed PC14PE6/SP-A and A549/SP-A, respectively). The empty vector was transduced as a control (termed PC14PE6/vector and A549/vector, respectively) (Figure 1A). To investigate the effect of SP-A on in vivo tumor growth, we initially injected male nude mice subcutaneously with these cells. For both cell lines, the growth of xenografts was significantly inhibited when cells were overexpressing SP-A compared with the vector control cells (Figure 1, B and C).
Figure 1.

The effect of SP-A on lung cancer subcutaneous xenografts in vivo. A: SP-A expression of human SP-A stable transfectants was confirmed by Western blot analysis. H441 cells were used as a positive control. ACTB, β-actin. Tumor growth (B) and weight (C) of xenografts produced by PC14PE6 (left panels; n = 6 per group) and A549 (right panels; n = 8 per group) cells transduced with SP-A or vector. Data are presented as means ± SEM (horizontal lines in C). ∗P < 0.05.
Direct Effect of SP-A on Lung Cancer Cell Proliferation
To explore the underlying mechanism by which SP-A suppressed the growth of xenografts, we performed immunohistochemical staining of Ki-67, CD31, or TUNEL (Supplemental Figure S1A). The number of Ki-67–positive cells was significantly decreased in tumors formed by SP-A overexpressing PC14PE6 cells, whereas no difference was seen in A549 cells. The number of TUNEL-positive cells was increased in both cell lines expressing SP-A. No difference was seen in the number of CD31-positive cells. These results led us to consider that SP-A might have a direct effect on cancer cell proliferation or the cell cycle. However, results of the in vitro MTT assay showed that SP-A–overexpressing cells had the same ability of cell proliferation with control cells in PC14PE6 and A549 cells (Supplemental Figure S1B). Propidium iodide staining revealed that the state of cell cycle and death was also similar by SP-A transduction in PC14PE6 cells (Supplemental Figure S1C). Moreover, the effect of SP-A on the cell cycle in PC14PE6 cells was also investigated by using the fluorescent ubiquitination-based cell-cycle indicator system, but no difference was observed (data not shown). Taken together, we considered that although SP-A inhibited tumor growth in vivo, its effect was not due to the direct effect on cell proliferation or the cell cycle or to the inhibition of tumor angiogenesis.
Effect of SP-A on the Recruitment of Tumor-Associated Macrophages
We next investigated whether cancer cell–produced SP-A might affect the tumor microenvironment. A variety of studies have shown that tumor-infiltrated tumor-associated macrophages (TAMs) play an important role in the progression of various types of cancers, including lung cancer.23,24 It is also known that activated macrophages are functionally polarized into either M1 (classically activated) or M2 (alternatively activated) macrophages. M1 macrophages produce large amounts of inflammatory cytokines, such as TNF-α and interferon-γ, and are essential for tumor suppression and host defense against bacteria.25 In contrast, M2 macrophages play important roles in tumor progression, tissue remodeling, and angiogenesis. M2 macrophages are characterized by their high expression of several factors, such as arginase-1, MRC-1, and IL-10. To determine whether SP-A affects the recruitment of TAMs, sections from resected tumors were subjected to IF staining. As shown in Figure 2A, the number of CD68-positive macrophages was significantly increased in tumors formed by SP-A–transduced cells. We then assessed whether M1 and M2 macrophage polarizations were altered by SP-A transduction. We performed IF double staining of CD68 and TNF-α for M1 and MRC-1 for M2 and determined M1 and M2 macrophages in the xenografts. In both SP-A–transduced cell lines, the number of M1 macrophages was significantly increased versus vector controls, whereas the number of M2 macrophages was not changed (Figure 2B). To confirm that the number of M1 macrophages was increased in the SP-A–expressing tumors, mRNA was extracted from the resected tumor, and the expression of M1 and M2 markers was determined by RT-PCR using mouse-specific primers. Multiple M1 markers were up-regulated in the SP-A–expressing tumors, whereas M2 markers were not changed compared with the vector control tumors (Figure 2C).
Figure 2.

The effect of SP-A on the recruitment of TAMs in xenografts. A: IF staining of CD68 (green). Nuclei were counterstained with DAPI (blue). B: IF double staining of CD68 (red) and TNF-α (green) or MRC-1 (green). C: Gene expression of mouse M1 and M2 markers in PC14PE6 xenografts determined by RT-PCR. The increased gene expression in PC14PE6/SP-A compared with control (vector) is shown as log2-fold changes. Data are the average of four to five mice per group. IFN-γ, interferon γ. Data are presented as means ± SEM. ∗P < 0.05. Scale bars: 200 μm (A); 100 μm (B).
Effect of SP-A on NK Cell Recruitment in Xenografts
We next focused on the other important immune cells, NK cells. Because cytokines/chemokines such as interferon-γ, CCL5, and IL-12 that were up-regulated in the SP-A–expressing tumors are known to be potent inducers of NK cells,26,27 we hypothesized that the in vivo tumor regression could be due to the recruitment of NK cells. Thus, we performed IF staining of NKp46 to determine the number of NK cells in the xenografts. As shown in Figure 3A, the number of recruited NK cells was significantly increased in the SP-A–expressing tumors. Moreover, the expression of mouse perforin 1 (Prf1) and granzyme B (GzmB), key factors produced by NK cells to exhibit cell killing, was strongly up-regulated in SP-A–expressing tumors (Figure 3B), suggesting that NK cell killing was activated by SP-A in the tumor microenvironment. Collectively, these results indicate that in the tumor microenvironment, SP-A led to increased numbers of activated M1 TAMs and NK cells, which, in turn, can inhibit tumor growth.
Figure 3.

The effect of SP-A on NK cell recruitment in xenografts. A: IF staining of NKp46 (green). Nuclei were counterstained with DAPI (blue). Scale bars: 200 μm. B: Gene expression of mouse Prf1 and GzmB in PC14PE6 xenografts determined by RT-PCR. Data are the average of four to five mice per group. Data are presented as means ± SEM. ∗P < 0.05.
Effect of SP-A on Macrophages and NK Cells in Vitro
Given the observation that the numbers of M1 macrophages and NK cells were increased in SP-A–expressing tumors, we performed in vitro experiments using thioglycollate-elicted mouse PMs, AMs, NK cells, and human peripheral blood monocytes to further examine the effect of SP-A on these cells. As shown in Figure 4A, exogenous SP-A treatment increased the expression of M1-related genes, such as CCL5, CCL2, TNF-α, and IL-1β, in mouse PMs. The expression of M2 markers did not change significantly. M1-related gene expression on human peripheral blood monocytes was also up-regulated by SP-A (Figure 4B). These results indicate that monocytes and macrophages can be directly targeted and activated toward the M1 phenotype by SP-A. Furthermore, the migration activity of PMs was increased by SP-A treatment (Figure 4C). Mouse AMs were not affected by exogenous SP-A treatment (Figure 4A), suggesting that AMs had developed to not overreact to SP-A exposure during their development and maturation in the pulmonary environment. The in vivo results also suggest that SP-A activated NK cells to demonstrate antitumor activity; however, exogenous treatment of SP-A did not directly affect Prf1 or GzmB gene expression in NK cells in vitro (Figure 4D). These results suggest that SP-A activated and attracted circulating monocytes/macrophages to obtain the M1 phenotype and that these increased M1 TAMs then recruited and activated NK cells to exhibit cell killing.
Figure 4.
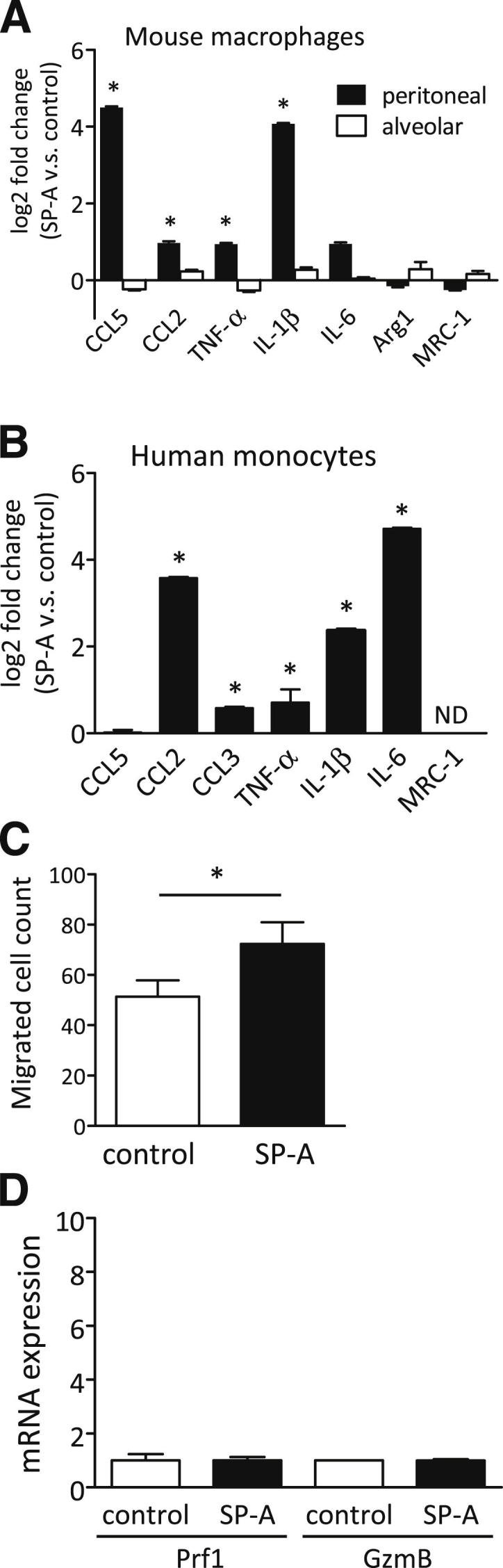
The effect of SP-A on various immune cells in vitro. After exogenous SP-A treatment, the expression of various genes was determined by RT-PCR in mouse PMs and AMs (A) and human peripheral monocytes (B). The increased gene expression in SP-A treatment compared with control (PBS) are shown as log2-fold changes (n = 3 per group). ND, not detected. C: The effect of exogenous SP-A on the migration of mouse PMs (n = 3 per group). D: Gene expressions of Prf1 and GzmB in mouse NK cells treated with exogenous SP-A (n = 3 per group). Data are presented as means ± SEM. ∗P < 0.05.
Effect of SP-A on the Lung Cancer Metastasis Model in Vivo
We next examined the effect of SP-A on lung metastasis induced by lung cancer cells. Intravenous injection of PC14PE6 or A549 cells into nude mice lead to the development of metastatic colonies in the lung. In addition, mice injected with PC14PE6 developed a large volume of pleural effusion.17,18 We compared the number of lung metastatic colonies, lung weight, and the amount of pleural effusion produced by vector control and SP-A–expressing cells. Mice injected with PC14PE6/SP-A cells produced significantly fewer lung metastatic colonies and a lower amount of pleural effusion than those injected with vector control cells (Table 2 and Figure 5A). Similarly, the lung metastasis formed by A549 cells was also suppressed by SP-A expression. Analogous to the results we obtained in the xenograft experiments, the numbers of CD68-positive macrophages, M1 macrophages, and NK cells were increased in the SP-A–expressing metastatic tumors compared with vector controls (Figure 5, B and C). The expression of multiple M1 markers, as well as Prf1 and GzmB, was up-regulated in the lung metastatic colonies formed by PC14PE6/SP-A, whereas the expression of M2 markers was not changed (Figure 5D).
Table 2.
Effect of SP-A on Lung Metastasis Produced by Lung Adenocarcinoma Cell Lines in Nude Mice
| Cell line | Lung |
Pleural effusion |
|||
|---|---|---|---|---|---|
| Weight (g) | Metastasis |
Incidence | Volume (μL) | ||
| Incidence | No. | ||||
| PC14PE6 | |||||
| Vector | 0.36 (0.30−0.42) | 6/6 | 117.7 (67–183) | 5/6 | 176.7 (0–300) |
| SP-A | 0.27 (0.22−0.31)∗ | 6/6 | 58.8 (21–111)∗ | 0/6 | All 0∗ |
| A549 | |||||
| Vector | 0.48 (0.30−0.85) | 9/9 | All >200 | ||
| SP-A | 0.26 (0.19−0.41)† | 10/10 | All >200 | ||
PC14PE6 or A549 cells (1 × 106 per mouse) were i.v. injected into nude mice, and lung metastasis and pleural effusion were evaluated. Values are means (ranges).
Statistically significant difference compared with PC14PE6/vector (P < 0.05).
Statistically significant difference compared with A549/vector (P < 0.05).
Figure 5.

The effect of SP-A on lung cancer metastasis. A: Representative images of lung metastasis produced by SP-A– or vector-transduced lung cancer cell lines. B: IF staining of CD68 or NKp46. Nuclei were counterstained with DAPI (blue). C: IF double staining of CD68 (red) and TNF-α (green) or MRC-1 (green) in PC14PE6 lung metastasis. D: Gene expression of mouse M1 and M2 markers in PC14PE6 metastatic lung nodules determined by RT-PCR. The increased gene expression in PC14PE6/SP-A compared with control (vector) is shown as log2-fold changes. Data are the average of four to five mice per group. IFN-γ, interferon γ. E: Representative images of the lung metastasis formed by PC14PE6/SP-A or vector in nude mice (NK+) or NK cell–depleted nude mice (NK−). Data are presented as means ± SEM. ∗P < 0.05. Scale bars: 200 μm (B); 100 μm (C).
Importance of NK Cells in the SP-A–Mediated Antitumor Effect in Lung Cancer Metastasis
To confirm that the activation of NK cells was essential in the antitumor effect of SP-A, we performed a lung metastasis experiment using nude mice depleted of NK cells.19 As shown in Table 3 and Figure 5E, PC14PE6/SP-A cells were confirmed to produce significantly fewer lung metastatic colonies and a lower volume of pleural effusion compared with control cells when injected into NK+ nude mice. However, when NK cells were depleted, no difference was observed in lung metastasis between PC14PE6/SP-A and control cells. These results indicate that the activation of NK cells was essential for SP-A–mediated suppression of lung cancer progression. Taken together, these findings suggest that SP-A in the tumor microenvironment displays antitumor activity by mediating the polarization of TAMs toward an M1-dominant phenotype, which, in turn, activates NK cells that then limit tumor progression.
Table 3.
Effect of SP-A on Lung Metastasis Produced by PC14PE6 Cells in Nude Mice Depleted of NK Cells
| PC14PE6 | Lung |
Pleural effusion |
|||
|---|---|---|---|---|---|
| Weight (g) | Metastasis |
Incidence | Volume (μL) | ||
| Incidence | No. | ||||
| Vector NK+ | 0.41 (0.24−0.65) | 6/6 | 113.0 (35–197) | 6/6 | 313.3 (20–900) |
| SP-A NK+ | 0.21 (0.15−0.26)∗ | 5/5 | 25.6 (6–34)∗ | 1/5 | 80.0 (0–400) |
| Vector NK– | 0.50 (0.26−0.89) | 7/7 | 148.6 (52–207) | 7/7 | 438.6 (20–1000) |
| SP-A NK– | 0.34 (0.21−0.48) | 5/5 | 101.2 (41–181) | 4/5 | 400.0 (0–1200) |
PC14PE6 cells were i.v. injected into nude mice with or without NK cell depletion, and lung metastasis and pleural effusion were evaluated. Values are means (ranges).
Statistically significant difference compared with vector NK+ (P < 0.05).
Discussion
In this study, we demonstrated that i) SP-A expression in cancer cells suppresses progression of lung adenocarcinoma in xenograft and lung metastasis models; ii) SP-A inhibits lung cancer progression not by its direct effect on tumor cells but by regulating the host microenvironment, including macrophages and NK cells; and iii) SP-A increases the number of M1 TAMs in the tumor microenvironment, resulting in NK cell recruitment and activation in tumor tissue. These results suggest new immunoregulatory functions of SP-A, which is frequently expressed in pulmonary adenocarcinoma.
Tumors comprise not only malignant cells but also many other nonmalignant cell types, and they produce a unique microenvironment that can modify the neoplastic properties of the tumor cells. Among the cells recruited in the tumor microenvironment, TAMs are one of the major players known to have pivotal roles in the progression and metastasis of tumors.23,24 Although partially contradictive, high numbers of TAMs often correlate with poor prognosis in various types of cancer.28 Therefore, a better understanding of the role of TAMs seems crucial to control cancer progression.
Considering the character of TAMs, it is now generally accepted that TAMs usually polarize to M2 and represent protumoral functions.23 Indeed, we have seen in this study that approximately 60% of TAMs had the M2 phenotype in PC14PE6 and A549 control (vector-transduced) tumors (Figures 2B and 5C). However, when tumor cells expressed SP-A, this M1/M2 balance was reversed, and M1 macrophages became dominant in both tumors. As far as we have investigated, the expression of multiple M1 markers was up-regulated in SP-A–expressing tumors, whereas the expression of M2 markers was not altered in xenograft and metastatic lung tumors. Together with the fact that the number of M1 TAMs was increased in SP-A–expressing tumors, these results indicate that SP-A aided in making the TAMs M1 dominant by increasing the number of recruited M1 macrophages rather than shifting the M2 TAMs into the M1 phenotype in the tumor microenvironment.
Numerous studies have shown that macrophages could be a target cell type that SP-A interacts with to regulate infectious inflammation, and, to date, diverse and contradictive functions of SP-A against monocytes/macrophages are reported.29–32 These studies all indicate that SP-A has various effects on inflammation induced by different agonists. Indeed, we also observed that the cytokine/chemokine expression profiles of PMs and human monocytes were different in response to SP-A (Figure 4), suggesting that SP-A may exert cell- and agonist-specific effects that contribute to the inflammation state of the host.
In addition to the observations investigating the role of SP-A during infection, we showed that SP-A activates and increases M1 macrophages in the tumor microenvironment and induces the production of inflammatory cytokines, suggesting that SP-A facilitates inflammation in the tumor to reduce tumor progression. The precise molecular mechanism by which SP-A activates M1 macrophages in the tumor remains unclear with the current observation; however, several mechanisms should be considered. First, SP-A may enhance the binding of cytokines to their respective receptors. SP-A is reported to bind to several receptors, including Toll-like receptors 2 and 4, and to regulate inflammatory responses induced by pathogen-derived products, such as peptidoglycan and lipopolysaccharide via Toll-like receptors.33–35 In addition to its role in Toll-like receptor–mediated cellular responses induced by infectious challenges, it is very possible that SP-A regulates the function of TAMs in the tumor microenvironment through the interaction with Toll-like receptors. Second, signal transduction in the TAMs could be regulated by SP-A. SP-A has been shown to trigger rapid tyrosine, but not serine or threonine, phosphorylation36 of macrophage proteins and could possibly enhance/accelerate the initial phosphorylation steps of the signal transduction pathway, which leads to the regulation of inflammation in the tumor. It was also possible that SP-A directly affects tumor cells and regulates cytokine expression. Thus, we compared the chemokine expression of SP-A–expressing PC14PE6 and control (vector-transduced) cells using the PCR array system. Of 92 chemokine and chemokine-related genes tested, none were altered by SP-A overexpression (data not shown), suggesting that SP-A did not directly affect chemokine expression of tumor cells.
Of note, we showed the effect of SP-A on M1 macrophage recruitment and tumor suppression in xenograft and lung metastasis models. This result indicated two important aspects of SP-A. First, the lung tumor–specific expression of SP-A is more important than the host SP-A in the lung to suppress lung cancer progression. Second, the recruited M1 TAMs by tumor-derived SP-A could be originated from circulating monocytes. This possibility was supported by the result that SP-A activated only circulating monocytes/macrophages (mouse PMs or human monocytes) and showed no effect on resident AMs in cytokine expression. In the lung, the sensitivity of resident AMs against SP-A is thought to be suppressed as they are continuously contacted by SP-A, which could be a plausible explanation because the host needs to be protected from the overzealous inflammation in the resting, normal, noninflamed lungs. The molecular mechanism of different SP-A sensitivities in different cells is not still clearly understood. However, as stated previously herein, the expression of inflammatory signaling molecules that might be regulated by SP-A could be different between AMs and other type of monocytes/macrophages. In addition, there could be an unknown SP-A receptor(s) that might be critical in regulating inflammation in macrophages, as SP-A is reported to bind to multiple receptors.2 Further studies are needed to understand the precise molecular mechanisms of the diverse and cell-specific function of SP-A against macrophages in the context of SP-A and lung cancer.
These results indicate that NK cell activation was the main mechanism by which SP-A lead to the reduction in tumor burden. The production of Prf1 and GzmB was strongly increased (15- to 30-fold), whereas the number of NK cells was increased only twofold to threefold in the SP-A–expressing tumor, suggesting the multiple pathways regulated by SP-A to recruit and activate NK cells. Because cytokines produced by M1 macrophages, such as interferon-γ and CCL2, are known to activate NK cells,37–39 it is likely that SP-A implicitly induces NK cell killing via activating M1 TAMs and increasing various inflammatory cytokines in the tumor microenvironment.
Two functional genes of SP-A were detected in a previous report40: SP-A1 and SP-A2. These genes were differentially regulated by development41 and have a minor difference in carbohydrate-binding activity.42 However, SP-A protein derived from SP-A1 and SP-A2 genes are reported to be functional and to enhance TNF-α secretion by the monocytic cell line.43 Thus, although we have transduced only the SP-A1 gene, we suspect that SP-A protein from the SP-A2 gene could also contribute to activation of the innate immune system and suppress tumor progression.
In conclusion, these findings demonstrate that SP-A regulates the tumor microenvironment by controlling the polarization of TAMs. SP-A expression by tumor cells leads to increased numbers and the activation of M1 TAMs. These activated M1 TAMs then recruit and activate NK cells that function in tumor suppression. These results indicate that SP-A plays an important protective role in the progression of lung cancer. Specifically targeting M1 TAMs (not bulk TAMs) to induce the activation of a proinflammatory program in the tumor, generating the pharmacologic modulators of SP-A for example, could be the therapeutic approach to improve the effect of anticancer therapy.
Acknowledgments
This work was performed in collaboration with the late Dr. Jo Rae Wright (Duke University, Durham, NC). We greatly appreciate and honor her contribution to this work.
We also thank Kathy Evans (Duke University) for the preparation of purified human SP-A, Tomoko Oka and the Support Center for Advanced Medical Sciences (The University of Tokushima) for technical assistance, and the Student Lab (The University of Tokushima) for helpful discussions.
Footnotes
Supported by the Ministry of Education, Culture, Sports, Science and Technology Grants-in Aid for Scientific Research (MEXT KAKENHI) grant 22790759 (H.G.) and NIH grants AI-81672 and HL-111151 (J.G.L.).
A.M. and H.G. contributed equally to this work.
Supplemental Data
A: Immunohistochemical analysis of subcutaneous tumors produced by SP-A– or vector-transduced lung cancer cell lines. Paraffin-embedded sections (4 μm thick) were stained with 1:50 dilution of mouse anti-human Ki-67 monoclonal antibody (Dako, Carpinteria, CA) or the DeadEnd fluorometric TUNEL system (Promega, Madison, WI). Frozen tissue sections (8 μm thick) were stained with rat anti-mouse CD31/PECAM-1 monoclonal antibody (BD Pharmingen). In each slide, the number of positive cells was counted in the five areas under fluorescent microscopy at ×100 magnification (lower panels). Scale bars: 100 μm. Data are the average of four to five mice per group. *P < 0.05. B: The cell proliferation of SP-A– or vector-transduced lung cancer cell lines was determined by MTT assay. C: Cell-cycle analysis of SP-A– or vector-transduced PC14PE6 cells. After staining with propidium iodide (PI), flow cytometry analysis was performed using FACSCaliber (Becton Dickinson Immunocytometry Systems, San Jose, CA). Data analysis was performed using CellQuest software version 5.1.1 (Becton Dickinson Immunocytometry Systems). Data are presented as means ± SEM.
References
- 1.Jemal A., Siegel R., Ward E., Hao Y., Xu J., Thun M.J. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Wright J.R. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. 2005;5:58–68. doi: 10.1038/nri1528. [DOI] [PubMed] [Google Scholar]
- 3.Borron P., McIntosh J.C., Korfhagen T.R., Whitsett J.A., Taylor J., Wright J.R. Surfactant-associated protein A inhibits LPS-induced cytokine and nitric oxide production in vivo. Am J Physiol Lung Cell Mol Physiol. 2000;278:840–847. doi: 10.1152/ajplung.2000.278.4.L840. [DOI] [PubMed] [Google Scholar]
- 4.LeVine A.M., Gwozdz J., Stark J., Bruno M., Whitsett J., Korfhagen T. Surfactant protein-A enhances respiratory syncytial virus clearance in vivo. J Clin Invest. 1999;103:1015–1021. doi: 10.1172/JCI5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mariencheck W.I., Savov J., Dong Q., Tino M.J., Wright J.R. Surfactant protein A enhances alveolar macrophage phagocytosis of a live, mucoid strain of P. aeruginosa. Am J Physiol. 1999;277:777–786. doi: 10.1152/ajplung.1999.277.4.L777. [DOI] [PubMed] [Google Scholar]
- 6.Giannoni E., Sawa T., Allen L., Wiener-Kronish J., Hawgood S. Surfactant proteins A and D enhance pulmonary clearance of Pseudomonas aeruginosa. Am J Respir Cell Mol Biol. 2006;34:704–710. doi: 10.1165/rcmb.2005-0461OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atochina E.N., Beck J.M., Preston A.M., Haczku A., Tomer Y., Scanlon S.T., Fusaro T., Casey J., Hawgood S., Gow A.J., Beers M.F. Enhanced lung injury and delayed clearance of Pneumocystis carinii in surfactant protein A-deficient mice: attenuation of cytokine responses and reactive oxygen-nitrogen species. Infect Immun. 2004;72:6002–6011. doi: 10.1128/IAI.72.10.6002-6011.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuronuma K., Sano H., Kato K., Kudo K., Hyakushima N., Yokota S., Takahashi H., Suzuki H., Kodama T., Abe S., Kuroki Y. Pulmonary surfactant protein A augments the phagocytosis of Streptococcus pneumoniae by alveolar macrophages through a casein kinase 2-dependent increase of cell surface localization of scavenger receptor A. J Biol Chem. 2004;279:21421–21430. doi: 10.1074/jbc.M312490200. [DOI] [PubMed] [Google Scholar]
- 9.Goto H., Ledford J.G., Mukherjee S., Noble P.W., Williams K.L., Wright J.R. The role of surfactant protein A in bleomycin-induced acute lung injury. Am J Respir Crit Care Med. 2010;181:1336–1344. doi: 10.1164/rccm.200907-1002OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pastva A.M., Walker J.K., Maddox L.A., Mukherjee S., Giamberardino C., Hsia B., Potts E., Zhu H., Degan S., Sunday M.E., Lawson B.L., Korfhagen T.R., Schwartz D.A., Eu J.P., Foster W.M., McMahon T.J., Que L., Wright J.R. Nitric oxide mediates relative airway hyperresponsiveness to lipopolysaccharide in surfactant protein A-deficient mice. Am J Respir Cell Mol Biol. 2011;44:175–184. doi: 10.1165/rcmb.2009-0284OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Bejarano P.A., Baughman R.P., Biddinger P.W., Miller M.A., Fenoglio-Preiser C., al-Kafaji B., Di Lauro R., Whitsett J.A. Surfactant proteins and thyroid transcription factor-1 in pulmonary and breast carcinomas. Mod Pathol. 1996;9:445–452. [PubMed] [Google Scholar]
- 12.Jiang F., Caraway N.P., Nebiyou Bekele B., Zhang H.Z., Khanna A., Wang H., Li R., Fernandez R.L., Zaidi T.M., Johnston D.A., Katz R.L. Surfactant protein A gene deletion and prognostics for patients with stage I non-small cell lung cancer. Clin Cancer Res. 2005;11:5417–5424. doi: 10.1158/1078-0432.CCR-04-2087. [DOI] [PubMed] [Google Scholar]
- 13.Tsutsumida H., Goto M., Kitajima S., Kubota I., Hirotsu Y., Yonezawa S. Combined status of MUC1 mucin and surfactant apoprotein A expression can predict the outcome of patients with small-size lung adenocarcinoma. Histopathology. 2004;44:147–155. doi: 10.1111/j.1365-2559.2004.01797.x. [DOI] [PubMed] [Google Scholar]
- 14.McIntosh J.C., Swyers A.H., Fisher J.H., Wright J.R. Surfactant proteins A and D increase in response to intratracheal lipopolysaccharide. Am J Respir Cell Mol Biol. 1996;15:509–519. doi: 10.1165/ajrcmb.15.4.8879185. [DOI] [PubMed] [Google Scholar]
- 15.Morita S., Kojima T., Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063–1066. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 16.Maekawa S., Tsukumo S., Chiba S., Hirai S., Hayashi Y., Okada H., Kishihara K., Yasutomo K. Delta1-Notch3 interactions bias the functional differentiation of activated CD4+ T cells. Immunity. 2003;19:549–559. doi: 10.1016/s1074-7613(03)00270-x. [DOI] [PubMed] [Google Scholar]
- 17.Yano S., Nokihara H., Yamamoto A., Goto H., Ogawa H., Kanematsu T., Miki T., Uehara H., Saijo Y., Nukiwa T., Sone S. Multifunctional interleukin-1β promotes metastasis of human lung cancer cells in SCID mice via enhanced expression of adhesion-, invasion- and angiogenesis-related molecules. Cancer Sci. 2003;94:244–252. doi: 10.1111/j.1349-7006.2003.tb01428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yano S., Shinohara H., Herbst R.S., Kuniyasu H., Bucana C.D., Ellis L.M., Fidler I.J. Production of experimental malignant pleural effusions is dependent on invasion of the pleura and expression of vascular endothelial growth factor/vascular permeability factor by human lung cancer cells. Am J Pathol. 2000;157:1893–1903. doi: 10.1016/S0002-9440(10)64828-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yano S., Nishioka Y., Izumi K., Tsuruo T., Tanaka T., Miyasaka M., Sone S. Novel metastasis model of human lung cancer in SCID mice depleted of NK cells. Int J Cancer. 1996;67:211–217. doi: 10.1002/(SICI)1097-0215(19960717)67:2<211::AID-IJC11>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 20.de Jonge H.J., Fehrmann R.S., de Bont E.S., Hofstra R.M., Gerbens F., Kamps W.A., de Vries E.G., van der Zee A.G., te Meerman G.J., ter Elst A. Evidence based selection of housekeeping genes. PLoS One. 2007;2:898. doi: 10.1371/journal.pone.0000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Utsugi T., Sone S. Comparative analysis of the priming effect of human interferon-gamma, -alpha, and -beta on synergism with muramyl dipeptide analog for anti-tumor expression of human blood monocytes. J Immunol. 1986;136:1117–1122. [PubMed] [Google Scholar]
- 22.Hyodo Y., Matsui K., Hayashi N., Tsutsui H., Kashiwamura S., Yamauchi H., Hiroishi K., Takeda K., Tagawa Y., Iwakura Y., Kayagaki N., Kurimoto M., Okamura H., Hada T., Yagita H., Akira S., Nakanishi K., Higashino K. IL-18 up-regulates perforin-mediated NK activity without increasing perforin messenger RNA expression by binding to constitutively expressed IL-18 receptor. J Immunol. 1999;162:1662–1668. [PubMed] [Google Scholar]
- 23.Pollard J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 24.Solinas G., Germano G., Mantovani A., Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86:1065–1073. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- 25.Benoit M., Desnues B., Mege J.L. Macrophage polarization in bacterial infections. J Immunol. 2008;181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- 26.Robertson M.J. Role of chemokines in the biology of natural killer cells. J Leukoc Biol. 2002;71:173–183. [PubMed] [Google Scholar]
- 27.Langers I., Renoux V.M., Thirty M., Delvenne P., Jacobs N. Natural killer cells: role in local growth and metastasis. Biologics. 2012;6:73–82. doi: 10.2147/BTT.S23976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bingle L., Brown N.J., Lewis C.E. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–265. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 29.Kremlev S.G., Phelps D.S. Surfactant protein A stimulation of inflammatory cytokine and immunoglobulin production. Am J Physiol. 1994;267:712–719. doi: 10.1152/ajplung.1994.267.6.L712. [DOI] [PubMed] [Google Scholar]
- 30.Kremlev S.G., Umstead T.M., Phelps D.S. Surfactant protein A regulates cytokine production in the monocytic cell line THP-1. Am J Physiol. 1997;272:996–1004. doi: 10.1152/ajplung.1997.272.5.L996. [DOI] [PubMed] [Google Scholar]
- 31.McIntosh J.C., Mervin-Blake S., Conner E., Wright J.R. Surfactant protein A protects growing cells and reduces TNF-alpha activity from LPS-stimulated macrophages. Am J Physiol. 1996;271:310–319. doi: 10.1152/ajplung.1996.271.2.L310. [DOI] [PubMed] [Google Scholar]
- 32.Stamme C., Walsh E., Wright J.R. Surfactant protein A differentially regulates IFN-gamma- and LPS-induced nitrite production by rat alveolar macrophages. Am J Respir Cell Mol Biol. 2000;23:772–779. doi: 10.1165/ajrcmb.23.6.4083. [DOI] [PubMed] [Google Scholar]
- 33.Sano H., Sohma H., Muta T., Nomura S., Voelker D.R., Kuroki Y. Pulmonary surfactant protein A modulates the cellular response to smooth and rough lipopolysaccharides by interaction with CD14. J Immunol. 1999;163:387–395. [PubMed] [Google Scholar]
- 34.Sato M., Sano H., Iwaki D., Kudo K., Konishi M., Takahashi H., Takahashi T., Imaizumi H., Asai Y., Kuroki Y. Direct binding of Toll-like receptor 2 to zymosan, and zymosan-induced NF-kappa B activation and TNF-alpha secretion are down-regulated by lung collectin surfactant protein A. J Immunol. 2003;171:417–425. doi: 10.4049/jimmunol.171.1.417. [DOI] [PubMed] [Google Scholar]
- 35.Henning L.N., Azad A.K., Parsa K.V., Crowther J.E., Tridandapani S., Schlesinger L.S. Pulmonary surfactant protein A regulates TLR expression and activity in human macrophages. J Immunol. 2008;180:7847–7858. doi: 10.4049/jimmunol.180.12.7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schagat T.L., Tino M.J., Wright J.R. Regulation of protein phosphorylation and pathogen phagocytosis by surfactant protein A. Infect Immun. 1999;67:4693–4699. doi: 10.1128/iai.67.9.4693-4699.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rolny C., Mazzone M., Tugues S., Laoui D., Johansson I., Coulon C., Squadrito M.L., Segura I., Li X., Knevels E., Costa S., Vinckier S., Dresselaer T., Akerud P., De Mol M., Salomaki H., Phillipson M., Wyns S., Larsson E., Buysschaert I., Botling J., Himmelreich U., Van Ginderachter J.A., De Palma M., Dewerchin M., Claesson-Welsh L., Carmeliet P. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19:31–44. doi: 10.1016/j.ccr.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 38.Schroder K., Hertzog P.J., Ravasi T., Hume D.A. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 39.Morrison B.E., Park S.J., Mooney J.M., Mehrad B. Chemokine-mediated recruitment of NK cells is a crucial host defense mechanisms in invasive aspergillosis. J Clin Invest. 2003;112:1862–1870. doi: 10.1172/JCI18125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.White R.T., Damm D., Miller J., Spratt K., Schilling J., Hawgood S., Benson B., Cordell B. Isolation and characterization of the human pulmonary surfactant apoprotein gene. Nature. 1985;317:361–363. doi: 10.1038/317361a0. [DOI] [PubMed] [Google Scholar]
- 41.McCormick S.M., Mendelson C.R. Human SP-A1 and SP-A2 genes are differentially regulated during development and by cAMP and glucocorticoids. Am J Physiol. 1994;266:367–374. doi: 10.1152/ajplung.1994.266.4.L367. [DOI] [PubMed] [Google Scholar]
- 42.Oberley R.E., Snyder J.M. Recombinant human SP-A1 and SP-A2 proteins have different carbohydrate-binding characteristics. Am J Physiol Lung Cell Mol Physiol. 2003;284:871–881. doi: 10.1152/ajplung.00241.2002. [DOI] [PubMed] [Google Scholar]
- 43.Wang G., Phelps D.S., Umstead T.M., Floros J. Human SP-A protein variants derived from one or both genes stimulate TNF-alpha production in the THP-1 cell line. Am J Physiol Lung Cell Mol Physiol. 2000;278:946–954. doi: 10.1152/ajplung.2000.278.5.L946. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A: Immunohistochemical analysis of subcutaneous tumors produced by SP-A– or vector-transduced lung cancer cell lines. Paraffin-embedded sections (4 μm thick) were stained with 1:50 dilution of mouse anti-human Ki-67 monoclonal antibody (Dako, Carpinteria, CA) or the DeadEnd fluorometric TUNEL system (Promega, Madison, WI). Frozen tissue sections (8 μm thick) were stained with rat anti-mouse CD31/PECAM-1 monoclonal antibody (BD Pharmingen). In each slide, the number of positive cells was counted in the five areas under fluorescent microscopy at ×100 magnification (lower panels). Scale bars: 100 μm. Data are the average of four to five mice per group. *P < 0.05. B: The cell proliferation of SP-A– or vector-transduced lung cancer cell lines was determined by MTT assay. C: Cell-cycle analysis of SP-A– or vector-transduced PC14PE6 cells. After staining with propidium iodide (PI), flow cytometry analysis was performed using FACSCaliber (Becton Dickinson Immunocytometry Systems, San Jose, CA). Data analysis was performed using CellQuest software version 5.1.1 (Becton Dickinson Immunocytometry Systems). Data are presented as means ± SEM.