

Abstract
Air pollutants and obesity are important factors that contribute to asthma. The aim of this study was to assess the airway responsiveness and inflammation in Otsuka-Long Evans Tokushima Fatty (OLETF) obese rats and Long Evans Tokushima-Otsuka (LETO) nonobese rats exposed to diesel exhaust particles (DEPs). Otsuka Long Evans Tokushima fatty rats and LETO rats were exposed intranasally to DEP and then challenged with aerosolized DEP on days 6 to 8. Body plethysmography, bronchoalveolar lavage (BAL), and histology were performed. Enhanced pause (Penh) was measured as an indicator of airway resistance on day 9 and samples were collected on day 10. After exposure to DEP, the OLETF group exhibited a greater increase in Penh compared to that in the LETO group. Moreover, the BAL fluid in mice showed an increase in the total and differential cell counts in the DEP-exposed OLETF group compared to that in the DEP-exposed LETO group. Histological assessment of lung tissue from each group revealed that the DEP-exposed OLETF group tended to have increased inflammatory cell infiltrations in the prebronchial area. Increased peroxisome proliferator-activated receptor γ, coactivator 1β messenger RNA was observed in the lungs of obese rats compared to that in nonobese rats following DEP exposure. These data indicate that the DEP-exposed OLETF group had increased airway responses and inflammation compared to the DEP-exposed LETO group, indicating that diesel particulates and obesity may be co-contributors to asthma.
Keywords: air pollution, asthma, obesity
Introduction
Asthma is an increasingly prevalent and important health problem.1,2 In addition, obesity is the most common metabolic disease worldwide, and its prevalence has increased over recent decades.3 The World Health Organization predicts that around 700 million adults will be obese (at least 10% of the projected global population) by 2015. Obesity is associated with increases in diabetes, malignancy, and cardiovascular and musculoskeletal diseases, and this rise in obesity is predicted to present an immense health care and economic burden. Although little attention has been focused on the impact of obesity on respiratory diseases, the effect of obesity on pulmonary function and inflammation is clear; thus, the increased incidence in obesity is likely to increase the prevalence and morbidity of lung disease. Cross-sectional and prospective studies have demonstrated the effects of weight loss or weight gain on a variety of asthma outcomes4–7; however, the precise mechanisms underlying the association between obesity and asthma have not yet been established.
Air pollution is a major risk factor in the development and exacerbation of respiratory diseases, and in particular asthma.8–12 Few studies have examined the associations among obesity, asthma, and air pollutants such as diesel exhaust particles (DEPs). This study aimed to evaluate whether air pollutants exacerbate airway responsiveness (AHR) and inflammation in obese rats by assessing the effect of DEP on airway inflammation and response in obese rats. Additionally, we aimed to develop an experimental model to further investigate the links between asthma and obesity.
Materials and Methods
Diesel Exhaust Particle Sources and Preparation
The DEPs used in this study were obtained from Hajime Takizawa and were collected using the following method. A 4JB1-type, light-duty (2740 cm), 4-cylinder diesel engine (Isuzu Automatic Co, Tokyo, Japan) was connected to an EDGY dynamometer (Meiden-Sya, Tokyo, Japan) and was operated using a standard diesel fuel at speeds of 1500 rpm under a torque load of 10 kg/m. The exhaust was introduced into a stainless-steel dilution tunnel (300 × 8400 mm) in a constant-volume sampler system attached at the end of the dilution tunnel. The temperature at the sampling point was below 50°C. The diameter of the particles was measured using a low-pressure-type Anderson Air Sampler (Anderson 2000 Inc, Atlanta, GA, USA); the mean diameter of the particles was 0.4 mm. Most of the shapes analyzed by scanning electron microscope were globular. Diesel exhaust particles were collected on filter samples for subsequent resuspension for the animal exposures.
Diesel Exhaust Particle Exposure
The endotoxin concentration of the DEP suspension was expected to be less than 0.064 ng/mL (0.32 EU/mL) based on a Limulus amebocyte lysate assay (QCL-1000; BioWhittaker, Inc Walkersville, Maryland).9,10,13 A spontaneously diabetic and obese rat was discovered by Charles River Canada (St Constant, Quebec, Canada) in an outbred colony of Long-Evans rats. A strain of rats developed from this rat by selective breeding has since been maintained at the Tokushima Research Institute (Otsuka Pharmaceutical, Tokushima, Japan) and named Otsuka Long-Evans Tokushima Fatty (OLETF). Otsuka Long-Evans Tokushima Fatty (120 g) obese rats and LETO nonobese rats (100 g)14 that were 5 to 6 weeks old and free of rat-specific pathogens were provided by Otsuka Co Ltd (Tokyo, Japan). The rats were housed throughout the experiments in a laminar flow cabinet and maintained on standard laboratory chow (Lab Diet OL79, Orient Bio, Korea). All experimental animals used in this study were treated according to the guidelines approved by the Institutional Animal Care and Use Committee of Soonchunhayng University Hospital. Diesel exhaust particle was resuspended in saline for 30 minutes before being administered. Diesel exhaust particle (300 μg) was administered intranasally for the first 5 days; then on days 6 to 8, the rats were exposed to 3 mg/m3 DEP for 1 hour in a closed system chamber attached to an ultrasonic nebulizer (NE-UO7; Omron Corporation, Tokyo, Japan) with an output of 1 mL/min and a particle size of 1 to 5 μm (Figure 1). The control rats were exposed to saline solution only.
Figure 1.
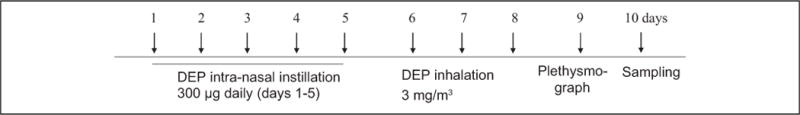
Experimental protocol. Rats were exposed intranasally with diesel exhaust particles (DEPs; 300 μg on days 1–5), and on days 6 to 8, the rats were exposed to 3 mg DEP/m3 or saline control for 1 hour using an ultrasonic nebulizer.
The rats were killed with an overdose of pentobarbital sodium (65 mg/kg body weight, administered intraperitoneally). The chest cavity was exposed, and a catheter was inserted carefully into the trachea and secured with ligatures. Bronchoalveolar lavage (BAL)9 was performed 4 times using 1 mL normal saline with gentle retrieval. Cell numbers were counted using a hemocytometer, and differential cell counts were performed on slides prepared by cytocentrifugation and Diff-Quik staining (Scientific Products, Gibbstowne, New Jersey). Supernatants were separated by centrifugation (500 × g, 5 minutes) and kept at −70°C until use. After ligation of the right main bronchus, the left lungs were fixed with 4% paraformaldehyde in phosphate-buffered saline and then embedded in paraffin. The right lungs were excised and immersed in TRI reagent (guanidium thiocyanate–phenol mixture; Molecular Research Center Inc, Cincinnati, Ohio) and immediately frozen in liquid nitrogen.
Determination of Airway Responsiveness to Methacholine
Airway responsiveness was measured in unrestrained, conscious rats 1 day afterthe last challenge, as describedpreviously.9,15 Rats were placed in a barometric plethysmographic chamber (All Medicus Co, Anyang, Korea) and baseline readings were taken and averaged for 3 minutes. Increasing concentrations of aerosolized methacholine, from 2.5 to 100 mg/mL, were nebulized through an inlet of the main chamber for 3 minutes. Readings were taken and averaged for 3 minutes after each nebulization, and the enhanced pause (Penh) was determined. Penh, calculated as (expiratory time/relaxation time − 1) × (peak expiratory flow/peak inspiratory flow), is a dimensionless value that represents a function of the proportion of maximal expiratory to maximal inspiratory box pressure signals and is a function of the timing of expiration. Penh was used as a measure of airway responsiveness to methacholine. Results were expressed as the percentage increase in Penh following challenge with each concentration of methacholine, where the baseline Penh (after saline challenge) was expressed as an actual value. Penh values averaged for 3 minutes after each nebulization were evaluated.
Histology of the Lung Tissue
Sections of fixed embedded tissues were cut in 4-μm-thick slices using a Leica model 2165 rotary microtome (Leica Microsystems Nussloch GmbH, Nussloch, Germany) and placed on glass slides. The tissues were processed, embedded in paraffin, sectioned, and stained with hematoxylin, eosin, and periodic acid–Schiff.
Measurement of Protein Levels in BAL Fluids
The levels of tumor necrosis factor (TNF) α, interleukin (IL) 6, IL-4, and IL-10 in BAL fluid were quantified in duplicate using a sandwich enzyme-linked immunosorbent assay kit according to the manufacturer’s protocol (Biosource International Inc, Camarillo, California). The lower detection limits for TNF-α, IL-6, IL10, and IL-4 were 5, 3, 3, and 5 pg/mL, respectively. Values below these limits were considered zero for statistical analysis. Inter- and intra-assay coefficients of variance were expected to be less than 10%.
Peroxisome Proliferator-Activated Receptor γ, Coactivator 1β Reverse Transcription-Polymerase Chain Reaction in Lung Extracts
Total RNA was extracted using TRI reagent (guanidium thiocyanate–phenol mixture; Molecular Research Center Inc) and chloroform according to the manufacturer’s instructions. The following primer sets used for polymerase chain reaction (PCR) were peroxisome proliferator-activated receptor γ, coactivator 1β (PPARGC1B): 5′-AAGACCCTGGAT-3′ and 5′-GCTACATGCATACCTACTG-3′, GAPDH: 5′-GGCATTGCTCTCAATGACAA-3′ and 5′-AGGGCCTCTCTCTTGCTCTC-3′. The RNA was reverse transcribed by incubation with 10 mmol/L deoxyribonucleotide triphosphate, 0.1 mol/L dithiothreitol, 1 μL of oligo (500 μg/mL), and 1 μL of Super-Script II (200 units/μL; Life Technologies, Grand Island, New York) at 42°C for 50 minutes and then heat inactivated at 70°C for 15 minutes. After reverse transcription, aliquots of the complementary DNA were transferred to tubes containing specific primer pairs for human PPARGC1B and mice GAPDH genes for PCR. The PPARGC1B amplification was performed in a thermocycler with an initial denaturation step at 94°C for 5 minutes, followed by 28 cycles at 94°C for 1 minute, 50°C for 1 minute, 72°C for 1 minute, 94°C for 1 minute, 55°C for 1 minute, and 72°C for 1 minute, followed by a final extension step at 72°C for 10 minutes. Amplified PCR products were electrophoresed on 1% agarose gel and visualized by ethidium bromide staining.
Statistical Analysis
Differences between independent samples were compared using the nonparametric Kruskal-Wallis H-test for continuous data. The Mann-Whitney U test was applied to compare significant differences between 2 samples. Differences were considered significant at P < 0.05. Results are expressed as means ± standard error of the mean, unless otherwise stated.
Results
The DEP-exposed OLETF group exhibited a greater increase in Penh compared to that exhibited by the DEP-exposed LETO group (Figure 2). Examination of the BAL fluid showed increased total and differential cell counts in the DEP-exposed OLETF group compared to cell counts in the DEP-exposed LETO group. Moreover, an increase in neutrophils, eosinophils, and lymphocytes was revealed in the DEP-exposed OLETF group compared to the DEP-exposed LETO group (Figure 3). Histological assessment of lung tissue samples from each group revealed that the DEP-exposed OLETF group tended to have increased infiltration of mononuclear cells and eosinophils in the prebronchial area (LETO control vs DEP-exposed LETO vs OLETF control vs DEP-exposed OLETF DEP; inflammatory index, 1.3 ± 0.7 vs 2.1 ± 0.9 vs 2.3 ± 1.3 vs 3.2 ± 0.9, respectively). The DEP-exposed OLETF group had a greater increase in goblet cell hyperplasia compared to that in the DEP-exposed LETO group (Figure 4), although it is recognized that lungs had previously been lavaged, and this could affect their histological appearance. Increased IL-6 and TNF-α were observed in the DEP-exposed OLETF group compared to the DEP-exposed LETO group, whereas decreased IL-10 was found in the DEP-exposed OLETF group compared to the DEP-exposed LETO group (Figure 5). Increased PPARGC1B messenger RNA (mRNA) was found in the lungs of DEP-exposed OLETF obese rats compared to that in the lungs of DEP-exposed LETO nonobese rats (Figure 6).
Figure 2.
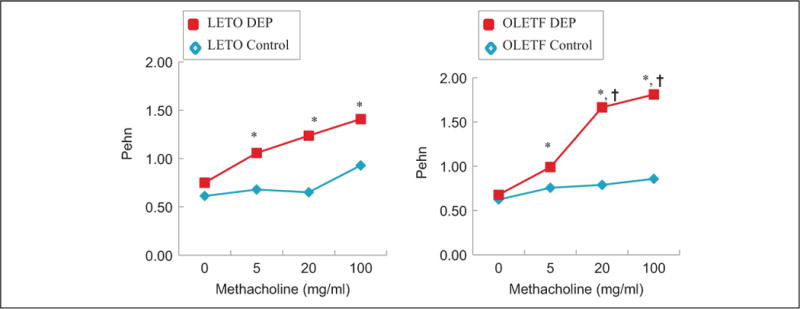
Whole-body plethysomography in DEP-exposed and challenged Otsuka Long Evans Tokushima fatty (OLETF) obese rats and Long Evans Tokushima-Otsuka (LETO) non-obese rats. Metacholine challenge was performed 24 h after DEP inhalation. Rats were placed in a plethysmograph and treated with saline followed by increasing doses of methacholine (0–100 mg/ml) for 3 min. Enhanced pause (Penh) was obtained for 3 min after each nebulization. *P < 0.05 LETF and OLETF following DEP was significantly greater than those following saline. †P < 0.05 OLETF following DEP was significantly greater than LETO following DEP. Data are means (n = 6 rats/group).
Figure 3.
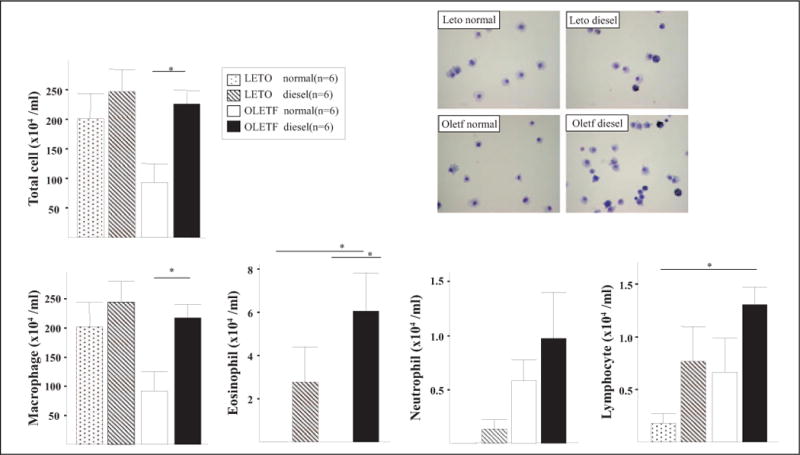
Cellular profile of bronchoalveolar lavage (BAL) fluids. The number of neutrophils and lymphocyte in BAL fluids was increased in the OLETF and DEP groups, respectively (*P < 0.05). DEP indicates diesel exhaust particles; OLETF, Otsuka Long Evans Tokushima Fatty.
Figure 4.
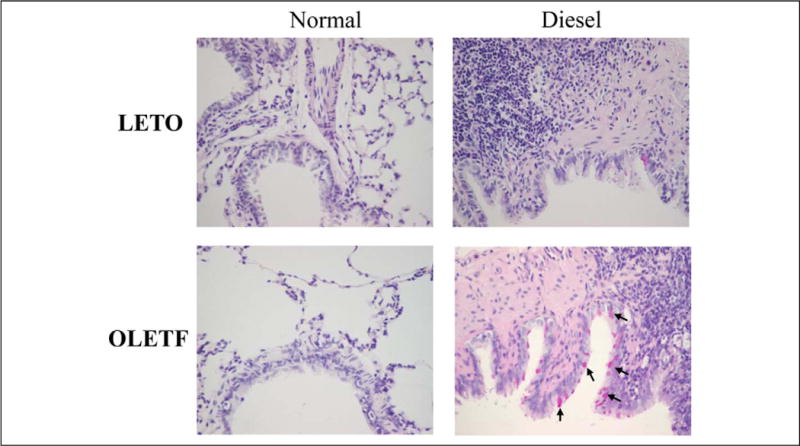
Goblet cell hyperplasia observed in the DEP-exposed OLETF group. Arrows indicate goblet cell hyperplasia. DEP indicates diesel exhaust particles; OLETF, Otsuka Long Evans Tokushima Fatty.
Figure 5.
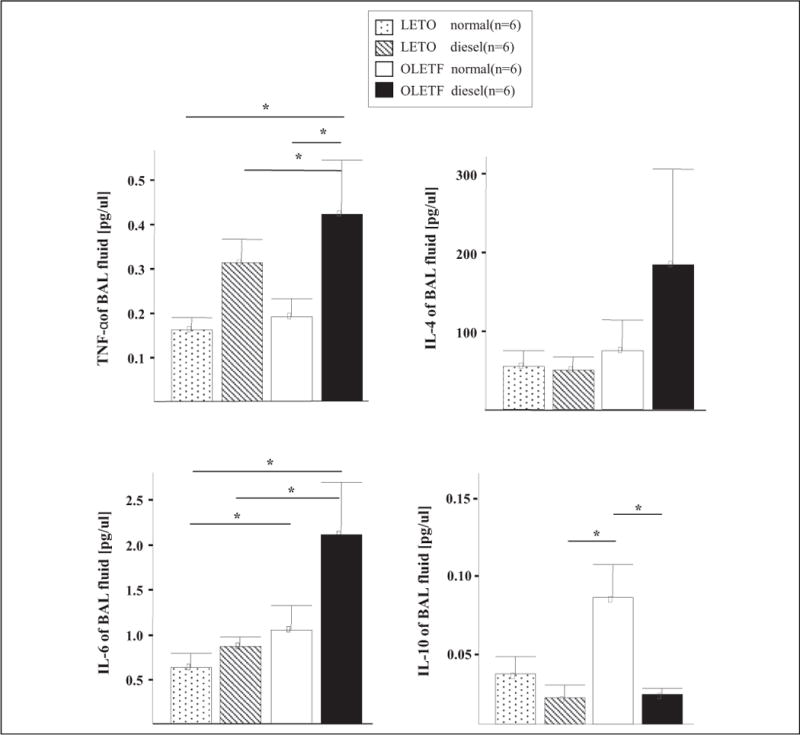
Cytokine profiles of BAL fluids of mice. Increased levels of interleukin (IL) 4, IL-6, and tumor necrosis factor (TNF) α, and decreased IL-10 levels were found in the DEP-exposed OLETF obese rats compared to the DEP-exposed LETO nonobese rats. Values are means ± SEM (n = 6). *P < 0.05 OLETF significantly different than LETO nonobese rats. DEP indicates diesel exhaust particles; OLETF, Otsuka Long Evans Tokushima Fatty; LETO, Long Evans Tokushima-Otsuka; SEM, standard error of the mean.
Figure 6.
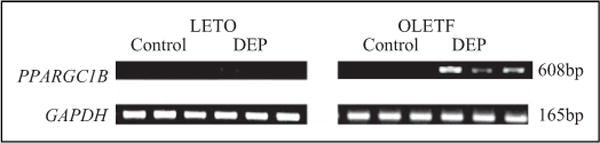
Expression of peroxisome proliferator-activated receptor γ, coactivator 1β (PPARGC1B) mRNA. Increased PPARGC1B mRNA levels were found in the lungs of DEP-exposed OLETF obese rats compared to those in the DEP-exposed LETO non-obese rats. RNA was isolated from lung tissue and analyzed by reverse-transcription polymerase chain reaction (RT-PCR). DEP indicates diesel exhaust particles; mRNA, messenger RNA; OLETF, Otsuka Long Evans Tokushima Fatty; LETO, Long Evans Tokushima-Otsuka.
Discussion
Several cross-sectional, case–control, and prospective studies have described an association between obesity and asthma.16–21 Moreover, recent reports have suggested that obesity precedes the development of asthma.22–24 Although the mechanisms underlying a putative relationship between obesity and asthma have not been fully established, numerous studies have suggested that obesity increases the risk of incident asthma.16–24 The basis for an increase of asthma in obesity is an area of growing interest, and the rising prevalence of obesity in both children and adults highlights the public health importance of this area of research. Several potential mechanisms have been postulated for this association of asthma and obesity including inflammation, mechanical factors, common genetic factors, and changes in airway structure and function.4 Some studies have demonstrated a dose–response relationship between weight and asthma, whereas others have shown a possible quadratic relationship, with the highest risk of asthma occurring at both extremes of body weight.3,18,25
Fat tissue in both humans and animals is a source of inflammatory mediators that may be implicated in asthma pathophysiology.26 These factors include leptin, adiponectin, and plasminogen activator inhibitor 1.27,28 The term “adipokine” refers to proteins synthesized by and secreted from adipose tissue.4 These adipokines include cytokines (IL-1, IL-6, TNF-α, IL-10, and visfatin), chemokines (IL-6, eotaxin, monocyte chemotactic protein 1, and macrophage inflammatory protein 1α), and hormones (leptin, adiponectin, and resistin) involved in energy regulation, as well as other factors (plasminogen activator inhibitor 1, angiotensinogen, vascular endothelial growth factor, complement B, C3, D, and IL-1 receptor antagonist). In our study, we assessed the cytokine profiles in the BAL fluids in rats. Increased levels of IL-4, IL-6, and TNF-α levels and decreased levels of IL-10 were observed in the DEP-exposed OLETF obese rats compared to the DEP-exposed LETO nonobese rats.
The OLETF rat is an outbred strain of the LETO rat that has been characterized previously as lacking cholecystokinin (CCK) 1 receptor expression due to a spontaneous mutation spanning the promoter and the first 2 exon regions in the CCK-1 receptor gene.29 These animals have been used as a model of insulin resistance because of their natural manifestation of hyperglycemia and noninsulin-dependent diabetes mellitus relative to age-matched LETO rats.30 In addition, OLETF rats are currently under investigation as a model of obesity. Therefore, we used the OLETF rats as obese subjects to examine the effect of obesity on airway response and inflammation.
Increased exposure to air pollutants such as DEP has been proposed as one mechanism to explain the rise in allergic disorders. Among inner city children with asthma, short-term increases in air pollutant concentrations below the National Ambient Air Quality Standards were associated with adverse respiratory health effects.31
Strong positive associations were found between the distance to the nearest main road and asthmatic bronchitis, hay fever, eczema, and sensitization, providing strong evidence for an increased risk in atopic diseases and allergic sensitization in children exposed to ambient particulate matter.32
The observations33 serve as a direct demonstration and explanation of the epidemiologic evidence that associates exposure to diesel traffic with the severity of asthma and of the symptoms that many patients with asthma report after exposure to DEP. A growing body of evidence suggests that the inhalation of DEP may have a role in the development or exacerbation of asthma and allergic diseases by altering airway immune responses.8–11 Prenatal and early-life exposure to air pollutants, such as carbon monoxide, particulate matter, and nitric dioxide have a negative effect on pulmonary function in subgroups of children with asthma.33 Moreover, extensive epidemiological studies have demonstrated that particulate air pollutants such as DEP are related to allergic diseases including asthma and allergic rhinitis.34 Although several animal and human studies have shown that DEPs act as an adjuvant during allergen exposure and affect acute asthma exacerbations,35–38 questions remain concerning the ability of DEP to induce asthma in the absence of an allergen.
Many studies35–38 have demonstrated that long-term exposure to DEP affects AHR and inflammation, depending on the dose, frequency, and time of DEP exposure. The precise mechanisms of any association between obesity and asthma have not yet been established. Studies linking obesity, asthma, and air pollutants such as DEP are sparse.
We hypothesize that obesity and air pollutants such as DEP play a role in the increasing asthma prevalence, although a causal relationship remains to be established. Using OLETF rats as an obesity model, we exposed and challenged the rats to DEP to investigate the effects of DEP exposure on airway inflammation and AHR.
Marked variation exists among individuals in their responses to air pollutants, suggesting that genetic factors influence the mechanisms of lung injury caused by air pollutants.39 Genetic association studies have compared the adverse effects of air pollutants between subjects with specific genotypes in biologically relevant genes. In addition to avoiding exposure in the most susceptible people, it is now a high priority to identify the most effective and safe chemopreventive agents for individuals who are genetically susceptible to the adverse effects of air pollution (eg, antioxidants to be taken during high ozone levels).
The PPARGC1B functions as an estrogen-related receptor ligand that contributes to the control of energy balance in vivo.40,41 Lin et al42 identified a homologous mouse gene termed Ppargc1b, which encodes a predicted protein of 1014 amino acids; the human and mouse PPARGC1B share 70% amino acid sequence identity. The Ppargc1b gene contains 3 N-terminal LXXLL motifs, 2 glutamic/aspartic acid-rich acidic domains, a binding site for host cell factor (HCF1; 300019), and a C-terminal RNA recognition motif. Northern blot analysis showed abundant expression of 9- and 5-kb mouse Ppargc1b transcripts in brown adipose tissue and heart, respectively, and moderate expression in the skeletal muscle, liver, and white adipose tissue. Variation in the PPARGC1B gene on chromosome 5 may contribute to the pathogenesis of obesity, with the widespread ala203 allele being a risk factor for the development of this common disorder.40–42 We selected the PPARGC1B gene as a candidate gene to clarify the effect of DEP on this gene in obese rats. In this study, increased PPARGC1B mRNA was found in the lungs of DEP-exposed OLETF obese rats than in DEP-exposed LETO obese rats, suggesting that air pollutants such as DEP act through PPARGC1B, influencing T cells, macrophages, and inflammatory cells in the lung inflammation.
In conclusion, these data indicate that the DEP-exposed OLETF group had increased airway responses and inflammation compared to the DEP-exposed LETO group, indicating that diesel particulates and obesity may be cocontributors to asthma.
Acknowledgments
This article is supported by Korea Ministry of Environment (2012001360001) as “The Environmental Health Action Program” and Soonchunhyang University Research Fund.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Mannino DM, Homa DM, Akinbami LJ, Moorman JE, Gwynn C, Redd SC. Surveillance for asthma–United States, 1980–1999. MMWR Surveill Summ. 2002;51(1):1–13. [PubMed] [Google Scholar]
- 2.Mokdad AH, Serdula MK, Dietz WH, Bowman BA, Marks JS, Koplan JP. The spread of the obesity epidemic in the United States, 1991–1998. JAMA. 1999;282(16):1519–1522. doi: 10.1001/jama.282.16.1519. [DOI] [PubMed] [Google Scholar]
- 3.McClean KM, Kee F, Young IS, Elborn JS. Obesity and the lung: 1. Epidemiology. Thorax. 2008;63(7):649–654. doi: 10.1136/thx.2007.086801. [DOI] [PubMed] [Google Scholar]
- 4.Aaron SD, Fergusson D, Dent R, Chen Y, Vandemheen KL, Dales RE. Effect of weight reduction on respiratory function and airway reactivity in obese women. Chest. 2004;125(6):2046–2052. doi: 10.1378/chest.125.6.2046. [DOI] [PubMed] [Google Scholar]
- 5.Ford ES. The epidemiology of obesity and asthma. J Allergy Clin Immunol. 2005;115(5):897–909. doi: 10.1016/j.jaci.2004.11.050. [DOI] [PubMed] [Google Scholar]
- 6.Shore SA, Johnston RA. Obesity and asthma. Pharmacol Ther. 2006;110(1):83–102. doi: 10.1016/j.pharmthera.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Simard B, Turcotte H, Marceau P, et al. Asthma and sleep apnea in patients with morbid obesity: outcome after bariatric surgery. Obes Surg. 2004;14(10):1381–1388. doi: 10.1381/0960892042584021. [DOI] [PubMed] [Google Scholar]
- 8.Bleck B, Tse DB, Jaspers I, Curotto de Lafaille MA, Reibman J. Diesel exhaust particle-exposed human bronchial epithelial cells induce dendritic cell maturation. J Immunol. 2006;176(12):7431–7437. doi: 10.4049/jimmunol.176.12.7431. [DOI] [PubMed] [Google Scholar]
- 9.Cha MH, Rhim T, Kim KH, Jang AS, Paik YK, Park CS. Proteomic identification of macrophage migration-inhibitory factor upon exposure to TiO2 particles. Mol Cell Proteomics. 2007;6(1):56–63. doi: 10.1074/mcp.M600234-MCP200. [DOI] [PubMed] [Google Scholar]
- 10.Song HM, Jang AS, Ahn MH, et al. Ym1 and Ym2 expression in a mouse model. exposed to diesel exhaust particles. Environ Toxicol. 2008;23(1):110–116. doi: 10.1002/tox.20319. [DOI] [PubMed] [Google Scholar]
- 11.Chang Y, Sénéchal S, de Nadai P, et al. Diesel exhaust exposure favors TH2 cell recruitment in nonatopic subjects by differentially regulating chemokine production. J Allergy Clin Immunol. 2006;118(2):354–360. doi: 10.1016/j.jaci.2006.04.050. [DOI] [PubMed] [Google Scholar]
- 12.van Zijverden M, van der Pijl A, Bol M, et al. Diesel exhaust, carbon black, and silica particles display distinct Th1/Th2 modulating activity. Toxicol Appl Pharmacol. 2000;168(2):131–139. doi: 10.1006/taap.2000.9013. [DOI] [PubMed] [Google Scholar]
- 13.Sahoo SK, Panyam J, Prabha S, Labhasetwar V. Residual polyvinyl alcohol associated with poly (D, L-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J Control Release. 2002;82(1):105–114. doi: 10.1016/s0168-3659(02)00127-x. [DOI] [PubMed] [Google Scholar]
- 14.Okauchi N, Mizuno A, Zhu M, et al. Effects of obesity and inheritance on the development of non-insulin-dependent diabetes mellitus in Otsuka-Long-Evans-Tokushima fatty rats. Diabetes Res Clin Pract. 1995;29(1):1–10. doi: 10.1016/0168-8227(95)01114-s. [DOI] [PubMed] [Google Scholar]
- 15.Hamelmann E, Schwarze J, Takeda K, et al. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography. Am J Respir Crit Care Med. 1997;156(3 pt 1):766–775. doi: 10.1164/ajrccm.156.3.9606031. [DOI] [PubMed] [Google Scholar]
- 16.Chinn S, Jarvis D, Burney P, European Community Respiratory Health Survey Relation of bronchial responsiveness to body mass index in the ECRHS. European community respiratory health survey. Thorax. 2002;57(12):1028–1033. doi: 10.1136/thorax.57.12.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gilliland FD, Berhane K, Islam T, et al. Obesity and the risk of newly diagnosed asthma in school-age children. Am J Epidemiol. 2003;158(5):406–415. doi: 10.1093/aje/kwg175. [DOI] [PubMed] [Google Scholar]
- 18.Guerra S, Sherrill DL, Bobadilla A, Martinez FD, Barbee RA. The relation of body mass index to asthma, chronic bronchitis, and emphysema. Chest. 2002;122(4):1256–1263. doi: 10.1378/chest.122.4.1256. [DOI] [PubMed] [Google Scholar]
- 19.Hancox RJ, Milne BJ, Poulton R, et al. Sex differences in the relation between body mass index and asthma and atopy in a birth cohort. Am J Respir Crit Care Med. 2005;171(5):440–445. doi: 10.1164/rccm.200405-623OC. [DOI] [PubMed] [Google Scholar]
- 20.Tantisira KG, Litonjua AA, Weiss ST, Fuhlbrigge AL, Childhood Asthma Management Program Research Group Association of body mass with pulmonary function in the childhood asthma management program (CAMP) Thorax. 2003;58(12):1036–1041. doi: 10.1136/thorax.58.12.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wickens K, Barry D, Friezema A, et al. Obesity and asthma in 11–12 year old New Zealand children in 1989 and 2000. Thorax. 2005;60(1):7–12. doi: 10.1136/thx.2002.001529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beckett WS, Jacobs DR, Jr, Yu X, Iribarren C, Williams OD. Asthma is associated with weight gain in females but not males, independent of physical activity. Am J Respir Crit Care Med. 2001;164(11):2045–2050. doi: 10.1164/ajrccm.164.11.2004235. [DOI] [PubMed] [Google Scholar]
- 23.Camargo CA, Jr, Weiss ST, Zhang S, Willett WC, Speizer FE. Prospective study of body mass index, weight change, and risk of adult-onset asthma in women. Arch Intern Med. 1999;159(21):2582–2588. doi: 10.1001/archinte.159.21.2582. [DOI] [PubMed] [Google Scholar]
- 24.Guerra S, Wright AL, Morgan WJ, Sherrill DL, Holberg CJ, Martinez FD. Persistence of asthma symptoms during adolescence: role of obesity and age at the onset of puberty. Am J Respir Crit Care Med. 2004;170(1):78–85. doi: 10.1164/rccm.200309-1224OC. [DOI] [PubMed] [Google Scholar]
- 25.Celedón JC, Palmer LJ, Litonjua AA, et al. Body mass index and asthma in adults in families of subjects with asthma in Anqing, China. Am J Respir Crit Care Med. 2001;164(10 pt 1):1835–1840. doi: 10.1164/ajrccm.164.10.2105033. [DOI] [PubMed] [Google Scholar]
- 26.Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev. 2000;21(6):697–738. doi: 10.1210/edrv.21.6.0415. [DOI] [PubMed] [Google Scholar]
- 27.Nawrocki AR, Scherer PE. The delicate balance between fat and muscle: adipokines in metabolic disease and musculoskeletal inflammation. Curr Opin Pharmacol. 2004;4(3):281–289. doi: 10.1016/j.coph.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 28.Rajala MW, Scherer PE. Minireview: The adipocyte–at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. 2003;144(9):3765–3773. doi: 10.1210/en.2003-0580. [DOI] [PubMed] [Google Scholar]
- 29.Takiguchi S, Takata Y, Funakoshi A, et al. Disrupted cholecystokinin type-A receptor (CCKAR) gene in OLETF rats. Gene. 1997;197(1–2):169–175. doi: 10.1016/s0378-1119(97)00259-x. [DOI] [PubMed] [Google Scholar]
- 30.Kawano K, Hirashima T, Mori S, Saitoh Y, Kurosumi M, Natori T. Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long-Evans Tokushima fatty (OLETF) strain. Diabetes. 1992;41(11):1422–1428. doi: 10.2337/diab.41.11.1422. [DOI] [PubMed] [Google Scholar]
- 31.O’Connor GT, Neas L, Vaughn B, et al. Acute respiratory health effects of air pollution on children with asthma in US inner cities. J Allergy Clin Immunol. 2008;121(5):1133–1139.e1. doi: 10.1016/j.jaci.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 32.Morgenstern V, Zutavern A, Cyrys J, et al. Atopic diseases, allergic sensitization, and exposure to traffic-related air pollution in children. Am J Respir Crit Care Med. 2008;177(12):1331–1337. doi: 10.1164/rccm.200701-036OC. [DOI] [PubMed] [Google Scholar]
- 33.Mortimer K, Neugebauer R, Lurmann F, Alcorn S, Balmes J, Tager I. Air pollution and pulmonary function in asthmatic children: effects of prenatal and lifetime exposures. Epidemiology. 2008;19(4):550–557. doi: 10.1097/EDE.0b013e31816a9dcb. [DOI] [PubMed] [Google Scholar]
- 34.Diaz-Sanchez D, Proietti L, Polosa R. Diesel fumes and the rising prevalence of atopy: an urban legend? Curr Allergy Asthma Rep. 2003;3(2):146–152. doi: 10.1007/s11882-003-0027-4. [DOI] [PubMed] [Google Scholar]
- 35.Ichinose T, Takano H, Miyabara Y, Sadakaneo K, Sagai M, Shibamoto T. Enhancement of antigen-induced eosinophilic inflammation in the airways of mast-cell deficient mice by dieselexhaust particles. Toxicology. 2002;180(3):293–301. doi: 10.1016/s0300-483x(02)00420-1. [DOI] [PubMed] [Google Scholar]
- 36.Nel AE, Diaz-Sanchez D, Ng D, Hiura T, Saxon A. Enhancement of allergic inflammation by the interaction between diesel exhaust particles and the immune system. J Allergy Clin Immunol. 1998;102(4 pt 1):539–554. doi: 10.1016/s0091-6749(98)70269-6. [DOI] [PubMed] [Google Scholar]
- 37.Ohta K, Yamashita N, Tajima M, et al. Diesel exhaust particulate induces airway hyperresponsiveness in a murine model: Essential role of GM-CSF. J Allergy Clin Immunol. 1999;104(5):1024–1030. doi: 10.1016/s0091-6749(99)70084-9. [DOI] [PubMed] [Google Scholar]
- 38.Walters DM, Breysse PN, Wills-Karp M. Ambient urban Baltimore particulate-induced airway hyperresponsiveness and inflammation in mice. Am J Respir Crit Care Med. 2001;164(8 pt 1):1438–1443. doi: 10.1164/ajrccm.164.8.2007121. [DOI] [PubMed] [Google Scholar]
- 39.Yang IA, Fong KM, Zimmerman PV, Holgate ST, Holloway JW. Genetic susceptibility to the respiratory effects of air pollution. Thorax. 2008;63(6):555–563. doi: 10.1136/thx.2007.079426. [DOI] [PubMed] [Google Scholar]
- 40.Andersen G, Wegner L, Yanagisawa K, et al. Evidence of an association between genetic variation of the coactivator PGC-1-beta and obesity. J Med Genet. 2005;42(5):402–407. doi: 10.1136/jmg.2004.026278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kamei Y, Ohizumi H, Fujitani Y, et al. PPAR-gamma coactivator 1-beta/ERR ligand 1 is an ERR protein ligand, whose expression induces a high-energy expenditure and antagonizes obesity. Proc Natl Acad Sci U S A. 2003;100(21):12378–12383. doi: 10.1073/pnas.2135217100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1-beta (PGC-1-beta), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem. 2002;277(3):1645–1648. doi: 10.1074/jbc.C100631200. [DOI] [PubMed] [Google Scholar]