

Abstract
The effectiveness of treatment of renal diseases is limited because the lack of diagnostic, prognostic and therapeutic markers. Despite the more than a decade of intensive investigation of urinary biomarkers, no new clinical biomarkers were approved. This is in part because the early expectations toward proteomics in biomarkers discovery were significantly higher than the capability of technology at the time. However, during the last decade, proteomic technology has made dramatic progress in both the hardware and software methods. In this review we are discussing modern quantitative methods of mass-spectrometry and providing several examples of their applications for discovery and validation of renal disease biomarkers. We are optimistic about future prospects for the development of novel of specific clinical urinary biomarkers.
Keywords: quantitative mass-spectrometry, proteomics, urinary biomarkers, renal disease
1. Introduction
Currently the effectiveness of treatment of renal diseases is limited by the lack of diagnostic, prognostic, and therapeutic markers. A renal biopsy is often necessary to establish a diagnosis, particularly in the case of glomerular diseases. Renal biopsy is a highly invasive method associated with high morbidity and mortality. In contrast, urine is an easily accessible biofluid and its protein content is derived mainly from the kidney and low urinary tract organs. Thus, urinary biomarkers are an attractive tool for development of clinical tests. Recently mass-spectrometry (MS) is playing an increasing role in the identification and quantification of biomarkers [1–6]. Despite its promise, the translation of urinary biomarkers into the clinic has been inefficient [7]. Part of the problem can be attributed to the underestimating of efforts required to discover novel biomarkers and underdevelopment of MS technology. There are several major obstacles for the development of clinically relevant urinary biomarkers [8]. Both the nature of urine and the MS techniques are responsible for generation of non-reproducible results. There is no standard protocol for urine collection and storage, concentration of samples, protein isolation and sample preparation for MS [9–11]. Urine has a high level of variability in volume and protein concentration. Urine composition depends on diet, circadian rhythms, age, gender and exercise [11–15]. Because MS-based methods are very sensitive and capable of detection of femtomoles of peptides, different methods for urine collection, concentration and protein isolation can yield distinct proteins discovery [16–18]. This question was intensively studied and discussed during a last decade, and it is not a major subject of this review. Human Proteomics Organisation (HUPO) has developed guidlines for patient data recording, urine collection and sample preparation for several MS based methods (http://www.hupo.org/initiatives/human-kidney-and-urine-proteome-project-hkupp/). A standard method still not commonly used by the proteomics community and most studies use protocols developed to their specific experiments. Urine samples can be diluted or concentrated depending on their water content, thus requiring normalization of biomarker concentrations. The most common normalization factor is urine creatinine (Cr), but its urinary concentration may vary depending on the level of muscle Cr generation (muscle mass) and renal tubular Cr secretion [19, 20]. The muscle mass depends on age, gender, race, fitness and muscle disease, and normalization of urinary samples using Cr can increase protein concentration variability even in the samples collected from healthy individuals. Specific gravity has also been used for normalization of urine samples [21]. Specific gravity is the ratio of the weight of a solution to the weight of an equal volume of distilled water. It is strongly influenced by both the number of particles in the solution and their size. Normalization of urinary proteins using specific gravity is problematic when large molecules are present in urine. Thus the best method for urine normalization is still under investigation. Because debris of spontaneously dying renal cells is released into the urine, uncontrolled amounts of intracellular and membrane proteins can be detected especially in highly concentrated samples collected from the patients with epithelial cell injury. Recently, urinary exosomes were used as a potential source of biomarkers of renal diseases [22–25]. Exosomes are low density inverted apical membrane vesicles normally secreted into the urine from all parts of nephron [22]. They are smaller than apoptotic vesicles, and can be separated from them by gradient centrifugation. They have been found to contain many disease-associated proteins including aquaporin-2, polycystin-1, podocin, non-muscle myosin II, angiotensin-converting enzyme, Na+ K+ 2Cl− cotransporter, thiazide-sensitive NaCl cotransporter, and epithelial sodium channel [22]. Exosomes may be usefull for determination of biomarkers for renal dysfunction and structural renal disease [23]. However, the lack of standard efficient methods for vesicle isolation and lysis, and the issue of protein normalization are major limitations for the quantitative proteomics of exosomes [26–28].
Despite all these shortcomings, urine is an attractive source of renal diseases biomarkers because of its noninvasiveness, large volume and because its proteins are originated from the kidney and low urinary tract organs.
In recent years the increased capability of the quantitative proteomics was based on the advances in both hardware and software methods. The increased performance capabilities, easy operation, and robustness of MS over other techniques have made it an ideal platform for quantitative proteomics. Novel MS-based quantitative methods offer the opportunity for faster, higher throughput, and a wider dynamic range protein analysis, and can be used for both stable-isotope labeling or label-free methods of protein quantification. While several quantitative proteomics approaches exist, each of them has its own advantages and limitations. In this review, we discuss modern quantitative proteomics approaches and their applications for the discovery and validation of urinary biomarkers of renal diseases. We do not describe all urinary biomarkers found by particular MS method but rather concentrate on modern quantitative MS methods and their application for urine proteomics. For each MS method we described only few examples that highlight the usefulness of it for urinary proteomics research.
2.1 Two-dimension gel electrophoresis (2DE)
The 2DE method is a primary technique that has been widely used in urinary proteomics [29–31]. In this gel-based method, urinary proteins are resolved in the first-dimension based on their isoelectric point (pI) followed by resolution based on molecular weight in the second-dimension. The gels are then stained by either Coomassie Brilliant Blue, silver stain or Sypro Ruby fluorescent stain to visualize the protein spots. The important step before the gel separation is urine concentration. Multiple protocols have been developed to concentrate and purify urinary proteins including lyophilization, precipitation, ultracentrifugation, and centrifugal filtration [11, 18, 29, 32–35]. Analysis of 2DE images is performed using computer-based platforms. Several commercial programs became recently available including Melanie (Geneva Bioinformatics), ImageMaster2D (GE Healthcare), PDQuest (Bio-Rad Laboratories), Dymension (Syngene), SameSpots (Totallab), BioNumerics (Applied Methods) and Delta2D (Decadon). The main steps in differential analysis of 2DE gels involve image noise substraction, protein spot detection, spot quantification, spot matching and statistical analysis. Most programs first detect spots, estimate spot boundaries, and calculate spot volumes for each individual gel, and then match the detected spots across different gels. This procedure may lead to spot mismatching and missing data, which require manual editing of data. Manual editing significantly increases time of analysis, decreases throughput and compromises the objectivity and reproducibility of the analysis [36]. Several novel software such as SameSpot (Totallab) and Pinnacle align the images before processing to reduce spot missmatching [37]. It significantly reduces time of analysis and increase reproducibility. After quantification analysis protein spots are extracted from the gel and identified by mass spectrometry (peptide mass fingerprinting) [38]. Matrix-assisted laser desorption ionization-time-of-flight (MALDI-TOF) mass spectrometry and electrospray ionization (ESI)-MS are most often used for the identification of the extracted proteins. This approach could lead to separation and identification of about 2000 unique spots [34, 39]. This approach was successfully used for identification of potential biomarkers of different renal diseases. High urinary levels of β2-microglobulin, retinol-binding protein, transferrin, hemopexin, haptoglobin, lactoferrin, and neutrophil gelatinase-associated lipocalin (NGAL) were identified as candidate biomarkers for HIV-associated nephropathy [40]. Retinol-binding protein was also identified as a candidate biomarker for acute tubular necrosis [41]. Retinol-binding protein 4, α-1-microglobulin, zinc-α2 glycoprotein, and α-1B glycoprotein were found to increase in the samples from micro-albuminuric patients with type 1 diabetes [42].
However, 2DE method has multiple limitations. Both the separation and the analysis are time consuming reducing number of urine samples. Gel to gel variability reduces reproducibility, and requires complex image analysis and manual correction. Importantly, because quantification of proteins is performed on the basis of in-gel proteins staining, it depends on the sensitivity of particular stain. The sensitivity of Coomassie Brilliant blue is about 50 ng of protein per spot or 20 ng per spot for colloidal Commassie Blue. Additional variability of results arises from destaining procedure and high background. The sensitivity of silver stain is higher than Coummassie Blue (about 1 ng per spot) but both stains demonstrate poor linear response. Sypro Ruby stain demonstrated similar with silver stain sensitivity (about 1 ng per spot) but less background and good linear response for various protein concentrations. But the sensitivity of in-gel methods is thousand times lower than sensitivity of MS-based methods. Thus low reproducibility and low relative quantification accuracy are additional obstacles [43]. Also, 2DE has a small dynamic range compared to MS-based methods being mostly suitable for major proteins. Though 2DE has its limitations, it remains a popular method of urinary protein analysis because of its robustness, simplicity and availability in most facilities [44, 45]. Moreover 2DE allows separating and studying proteins isoforms, modified proteins and degradated peptides specific for urine that is difficult to do by MS-based methods.
2.2 Two-dimensional difference gel electrophoresis (2D-DIGE)
The 2D-DIGE method is an improved version of 2DE. In this method, two different protein samples (control and a disease) and one internal control (pooled mixture of controls and disease samples in equal proportion) are labeled with three different fluorophores: Cy2, Cy3, or Cy5 before in gel separation. These fluorophores have the identical charge and molecular mass but unique emission wavelengths that allows identification of those fluorophores using appropriate optical filters [46–48]. The labeled samples are then mixed together and separated on a 2DE. The same internal control is used for all samples for normalization. The gel is scanned at three different wavelengths: 488 nm (Cy2), 532 nm (Cy3), and 633 nm (Cy5) and relative abundance of proteins are quantified using computer software such as DeCyder (GE Healthcare Life Science), Melanie (Geneva Bioinformatics) and PDQuest (Bio-Rad). The sensitivity for each fluorescent dye is similar to Sypro Ruby fluorescent dye (about 1 ng per spot). Addition of internal standards to each gel allows protein normalization and quantification of protein amounts as ratios and not as volumes. This method reduces gel-to-gel variation and separates experimental variability form biological one. The quantification accuracy of 2D-DIGE is higher than 2DE method. This technique has been routinely used for the discovery of candidate urinary biomarkers of renal disease in patient and animal models [49–52]. 2D-DIGE-SELDI-TOF (surface-enhanced laser desorption ionization -time of flight) was used for the detection of early stage tubular injury in canine model of progressive glomerular disease [50]. Alpha 1 antitrypsin was discovered as a diagnostic biomarker for diabetic nephropathy [52]. A number of highly abundant proteins in urine such as albumin fragments have also been identified by gel-based proteomics approaches, and these abundant proteins were considered disease-biomarker candidates [53–55]. Major limitations of this method are time-consuming separation and analysis steps that restricts its use used for high throughput screening. When the number of urine samples is large, cost of fluorescent dyes is also an additional limitation. Both 2DE and 2D-DIGE methods have less sensitivity and small dynamic range compared to MS-based methods and are mostly suitable for major proteins.
While 2DE and 2D-DIGE methods employ in-gel quantification based on the protein staining techniques, all other methods described below are MS-based quantification techniques (see Table 1).
Table 1.
Quantitative methods to analyze urinary biomarkers
| Method | Quantification | Advantages | Limitation |
|---|---|---|---|
|
Biomarkers Identification
| |||
|
2DE Two-dimension electrophoresis |
In-gel Coomassie Brilliant Blue, silver staining or Sypro Ruby | Robust, simple, cheap. Suitable for protein isoforms, modifications and degradation analysis. |
Low reproducibility and relative quantification accuracy, small dynamic range |
|
2D-DIGE Two Dimension Differences Gel electrophoresis |
In gel fluorescence intensity of Cy2, Cy3 and Cy5 fluorophores | Reduces gel-to-gel variation and enhances sensitivity Suitable for protein isoforms, modifications and degradation analysis |
Variability in labeling efficiencies, small dynamic range comparing to MS based methods, expensive |
|
SILAC Stable-isotope labeling by amino acids |
MS based on metabolic labeling of proteins with heavy isotopes in vivo | Independent of the degree of resolution and instrument sensitivity, accurate for low abundant protein | Difficult and time-consuming to establish, expensive, complicated data analysis, not suitable for human samples |
|
iTRAQ Isobaric Tags for Relative and Absolute Quantitation |
MS based on in vitro peptides labeling with eight isobaric tags | Eight samples can be pooled and relative abundance can be quantified in one MS/MS run | Variability in labeling efficiencies, loss of peptides during chromatography, expensive |
| Label-free method | MS based on peptide peak areas and the spectral counting | High throughput, cheap, simple in sample preparation, less complicated MS analysis. | Less accuracy than tag methods, semi quantitative in nature, not suitable for low abundant and short proteins. |
|
Biomarkers Validation | |||
|
SRM and MRM selected reaction monitoring and multiple-reaction monitoring |
MS-based on counting the ions for transition pairs | Good linearity and excellent precision, wide range | Targeted approach focused on a limited set of pre-detected proteins |
3.1 Stable-isotope labeling by amino acids (SILAC)
This method is based on metabolic labeling of proteins with heavy isotopes (H2, C13, and N15) incorporated into amino acids [56]. A number of amino acids such as arginine, leucine, and lysine with stable isotope are suitable for use in SILAC, but lysine and arginine are the most often used amino acids, because trypsin-digested peptides contain at least one arginine or lysine making all peptides eligible for quantification [57, 58]. Originally this method was developed for in vitro cell culture [56]. In this method either two different lines of cells (experimental and control) are cultured under similar conditions with addition of labeled amino acid to experimental cell line, or cells are cultured under different conditions with addition of labeled amino acids to experimental group. Cells are collected after five to seven passages to ensure >95% labeling, lysates are prepared, and then experimental and control samples are combined in a 1:1 stoichiometric ratio [56]. Combined samples are separated either on 1DE or 2DE following by in-gel digestion, peptides extraction and LC-MS/MS analysis. Alternatively, the samples are digested in-solution and analyzed by LC-MS/MS. Labeled amino acid induces a shift in the mass/charge (m/z) ratio comparing to the unlabelled amino acid. This shift allows to discriminate peptides between experimental and control samples, and to quantify relative changes in protein concentration (Fig. 1 A). Combining the differentially labeled samples before any purification and fractionation steps minimizes the possible quantitative error caused by handling different samples in parallel [56].
Figure 1. MS- based quantification methods.
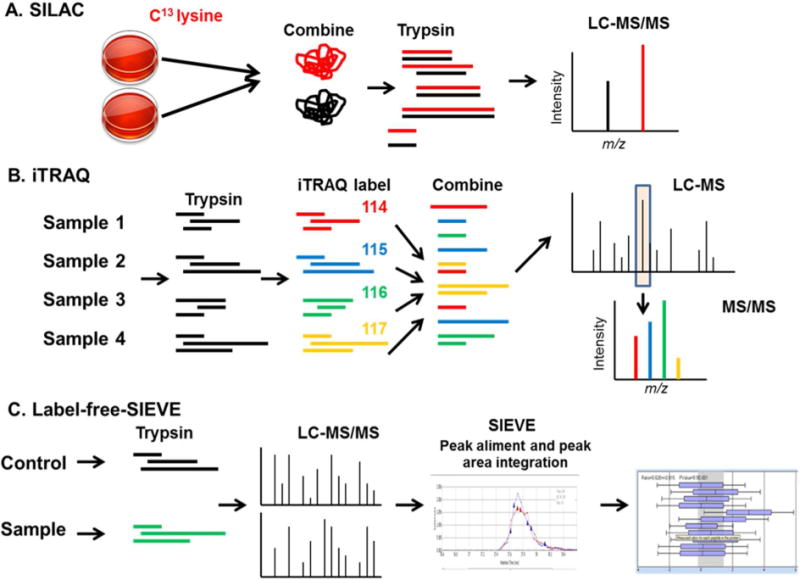
(A). SILAC- Stable-isotope labeling by amino acids. Cells are differentially labeled by growing them in medium with normal lysine (black color) or with heavy lysine (C13-lysine, red color). Both samples are combined, trypsinized and LC-MS/MS is performed. Metabolic incorporation of the amino acids into the proteins results in a mass shift of the corresponding peptides. (B) iTRAQ- Isobaric Tags for Relative and Absolute Quantitation. Samples are trypsinized, and peptide are labeled in vitro with iTRAQ tags with different mass (114-red, 115-blue, 116, green and 117-yellow color). Samples are combined together and LC-MS/MS is performed. Identical peptides labeled with the different iTRAQ tags produce the same peak in MS spectra (shown in rectangle). MS/MS fragmentation of ion produces unique peak for each tag that allowed comparison of relative intensity. (C) Label-free quantification using SIEVE program. Sample (red color) and control (blue color) are processed separately and LC-MS/MS is performed. SIEVE program from Thermo Electron perform aliment of peaks, peak area integration and spectral counting, that quantify relative amount of protein in sample.
Recently, this method has been extended to the animal models [59–61]. Feeding mice with diet containing a heavy isotope C13-lysine for one generation leads to a complete exchange of the natural (light) isotope (12)C6-lysine. Blood, tissue, and organs are labeled, and can be used for global proteomics [62–64].
Additionally SILAC can be used for an indirect ‘spike-in’ approach where cell line is used to produce a heavy-labelled reference sample, which is added as an internal standard to the tissue or organ samples [65].
SILAC’s advantage is that this method does not require a targeted analysis of specific proteins or peptides because every peptide is labeled and can be quantified independently of the degree of resolution and instrument sensitivity. It is also more robust and accurate than other quantitative techniques such as iTRAQ and label-free method [66]. However, SILAC also has several disadvantages. It is difficult and time-consuming to establish this method in new model organisms. The medium composition has to be controlled and the reagents are expensive. The data analysis is also challenging due to incomplete incorporation of labeled amino acids and arginine-to-proline conversion by arginase [67]. Because arginase II is highly expressed in renal cells, labeled proline incorporation into the proteins increases complexisity of data analysis. Moreover, SILAC cannot be used directly in human samples and has not been used for discovery of human urinary biomarkers.
Investigation of renal cell secretome is a potential step in the urinary biomarker discovery. Treatment of HEK-293 renal cells with cyclosporine demonstrated up-regulation of secreted cyclophilins A and B, macrophage inhibition factor and phosphatidylethanolamine-binding protein 1 [68]. Recently, the SILAC-labeled mouse serum was used for ‘spike-in’ quantification of human serum and urine [69]. SILAC mouse serum was mixed with human serum and urine, and multidimensional separation and LC-MS/MS analysis was performed. The shared peptides between two species were quantified by SILAC pairs. Analysis of urine from immunoglobulin A nephropathy patients identified novel biomarker candidates, such as Complement C3, Albumin, VDBP, ApoA1, and IGFBP7 [69]. Thus, despite the fact that SILAC cannot be used directly in human samples, its application in renal cell secretome and animal models can potentially lead to the discovery candidates biomarkers of renal disease.
3.2 Isobaric Tags for Relative and Absolute Quantitation (iTRAQ)
iTRAQ is a method of in vitro peptides labeling after trypsin digestion of proteins that allowed to compare multiple samples in one MS/MS run [70–72]. iTRAQ label consists of a reporter group with a defined molecular weight, a balance group, and an amine-reactive group that reacts at lysine side chains and NH2-terminal amino acid. Recently, eight iTRAQ reagents became available, with the following reporter/balance group masses: 113/192, 114/191, 115/190, 116/189, 117/188, 118/187, 119/186, and 121/184 Da. The combined mass remains constant (305 Da) for each of the eight reagents. The iTRAQ labels are generated using heavy weight isotops of 13C, 15N, and 18O atoms in such way that all peptides with different iTRAQ labels attached are isobaric (same mass) and indistinguishable in chromatographic separation and MS. The function of balance groups is to make all iTRAQ tags isobaric so the combined mass of reporter group and balance group remains constant. Following fragmentation in MS/MS the iTRQ label looses the balance group, while the charge is retained by the reporter group. The eight reporter group ions appear as distinct masses in MS/MS that can be used to identify and quantify individual members of the multiplex set [70]. In iTRAQ, up to eight (8-plex) samples are labeled after trypsin digestion with iTRAQ reagents. The samples then are pooled together, the labeled peptides are separated by strong cation exchange chromatography, and the isolated labeled peptides are separated by LC-MS/MS [73]. Different samples can be run together in the single MS/MS run. The isobaric nature of the tags allows the protein samples to be pooled together after labeling without increasing the complexity of the MS analysis. Identical peptides labeled with the different iTRAQ reagents produce the same peak (ion) in MS spectra. Upon MS/MS fragmentation of the parent ion, unique signature ions are generated which distinguish the individual samples and allow to compare the relative amount of each sample (Fig. 1B). iTRAQ method can also be used for absolute quantification of peptides by adding an internal standard peptide. The advantage of iTRAQ labeling is that the signal obtained from combined peptides enhances the sensitivity of detection in MS/MS. However, the variability in labeling efficiencies and the costly reagents are major limitations of this method [74]. Labeling also increases complexity of the samples and can reduce number of the identified peptides during MS/MS run. Some peptides are lost during the separation on SCX chromatography. Recently, electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) have been developed as an alternative to the SCX chromatography [75]. ERLIC method separates peptides on the basis of electrostatic repulsion and hydrophilic interaction and is found to increase the proteome coverage.
The use of this powerful technique is gradually becoming the method of choice in the field of biomarker discovery [3, 76–78]. This method allowed discovering P- and E-cadherins as urinary biomarkers of idiopathic nephrotic syndrome [76]. Alpha-1-antitrypsin, alpha-1-acid glycoprotein 1, and prostate stem cell antigen has been discovered as candidate biomarkers for diabetic nephropathy [77]. Uromodulin, SERPINF1, and CD44 were identified and verified in an independent cohort as urinary biomarkers to differentiate patients with early acute kidney transplant rejection from other groups [78].
3.3 Label-free quantitative methods
To overcome the problems in the labeling techniques such as high cost of the reagents, higher concentration of sample requirement, and incomplete labeling, label-free shotgun proteomic technologies have been developed. These methods are based on the assumption that the peak area of a peptide in the chromatogram is directly proportional to its concentration [79–81]. Label-free protein quantification approach is based on two types of measurements; the measurement of ion intensity by quantification of peptide peak areas or peak heights in chromatogram, and the spectral counting in the MS/MS analysis. For spectral counting, peptides from the same protein are identified, chromatographic peaks aligned and normalized (Fig. 1C). There are several commercially available software packages for label-free analysis (Decyder MS from GE Healthcare, Protein Lynx from Waters, and SIEVE from Thermo Electron). This approach is primarily used for the analysis of human samples and has been applied to the analysis of urinary proteome [1, 82, 83]. It is a very high throughput technique that increases opportunities in the discovery of candidate biomarkers. There are several advantages in label-free quantification approach. It is a cheap method comparing to the labeling techniques. It is simpler in terms of sample preparation, and less complicated in terms of MS/MS analysis [81]. The limitation of this method is redundancy in peak detection which arises from the peptides which are similar for several proteins [84]. Other limitations of label-free quantification methods are less accuracy, semi-quantitative nature, and unsuitability for low abundance and small proteins [85]. Small proteins or proteins of low abundance could still be present in the sample in spite of the spectral count being zero, larger proteins generate more tryptic digest products, and more spectral counts. Another limitation of the method is a spectra normalization. In contrast to SILAC and iTRAQ methods, in label-free method the spectra are generated in separate MS/MS runs that are different in many factors like efficiency of fragmentation and ionization [85]. Label-free quantification methods overcome those limitations by additional computational calculations. There are several algorithms available that take into account the sequence and length of the peptides and compute the predicted abundance of proteins in the sample [86–88]. Protein abundance index (PAI) is defined as the number of identified peptides divided by the number of theoretically observable tryptic peptides for each protein. Absolute quantification of proteins is based on exponentially modified PAI values with or without added standards [79, 85].
Label-free quantitative analysis of urinary exosomes in diabetic nephropathy resulted in the discovery of three proteins AMBP, MLL3 and VDAC1 as candidate biomarkers [24]. Another group of proteins (Tamm-Horsfall glycoprotein, progranulin, clusterin and α-1 acid glycoprotein) were determined as candidate biomarkers for microalbuminuria progression in diabetic nephropathy [89].
4. MS-based absolute quantification methods for biomarkers validation
The methods described above have been used mostly for urinary biomarkers discovery. Traditional methods such as Western blot and Elisa are the first choice for validation of biomarkers, but novel stable isotope dilution MS (SID-MS) quantification methods suitable for validation have been developed. Two methods (selected reaction monitoring (SRM), and multiple-reaction monitoring (MRM)) have been used for absolute quantification of proteins in combination with stable isotope dilution. These methods are based on the addition of known quantities of isotope-labeled standards, which have similar chromatographic properties to the target compounds but can be distinguished from them by their difference in m/z [90, 91]. The isotope dilution method is a targeted approach focused on a limited set of proteins. The identification of candidate proteins requires the prior generation of isotope-labeled standards [92, 93]. Quantification is performed by comparing the peak height or peak area of the isotope-labeled and the native forms of a peptide of interest. SRM is a non-scanning mass spectrometry technique, performed on triple quadrupole instruments. In SRM experiments, two mass analyzers are used as static mass filters, to monitor a particular fragment ion of a selected precursor ion. The specific pair of m/z values associated with the precursor and fragment ions selected are referred to as a “transition” [94]. Unlike common MS based proteomics, no mass spectra are recorded in a SRM analysis. Instead, the detector acts as a counting device for the ions matching the selected transition thereby returning an intensity value over time. In MRM experiment, multiple transitions can be measured within the same experiment on the chromatographic time scale by rapidly shifting between the different precursor/fragment pairs. Typically, a triple quadrupole instrument cycles through a series of transitions and records the signal of each transition as a function of the elution time.
The major advantage of these methods is good linearity and excellent precision, but the accuracy and ability to determine the true abundance of target protein strongly depends on the choice of selected peptides and the purity of internal standards [95, 96]. This method covers a complete dynamic range of cellular proteome, with a low limit of detection below. 50 copies of protein per cell [97]. The disadvantage of these methods is that they are limited to a small number of proteins because suitable internal standards have to be purchased or synthesized. SID-MS based quantification is filling the gap between the discovery and validation of biomarkers that may promote candidate biomarkers towards clinical trials and established them as diagnostic tools. However, developing and validating SID-MS-based assays is an expensive and time consuming process, requiring a coordinated and collaborative effort by the scientific community through the sharing of publicly accessible data and datasets, bioinformatic tools, standard operating procedures, and well characterized reagents [98].
There are several examples of recent coordinated efforts for development of urinary biomarkers for renal diseases. The Nephrotoxicity Working Group of the Predictive Safety Testing Consortium have selected 23 previously discovered urinary biomarkers and evaluated them in rat models of acute kidney injury (AKI) [99–103]. Seven markers were selected for further preclinical studies, including: kidney injury molecule-1 (kim-1), albumin, total protein, β2-microglobulin, cystatin C, clusterin, and trefoil factor-3. Chronic kidney disease (CKD) consortium (www.ckdbiomarkersconsortium.org) have identified fourteen candidate biomarkers for CDK progression and twelve biomarkers for early stage CKD in diabetes and lupus nephrology [104, 105]. Future coordinated efforts from scientific community will validate recently discovered biomarkers of renal diseases.
5. Concluding Remarks
Nephrology is in a dire need for improved diagnostic and therapeutic markers. Despite the more than a decade of intensive investigation of urinary biomarkers no new clinical biomarkers were approved [106]. Similar to the early genomic studies, expectations toward proteomics in biomarkers discovery were significantly higher than the ability of the technology a decade ago. The technology was underdeveloped with limited analytical and quantification capability. Thus early investigations in this area were largely confined to measurement of major urinary proteins without association with disease mechanisms. Now it is clear that the most promising biomarkers have been found in well-designed studies guided by specific research questions. Moreover, during the last decade, proteomic technology has made dramatic progress in both the hardware and software methods [107]. Advances in quantitative proteomics and development of SRM and MRM methods let the protein-quantification data stand by their own without validation from other protein quantification methods as Western blot and Elisa [108]. This progress opens a new era in the discovery and validation of urinary biomarkers of renal disease. Collaborative efforts by the scientific community are needed for the development of standardized protocols for sample preparation methods suitable for examination of lowabundance urinary proteins. Addition of other indirect approaches, such as cell cultures and animals models, may be useful for the discovery of potential biomarker candidates that could be subsequently found in urines. Uncovering of disease molecular mechanisms may predict new candidate urinary biomarkers.
Acknowledgments
This project was supported by NIH Research Grants 1P50HL118006 and 8G12MD007597 from the Research Centers in Minority Institutions (RCMI) Program (Division of Research Infrastructure, National Center for Research Resources, NIH). The content is solely the responsibility of the authors and does not necessarily represents the official views of the National Institute of Health.
Abbreviations
- MS
mass spectrometry
- 2DE
two dimension gel electrophoresis
- MALDI-TOF
matrix-assisted laser desorption ionization-time-of-flight
- 2D-DIGE
two dimension differences gel electrophoresis
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- SILAC
stable-isotope labeling by amino acids
- iTRAQ
isobaric tags for relative and absolute quantification
- SCX
strong cation exchange chromatography
- ERLIC
electrostatic repulsion:hydrophilic interaction chromatography
- PAI
protein abundance index
- SRM
selected reaction monitoring
- MRM
multiple-reaction monitoring
- AKI
acute kidney injury
- Cr
creatinine
- BUN
blood urine nitrogen
- CKD
chronic kidney disease
References
- 1.Kalantari S, Nafar M, Samavat S, Rezaei-Tavirani M, Rutishauser D, Zubarev R. Nephrourol Mon. 2014;6:e16806. doi: 10.5812/numonthly.16806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nafar M, Kalantari S, Samavat S, Rezaei-Tavirani M, Rutishuser D, Zubarev RA. Int J Nephrol. 2014;2014:574261. doi: 10.1155/2014/574261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sigdel TK, Salomonis N, Nicora CD, Ryu S, He J, Dinh V, Orton DJ, Moore RJ, Hsieh SC, Dai H, Thien-Vu M, Xiao W, Smith RD, Qian WJ, Camp DG, 2nd, Sarwal MM. Mol Cell Proteomics. 2014;13:621–631. doi: 10.1074/mcp.M113.030577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Su L, Zhou R, Liu C, Wen B, Xiao K, Kong W, Tan F, Huang Y, Cao L, Xie L. J Trauma Acute Care Surg. 2013;74:940–945. doi: 10.1097/TA.0b013e31828272c5. [DOI] [PubMed] [Google Scholar]
- 5.Kistler AD, Serra AL, Siwy J, Poster D, Krauer F, Torres VE, Mrug M, Grantham JJ, Bae KT, Bost JE, Mullen W, Wuthrich RP, Mischak H, Chapman AB. PLoS One. 2013;8:e53016. doi: 10.1371/journal.pone.0053016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zurbig P, Jerums G, Hovind P, Macisaac RJ, Mischak H, Nielsen SE, Panagiotopoulos S, Persson F, Rossing P. Diabetes. 2012;61:3304–3313. doi: 10.2337/db12-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mischak H, Ioannidis JP, Argiles A, Attwood TK, Bongcam-Rudloff E, Broenstrup M, Charonis A, Chrousos GP, Delles C, Dominiczak A, Dylag T, Ehrich J, Egido J, Findeisen P, Jankowski J, Johnson RW, Julien BA, Lankisch T, Leung HY, Maahs D, Magni F, Manns MP, Manolis E, Mayer G, Navis G, Novak J, Ortiz A, Persson F, Peter K, Riese HH, Rossing P, Sattar N, Spasovski G, Thongboonkerd V, Vanholder R, Schanstra JP. A Vlahou, Eur J Clin Invest. 2012;42:1027–1036. doi: 10.1111/j.1365-2362.2012.02674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mischak H, Vlahou A, Ioannidis JP. Clin Biochem. 2013;46:432–443. doi: 10.1016/j.clinbiochem.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 9.Mischak H, Allmaier G, Apweiler R, Attwood T, Baumann M, Benigni A, Bennett SE, Bischoff R, Bongcam-Rudloff E, Capasso G, Coon JJ, D’Haese P, Dominiczak AF, Dakna M, Dihazi H, Ehrich JH, Fernandez-Llama P, Fliser D, Frokiaer J, Garin J, Girolami M, Hancock WS, Haubitz M, Hochstrasser D, Holman RR, Ioannidis JP, Jankowski J, Julian BA, Klein JB, Kolch W, Luider T, Massy Z, Mattes WB, Molina F, Monsarrat B, Novak J, Peter K, Rossing P, Sanchez-Carbayo M, Schanstra JP, Semmes OJ, Spasovski G, Theodorescu D, Thongboonkerd V, Vanholder R, Veenstra TD, Weissinger E, Yamamoto T. A Vlahou, Sci Transl Med. 2010;2:46ps42. doi: 10.1126/scitranslmed.3001249. [DOI] [PubMed] [Google Scholar]
- 10.Court M, Selevsek N, Matondo M, Allory Y, Garin J, Masselon CD, Domon B. Proteomics. 2011;11:1160–1171. doi: 10.1002/pmic.201000566. [DOI] [PubMed] [Google Scholar]
- 11.Thongboonkerd V. J Proteome Res. 2007;6:3881–3890. doi: 10.1021/pr070328s. [DOI] [PubMed] [Google Scholar]
- 12.Lafitte D, Dussol B, Andersen S, Vazi A, Dupuy P, Jensen ON, Berland Y, Verdier JM. Clin Biochem. 2002;35:581–589. doi: 10.1016/s0009-9120(02)00362-4. doi: S0009912002003624 [pii] [DOI] [PubMed] [Google Scholar]
- 13.Schaub S, Rush D, Wilkins J, Gibson IW, Weiler T, Sangster K, Nicolle L, Karpinski M, Jeffery J, Nickerson P. J Am Soc Nephrol. 2004;15:219–227. doi: 10.1097/01.asn.0000101031.52826.be. [DOI] [PubMed] [Google Scholar]
- 14.Hortin GL, Sviridov D. Pharmacogenomics. 2007;8:237–255. doi: 10.2217/14622416.8.3.237. [DOI] [PubMed] [Google Scholar]
- 15.Zurbig P, Decramer S, Dakna M, Jantos J, Good DM, Coon JJ, Bandin F, Mischak H, Bascands JL, Schanstra JP. Proteomics. 2009;9:2108–2117. doi: 10.1002/pmic.200800560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schaub S, Wilkins J, Weiler T, Sangster K, Rush D, Nickerson P. Kidney Int. 2004;65:323–332. doi: 10.1111/j.1523-1755.2004.00352.x. [DOI] [PubMed] [Google Scholar]
- 17.Havanapan PO, Thongboonkerd V. J Proteome Res. 2009;8:3109–3117. doi: 10.1021/pr900015q. [DOI] [PubMed] [Google Scholar]
- 18.Thongboonkerd V, Chutipongtanate S, Kanlaya R. J Proteome Res. 2006;5:183–191. doi: 10.1021/pr0502525. [DOI] [PubMed] [Google Scholar]
- 19.Cirillo M. J Nephrol. 2010;23:125–132. [PubMed] [Google Scholar]
- 20.Hibi Y, Uemura O, Nagai T, Yamakawa S, Yamasaki Y, Yamamoto M, Nakano M, Kasahara K. Pediatr Int. 2014 doi: 10.1111/ped.12470. [DOI] [PubMed] [Google Scholar]
- 21.Burton C, Shi H, Ma Y. Clin Chim Acta. 2014;435:42–47. doi: 10.1016/j.cca.2014.04.022. [DOI] [PubMed] [Google Scholar]
- 22.Pisitkun T, Shen RF, Knepper MA. Proc Natl Acad Sci U S A. 2004;101:13368–13373. doi: 10.1073/pnas.0403453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou H, Pisitkun T, Aponte A, Yuen PS, Hoffert JD, Yasuda H, Hu X, Chawla L, Shen RF, Knepper MA, Star RA. Kidney Int. 2006;70:1847–1857. doi: 10.1038/sj.ki.5001874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zubiri I, Posada-Ayala M, Sanz-Maroto A, Calvo E, Martin-Lorenzo M, Gonzalez-Calero L, de la Cuesta F, Lopez JA, Fernandez-Fernandez B, Ortiz A, Vivanco F, Alvarez-Llamas G. J Proteomics. 2014;96:92–102. doi: 10.1016/j.jprot.2013.10.037. [DOI] [PubMed] [Google Scholar]
- 25.Zubiri I, Vivanco F, Alvarez-Llamas G. Methods Mol Biol. 2013;1000:209–220. doi: 10.1007/978-1-62703-405-0_16. [DOI] [PubMed] [Google Scholar]
- 26.Raj DA, Fiume I, Capasso G, Pocsfalvi G. Kidney Int. 2012;81:1263–1272. doi: 10.1038/ki.2012.25. [DOI] [PubMed] [Google Scholar]
- 27.Zhou H, Yuen PS, Pisitkun T, Gonzales PA, Yasuda H, Dear JW, Gross P, Knepper MA, Star RA. Kidney Int. 2006;69:1471–1476. doi: 10.1038/sj.ki.5000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheruvanky A, Zhou H, Pisitkun T, Kopp JB, Knepper MA, Yuen PS, Star RA. Am J Physiol Renal Physiol. 2007;292:F1657–1661. doi: 10.1152/ajprenal.00434.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin-Lorenzo M, Gonzalez-Calero L, Zubiri I, Diaz-Payno PJ, Sanz-Maroto A, Posada-Ayala M, Ortiz A, Vivanco F, Alvarez-Llamas G. Electrophoresis. 2014;35:2634–2641. doi: 10.1002/elps.201300601. [DOI] [PubMed] [Google Scholar]
- 30.Meleady P. Methods Mol Biol. 2011;784:123–137. doi: 10.1007/978-1-61779-289-2_9. [DOI] [PubMed] [Google Scholar]
- 31.He W, Huang C, Luo G, Dal Pra I, Feng J, Chen W, Ma L, Wang Y, Chen X, Tan J, Zhang X, Armato U, Wu J. Proteomics. 2012;12:1059–1072. doi: 10.1002/pmic.201100400. [DOI] [PubMed] [Google Scholar]
- 32.Lee RS, Monigatti F, Briscoe AC, Waldon Z, Freeman MR, Steen H. J Proteome Res. 2008;7:4022–4030. doi: 10.1021/pr800301h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orenes-Pinero E, Corton M, Gonzalez-Peramato P, Algaba F, Casal I, Serrano A, Sanchez-Carbayo M. J Proteome Res. 2007;6:4440–4448. doi: 10.1021/pr070368w. [DOI] [PubMed] [Google Scholar]
- 34.Pieper R, Gatlin CL, McGrath AM, Makusky AJ, Mondal M, Seonarain M, Field E, Schatz CR, Estock MA, Ahmed N, Anderson NG, Steiner S. Proteomics. 2004;4:1159–1174. doi: 10.1002/pmic.200300661. [DOI] [PubMed] [Google Scholar]
- 35.Wisniewski JR, Zougman A, Nagaraj N, Mann M. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 36.Wheelock AM, Buckpitt AR. Electrophoresis. 2005;26:4508–4520. doi: 10.1002/elps.200500253. [DOI] [PubMed] [Google Scholar]
- 37.Morris JS, Clark BN, Wei W, Gutstein HB. J Proteome Res. 2010;9:595–604. doi: 10.1021/pr9005603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bertucci F, Birnbaum D, Goncalves A. Mol Cell Proteomics. 2006;5:1772–1786. doi: 10.1074/mcp.R600011-MCP200. [DOI] [PubMed] [Google Scholar]
- 39.Lemkin PF, Myrick JE, Upton KM. Appl Theor Electrophor. 1993;3:163–172. [PubMed] [Google Scholar]
- 40.Soler-Garcia AA, Johnson D, Hathout Y, Ray PE. Clin J Am Soc Nephrol. 2009;4:763–771. doi: 10.2215/CJN.0200608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Varghese SA, Powell TB, Janech MG, Budisavljevic MN, Stanislaus RC, Almeida JS, Arthur JM. J Investig Med. 2010;58:612–620. doi: 10.231/JIM.0b013e3181d473e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soggiu A, Piras C, Bonizzi L, Hussein HA, Pisanu S, Roncada P. Acta Diabetol. 2012;49:453–464. doi: 10.1007/s00592-012-0407-0. [DOI] [PubMed] [Google Scholar]
- 43.Rabilloud T, Lelong C. J Proteomics. 2011;74:1829–1841. doi: 10.1016/j.jprot.2011.05.040. [DOI] [PubMed] [Google Scholar]
- 44.Rogowska-Wrzesinska A, Le Bihan MC, Thaysen-Andersen M, Roepstorff P. J Proteomics. 2013;88:4–13. doi: 10.1016/j.jprot.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 45.Timms JF, Cramer R. Proteomics. 2008;8:4886–4897. doi: 10.1002/pmic.200800298. [DOI] [PubMed] [Google Scholar]
- 46.Yan JX, Devenish AT, Wait R, Stone T, Lewis S, Fowler S. Proteomics. 2002;2:1682–1698. doi: 10.1002/1615-9861(200212)2:12<1682::AID-PROT1682>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 47.Gao W. Methods Mol Biol. 2014;1105:17–30. doi: 10.1007/978-1-62703-739-6_2. [DOI] [PubMed] [Google Scholar]
- 48.Banon-Maneus E, Diekmann F, Carrascal M, Quintana LF, Moya-Rull D, Bescos M, Ramirez-Bajo MJ, Rovira J, Gutierrez-Dalmau A, Sole-Gonzalez A, Abian J, Campistol JM. Transplantation. 2010;89:548–558. doi: 10.1097/TP.0b013e3181c690e3. [DOI] [PubMed] [Google Scholar]
- 49.Nabity MB, Lees GE, Dangott LJ, Cianciolo R, Suchodolski JS, Steiner JM. Vet Clin Pathol. 2011;40:222–236. doi: 10.1111/j.1939-165X2011.00307.x. [DOI] [PubMed] [Google Scholar]
- 50.Li F, Chen DN, He CW, Zhou Y, Olkkonen VM, He N, Chen W, Wan P, Chen SS, Zhu YT, Lan KJ, Tan WL. J Proteomics. 2012;77:225–236. doi: 10.1016/j.jprot.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 51.Sharma K, Lee S, Han S, Lee S, Francos B, McCue P, Wassell R, Shaw MA, RamachandraRao SP. Proteomics. 2005;5:2648–2655. doi: 10.1002/pmic.200401288. [DOI] [PubMed] [Google Scholar]
- 52.Magistroni R, Ligabue G, Lupo V, Furci L, Leonelli M, Manganelli L, Masellis M, Gatti V, Cavazzini F, Tizzanini W, Albertazzi A. Nephrol Dial Transplant. 2009;24:1672–1681. doi: 10.1093/ndt/gfp020. [DOI] [PubMed] [Google Scholar]
- 53.Buhimschi IA, Zhao G, Funai EF, Harris N, Sasson IE, Bernstein IM, Saade GR, Buhimschi CS. Am J Obstet Gynecol. 2008;199:551 e551–516. doi: 10.1016/j.ajog.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Candiano G, Bruschi M, Petretto A, Santucci L, Del Boccio P, Urbani A, Bertoni E, Gusmano R, Salvadori M, Scolari F, Ghiggeri GM. Proteomics Clin Appl. 2008;2:956–963. doi: 10.1002/prca.200780157. [DOI] [PubMed] [Google Scholar]
- 55.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 56.Ong SE, Kratchmarova I, Mann M. J Proteome Res. 2003;2:173–181. doi: 10.1021/pr0255708. [DOI] [PubMed] [Google Scholar]
- 57.Ibarrola N, Kalume DE, Gronborg M, Iwahori A, Pandey A. Anal Chem. 2003;75:6043–6049. doi: 10.1021/ac034931f. [DOI] [PubMed] [Google Scholar]
- 58.Kruger M, Moser M, Ussar S, Thievessen I, Luber CA, Forner F, Schmidt S, Zanivan S, Fassler R, Mann M. Cell. 2008;134:353–364. doi: 10.1016/j.cell.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 59.Konzer A, Ruhs A, Braun T, Kruger M. Methods Mol Biol. 2013;1005:39–52. doi: 10.1007/978-1-62703-386-2_4. [DOI] [PubMed] [Google Scholar]
- 60.Zanivan S, Krueger M, Mann M. Methods Mol Biol. 2012;757:435–450. doi: 10.1007/978-1-61779-166-6_25. [DOI] [PubMed] [Google Scholar]
- 61.Hathout Y, Marathi RL, Rayavarapu S, Zhang A, Brown KJ, Seol H, Gordish-Dressman H, Cirak S, Bello L, Nagaraju K, Partridge T, Hoffman EP, Takeda S, Mah JK, Henricson E, McDonald C. Hum Mol Genet. 2014;23:6458–6469. doi: 10.1093/hmg/ddu366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robles MS, Cox J, Mann M. PLoS Genet. 2014;10:e1004047. doi: 10.1371/journal.pgen.1004047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rayavarapu S, Coley W, Cakir E, Jahnke V, Takeda S, Aoki Y, Grodish-Dressman H, Jaiswal JK, Hoffman EP, Brown KJ, Hathout Y, Nagaraju K. Mol Cell Proteomics. 2013;12:1061–1073. doi: 10.1074/mcp.M112.023127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Geiger T, Wisniewski JR, Cox J, Zanivan S, Kruger M, Ishihama Y, Mann M. Nat Protoc. 2011;6:147–157. doi: 10.1038/nprot.2010.192. [DOI] [PubMed] [Google Scholar]
- 65.Mann M. Nat Rev Mol Cell Biol. 2006;7:952–958. doi: 10.1038/nrm2067. [DOI] [PubMed] [Google Scholar]
- 66.Park SS, Wu WW, Zhou Y, Shen RF, Martin B, Maudsley S. J Proteomics. 2012;75:3720–3732. doi: 10.1016/j.jprot.2012.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lamoureux F, Gastinel LN, Mestre E, Marquet P, Essig M. J Proteomics. 2012;75:3674–3687. doi: 10.1016/j.jprot.2012.04.024. [DOI] [PubMed] [Google Scholar]
- 68.Zhao S, Li R, Cai X, Chen W, Li Q, Xing T, Zhu W, Chen YE, Zeng R, Deng Y. Evid Based Complement Alternat Med. 2013;2013:275390. doi: 10.1155/2013/275390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 70.Evans C, Noirel J, Ow SY, Salim M, Pereira-Medrano AG, Couto N, Pandhal J, Smith D, Pham TK, Karunakaran E, Zou X, Biggs CA, Wright PC. Anal Bioanal Chem. 2012;404:1011–1027. doi: 10.1007/s00216-012-5918-6. [DOI] [PubMed] [Google Scholar]
- 71.Luo R, Zhao H. Stat Interface. 2012;5:99–107. doi: 10.4310/sii.2012.v5.n1.a9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen YT, Chen CL, Chen HW, Chung T, Wu CC, Chen CD, Hsu CW, Chen MC, Tsui KH, Chang PL, Chang YS, Yu JS. J Proteome Res. 2010;9:5803–5815. doi: 10.1021/pr100576x. [DOI] [PubMed] [Google Scholar]
- 73.DeSouza LV, Romaschin AD, Colgan TJ, Siu KW. Anal Chem. 2009;81:3462–3470. doi: 10.1021/ac802726a. [DOI] [PubMed] [Google Scholar]
- 74.Hao P, Guo T, Li X, Adav SS, Yang J, Wei M, Sze SK. J Proteome Res. 2010;9:3520–3526. doi: 10.1021/pr100037h. [DOI] [PubMed] [Google Scholar]
- 75.Andersen RF, Palmfeldt J, Jespersen B, Gregersen N, Rittig S. Proteomics Clin Appl. 2012;6:382–393. doi: 10.1002/prca.201100081. [DOI] [PubMed] [Google Scholar]
- 76.Jin J, Ku YH, Kim Y, Kim Y, Kim K, Lee JY, Cho YM, Lee HK, Park KS, Kim Y. Exp Diabetes Res. 2012;2012:168602. doi: 10.1155/2012/168602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sigdel TK, Kaushal A, Gritsenko M, Norbeck AD, Qian WJ, Xiao W, Camp DG, 2nd, Smith RD, Sarwal MM. Proteomics Clin Appl. 2010;4:32–47. doi: 10.1002/prca.200900124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu W, Smith JW, Huang CM. J Biomed Biotechnol. 2010;2010:840518. doi: 10.1155/2010/840518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chelius D, Bondarenko PV. J Proteome Res. 2002;1:317–323. doi: 10.1021/pr025517j. [DOI] [PubMed] [Google Scholar]
- 80.Bondarenko PV, Chelius D, Shaler TA. Anal Chem. 2002;74:4741–4749. doi: 10.1021/ac0256991. [DOI] [PubMed] [Google Scholar]
- 81.Sedic M, Gethings LA, Vissers JP, Shockcor JP, McDonald S, Vasieva O, Lemac M, Langridge JI, Batinic D, Pavelic SK. Biochem Biophys Res Commun. 2014;452:21–26. doi: 10.1016/j.bbrc.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 82.Kalantari S, Rutishauser D, Samavat S, Nafar M, Mahmudieh L, Rezaei-Tavirani M, Zubarev RA. PLoS One. 2013;8:e80830. doi: 10.1371/journal.pone.0080830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lukasse PN, America AH. J Proteome Res. 2014;13:3191–3199. doi: 10.1021/pr401072g. [DOI] [PubMed] [Google Scholar]
- 84.Arike L, Peil L. Methods Mol Biol. 2014;1156:213–222. doi: 10.1007/978-1-4939-0685-7_14. [DOI] [PubMed] [Google Scholar]
- 85.Mirza SP, Olivier M. Physiol Genomics. 2008;33:3–11. doi: 10.1152/physiolgenomics.00292.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rosenberger G, Ludwig C, Rost HL, Aebersold R, Malmstrom L. Bioinformatics. 2014;30:2511–2513. doi: 10.1093/bioinformatics/btu200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Neilson KA, Keighley T, Pascovici D, Cooke B, Haynes PA. Methods Mol Biol. 2013;1002:205–222. doi: 10.1007/978-1-62703-360-2_17. [DOI] [PubMed] [Google Scholar]
- 88.Schlatzer D, Maahs DM, Chance MR, Dazard JE, Li X, Hazlett F, Rewers M, Snell-Bergeon JK. Diabetes Care. 2012;35:549–555. doi: 10.2337/dc11-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Prakash A, Tomazela DM, Frewen B, Maclean B, Merrihew G, Peterman S, Maccoss MJ. J Proteome Res. 2009;8:2733–2739. doi: 10.1021/pr801028b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gerber SA, Rush J, Stemman O, Kirschner MW, Gygi SP. Proc Natl Acad Sci U S A. 2003;100:6940–6945. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mallick P, Schirle M, Chen SS, Flory MR, Lee H, Martin D, Ranish J, Raught B, Schmitt R, Werner T, Kuster B, Aebersold R. Nat Biotechnol. 2007;25:125–131. doi: 10.1038/nbt1275. [DOI] [PubMed] [Google Scholar]
- 92.Mohammed Y, Domanski D, Jackson AM, Smith DS, Deelder AM, Palmblad M, Borchers CH. J Proteomics. 2014;106:151–161. doi: 10.1016/j.jprot.2014.04.018. [DOI] [PubMed] [Google Scholar]
- 93.Lange V, Picotti P, Domon B, Aebersold R. Mol Syst Biol. 2008;4:222. doi: 10.1038/msb.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kiyonami R, Schoen A, Prakash A, Peterman S, Zabrouskov V, Picotti P, Aebersold R, Huhmer A, Domon B. Mol Cell Proteomics. 2011;10:M110 002931. doi: 10.1074/mcp.M110.002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chang CY, Picotti P, Huttenhain R, Heinzelmann-Schwarz V, Jovanovic M, Aebersold R, Vitek O. Mol Cell Proteomics. 2012;11:M111 014662. doi: 10.1074/mcp.M111.014662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Picotti P, Bodenmiller B, Mueller LN, Domon B, Aebersold R. Cell. 2009;138:795–806. doi: 10.1016/j.cell.2009.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Boja ES, Rodriguez H. Proteomics. 2012;12:1093–1110. doi: 10.1002/pmic.201100387. [DOI] [PubMed] [Google Scholar]
- 98.Mattes WB, Walker EG, Abadie E, Sistare FD, Vonderscher J, Woodcock J, Woosley RL. Nat Biotechnol. 2010;28:432–433. doi: 10.1038/nbt0510-432. [DOI] [PubMed] [Google Scholar]
- 99.Warnock DG, Peck CC. Nat Biotechnol. 2010;28:444–445. doi: 10.1038/nbt0510-444. [DOI] [PubMed] [Google Scholar]
- 100.Bonventre JV, Vaidya VS, Schmouder R, Feig P, Dieterle F. Nat Biotechnol. 2010;28:436–440. doi: 10.1038/nbt0510-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dieterle F, Sistare F, Goodsaid F, Papaluca M, Ozer JS, Webb CP, Baer W, Senagore A, Schipper MJ, Vonderscher J, Sultana S, Gerhold DL, Phillips JA, Maurer G, Carl K, Laurie D, Harpur E, Sonee M, Ennulat D, Holder D, Andrews-Cleavenger D, Gu YZ, Thompson KL, Goering PL, Vidal JM, Abadie E, Maciulaitis R, Jacobson-Kram D, Defelice AF, Hausner EA, Blank M, Thompson A, Harlow P, Throckmorton D, Xiao S, Xu N, Taylor W, Vamvakas S, Flamion B, Lima BS, Kasper P, Pasanen M, Prasad K, Troth S, Bounous D, Robinson-Gravatt D, Betton G, Davis MA, Akunda J, McDuffie JE, Suter L, Obert L, Guffroy M, Pinches M, Jayadev S, Blomme EA, Beushausen SA, Barlow VG, Collins N, Waring J, Honor D, Snook S, Lee J, Rossi P, Walker E, Mattes W. Nat Biotechnol. 2010;28:455–462. doi: 10.1038/nbt.1625. [DOI] [PubMed] [Google Scholar]
- 102.Ozer JS, Dieterle F, Troth S, Perentes E, Cordier A, Verdes P, Staedtler F, Mahl A, Grenet O, Roth DR, Wahl D, Legay F, Holder D, Erdos Z, Vlasakova K, Jin H, Yu Y, Muniappa N, Forest T, Clouse HK, Reynolds S, Bailey WJ, Thudium DT, Topper MJ, Skopek TR, Sina JF, Glaab WE, Vonderscher J, Maurer G, Chibout SD, Sistare FD, Gerhold DL. Nat Biotechnol. 2010;28:486–494. doi: 10.1038/nbt.1627. [DOI] [PubMed] [Google Scholar]
- 103.Good DM, Zurbig P, Argiles A, Bauer HW, Behrens G, Coon JJ, Dakna M, Decramer S, Delles C, Dominiczak AF, Ehrich JH, Eitner F, Fliser D, Frommberger M, Ganser A, Girolami MA, Golovko I, Gwinner W, Haubitz M, Herget-Rosenthal S, Jankowski J, Jahn H, Jerums G, Julian BA, Kellmann M, Kliem V, Kolch W, Krolewski AS, Luppi M, Massy Z, Melter M, Neususs C, Novak J, Peter K, Rossing K, Rupprecht H, Schanstra JP, Schiffer E, Stolzenburg JU, Tarnow L, Theodorescu D, Thongboonkerd V, Vanholder R, Weissinger EM, Mischak H, Schmitt-Kopplin P. Mol Cell Proteomics. 2010;9:2424–2437. doi: 10.1074/mcp.M110.001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Siwy J, Schanstra JP, Argiles A, Bakker SJ, Beige J, Boucek P, Brand K, Delles C, Duranton F, Fernandez-Fernandez B, Jankowski ML, Al Khatib M, Kunt T, Lajer M, Lichtinghagen R, Lindhardt M, Maahs DM, Mischak H, Mullen W, Navis G, Noutsou M, Ortiz A, Persson F, Petrie JR, Roob JM, Rossing P, Ruggenenti P, Rychlik I, Serra AL, Snell-Bergeon J, Spasovski G, Stojceva-Taneva O, Trillini M, von der Leyen H, Winklhofer-Roob BM, Zurbig P, Jankowski J. Nephrol Dial Transplant. 2014;29:1563–1570. doi: 10.1093/ndt/gfu039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Blonder J, Issaq HJ, Veenstra TD. Electrophoresis. 2011;32:1541–1548. doi: 10.1002/elps.201000585. [DOI] [PubMed] [Google Scholar]
- 106.Schirle M, Bantscheff M, Kuster B. Chem Biol. 2012;19:72–84. doi: 10.1016/j.chembiol.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 107.Aebersold R, Burlingame AL, Bradshaw RA. Mol Cell Proteomics. 2013;12:2381–2382. doi: 10.1074/mcp.E113.031658. [DOI] [PMC free article] [PubMed] [Google Scholar]