

Abstract
Background
Shiga toxin producing Escherichia coli O157 can cause severe bloody diarrhea and haemolytic uraemic syndrome. Phage typing of E. coli O157 facilitates public health surveillance and outbreak investigations, certain phage types are more likely to occupy specific niches and are associated with specific age groups and disease severity. The aim of this study was to analyse the genome sequences of 16 (fourteen T4 and two T7) E. coli O157 typing phages and to determine the genes responsible for the subtle differences in phage type profiles.
Results
The typing phages were sequenced using paired-end Illumina sequencing at The Genome Analysis Centre and the Animal Health and Veterinary Laboratories Agency and bioinformatics programs including Velvet, Brig and Easyfig were used to analyse them. A two-way Euclidian cluster analysis highlighted the associations between groups of phage types and typing phages. The analysis showed that the T7 typing phages (9 and 10) differed by only three genes and that the T4 typing phages formed three distinct groups of similar genomic sequences: Group 1 (1, 8, 11, 12 and 15, 16), Group 2 (3, 6, 7 and 13) and Group 3 (2, 4, 5 and 14). The E. coli O157 phage typing scheme exhibited a significantly modular network linked to the genetic similarity of each group showing that these groups are specialised to infect a subset of phage types.
Conclusion
Sequencing the typing phage has enabled us to identify the variable genes within each group and to determine how this corresponds to changes in phage type.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1470-z) contains supplementary material, which is available to authorized users.
Background
Escherichia coli O157:H7 is the most prevalent Shiga toxin producing E. coli (STEC) serotype in the UK and has the most severe impact on human health [1]. STEC O157 symptoms can range from mild gastroenteritis to severe bloody diarrhoea and in more extreme cases haemolytic uraemic syndrome (HUS) [2]. The very young, elderly and immune-compromised are particularly at risk of HUS. A recent Public Health England (PHE) study found incidence to be as high as 1.78 per 100,000 person-years with up to 33% of cases being hospitalised (Gastrointestinal Bacterial Reference Unit (GBRU) in house data). The GBRU at PHE receives approximately 1000 STEC O157 samples per year. Recent outbreaks in the UK have been foodborne or linked to petting farms [3-5]. For purposes of public health surveillance and outbreak investigations, STEC strains are differentiated by phage typing and multilocus variable number tandem repeat analysis [6].
Bacteriophages are viruses that infect bacteria and cause bacterial lysis and cell death, but can also promote horizontal gene transfer between bacteria, play an important role in dynamic bacterial genome evolution and can regulate the abundance and diversity of bacterial communities through co-evolution [7]. There are a range of phages that infect Escherichia coli that progress either to a lytic or lysogenic phase after infection. A lytic phase will cause cell lysis whereas in lysogenic phase the phage becomes integrated into the host genome and becomes a prophage. Prophages are important as they often encode additional factors not directly linked to phage production that may provide an evolutionary advantage to the bacterial host enabling survival of the embedded prophage. These include factors that promote colonisation of animal hosts as well as their regulators [8,9]. Bacteriophage specificity is, in part, dependent on the ability of tail fiber proteins to bind to specific receptors on the bacterial host [10].
Phage-typing of STEC O157 is a scheme based on the use of 16 bacteriophages that produce a phage infection profile for a strain based on the level of lysis achieved by each phage [11] and has been used to categorize outbreaks and sporadic cases. Today 80% of all STEC O157 strains typed are PT 8, 21/28, 2, 4 or 32 in the UK (GBRU in house data). Certain PTs are more likely to be associated with human infection and so far there is little understanding of the basis for this. While ongoing work is focused on sequencing and analysis of the bacterial strains, we propose that further insight into relevant strain differences can be gained by also understanding the typing phages themselves and the basis of their infection selectivity. A longer term aim of the work is to understand the factors that mediate resistance and susceptibility in the phage-bacterium relationship.
Little is known about the molecular basis for the interaction between phages and different strains of different phage types, however we can interrogate the phage infection profile of who-infects-whom as a bipartite (two-mode) network. Two common methods for analysing community structure in bipartite data are nestedness and modularity. Nestedness is a way of measuring the ranges of both host resistance and phage infectivity across a specialist to generalist gradient. Specialists are assumed to have strategies that are subsets of those which are more generalised. Modularity is the degree to which a network can be split into distinct modular groupings of phage and bacteria such that there are many infections within rather than between groups [12].
The 16 phages in the STEC phage-typing scheme are made up of 14 T4 phages and 2 T7 phages. An example of a T7 phage has been sequenced previously and T7 are known to consist of a single ‘chromosome’ carrying about 30 genes [13]. The 5’ end genes of the chromosome are expressed at an early stage of infection and their products are involved in the induction of host RNA polymerase for transcription and control the expression of other phage genes in a positive feedback mechanism. Genes that are expressed later are involved in the metabolism of phage DNA and code for capsid proteins or are involved in the assembly of infective progeny particles [13]. T4 phages have much larger genomes with 300 putative genes, only 62 of these have been found to be ‘essential’ under laboratory conditions [14]. The order of expression works in a similar way to T7 phage.
The STEC O157 typing phages 5, 7 and 10 from the typing scheme have previously been sequenced [15-17]. Our sequencing results are consistent with previously published sequences. We build on this data by placing the previously sequenced phages into similarity groups within the typing phages. The aim of this study was to analyse the genome sequences of 16 (fourteen T4 and two T7) STEC O157 typing phages (TPs) and to identify genes that may account for differences in infectivity between related phages.
Methods
Phage propagation and DNA extraction
The typing phages were obtained as a gift from the National Microbiology Laboratory, Winnipeg, MN, Canada to GBRU in the late 1980s. To propagate the phage, 0.1 ml of the propagating strain (Additional file 1: Table S1, Figure 1) was inoculated into 2 × 20 ml of single strength Difco nutrient broth and 0.1 ml of test phage was added to one and the other kept as a control. The bottles were incubated and turbidity was monitored. When lysis was judged to be at its maximum compared to the control, a small amount of the phage solution was centrifuged at 2,200 g for 20 min. The supernatant was removed and spotted onto a flooded plate of propagating strain as a test; the plate was dried and incubated at 37°C overnight. The plates were examined for lysis and if positive the phage lysate was sterilized by filtration and stored at 4°C.
Figure 1.

Two-way cluster analysis dendrogram of 66 phage types and 16 typing phages. The matrix of shaded squares represents the phage type × typing phage matrix, while the dendrograms show the clustering. The dendrograms are scaled by Wishart ‘s (1969) objective function, expressed as the percentage of information remaining at each level of grouping (McCune and Grace, 2002). Each square represents the presence (black) and absence (white) of a reaction with a given typing phage. The three phage type clusters and the 4 typing phage clusters are indicated at the node with numbers.
All phages were filtered before extraction took place. Eleven (phages 1, 3, 4, 5, 6, 7, 8, 9, 12, 13 and 14) of the 16 phages were extracted using the QIAamp UltraSens Virus kit (Qiagen, UK) following the manufacturer’s instructions. This method failed to produce a high enough concentration of DNA for the remaining phages (2, 10, 11, 15 and 16) and these were extracted using a Zinc Chloride protocol [18]. Briefly, 20 μl of a 2 M Zinc chloride solution was added to 1 ml of sample and incubated for 5 min at 37°C. The sample was then centrifuged at 10000 rpm and the supernatant was removed. The pellet was resuspended in 500 μl of TES buffer (0.1 M Tris–HCl, pH8; 0.1 M EDTA and 0.3% SDS) and then incubated at 60°C for 15 min. Subsequently, 60 μl of a 3 M potassium acetate solution was added and the sample left on ice for 10 to 15 min. Following the formation of a white, dense precipitation the sample was centrifuged for 1 min at 12000 rpm and the supernatant removed to a new tube. To this an equal volume of isopropanol was added, the solution vortexed and left on ice for 5 min. The solution was centrifuged and evaporated simultaneously using a Speedy-Vac machine and the pellet washed with 70% ethanol before being resuspended in 20–100 μl TE (10 mM Tris–HCl, pH8; ImM EDTA). Samples were pooled by five extractions to give a higher yield of DNA. This method also failed to produce high enough concentration of DNA for sequencing TP 2 and 16 and we were ultimately unable to obtain sequencing data for these two TPs.
Sequencing
The first set of phages (1, 3, 4, 5, 6, 7, 8, 9, 12, 13 and 14) was sequenced at The Genome Analysis Centre (TGAC) on an Illumina MiSeq. Illumina TruSeq DNA library construction was performed and sequencing of the libraries was pooled on one run using 150 bp paired-end reads, this generated greater than 1 Gbp of data for the run. Data was then quality controlled, basecalling was performed and it was formatted. The second set of phages (10, 11 and 15) was sequenced at the Animal Health and Veterinary Laboratories Agency on an Illumina GAII. The library construction was performed using a Nextera DNA sample preparation kit (Illumina) and then sequenced in the same manner as the other set.
Bioinformatic sequencing analysis
Reads for all phages apart from TP 15 were de novo assembled into whole genomes using Velvet optimizer with a range of k-mer values from 90–120 [19] and annotated using Prokka 1.5.2 and output as GenBank files [20]. The genomes were visualised in the multiple genome alignment tool Mauve with a progressive alignment to visualise similarities and differences between them based on sequence content. The reads assembled into between 1 and 7 contigs for each phage.
TP15 could not be assembled correctly because the propagation process had induced other temperate phages in the genome of the propagating strain and the DNA had been co-extracted. Subsampling to x150 coverage and the genome assembler SPAdes with a better low frequency k-mer elimination step [21] was used to overcome this issue and resolve 15 true typing phage 15 contigs from the assemblies. The sequencing data has been made publicly available in the Short Read Archive under study alias PRJNA252693 and Genbank accession numbers for each phage can be found in the availability of supporting data section.
Euclidian tree
Data from PHE on the protocol used to identify phage types (Additional file 1: Table S3, Additional file 1: Table S2) was converted into binary (presence/absence) format. In the original scheme there were 66 established phage types (PT) and 16 typing phages (TP). This set of data was analysed using a two-way cluster hierarchical agglomerative analysis in PC-ORD software version 6.08 (MJM software Design, Gleneden Beach, OR). The clustering was performed with Euclidian distance matrix and Ward linkage method.
The optimal number of groups of plots was first evaluated with multiresponse permutation procedure, seeking the solution with fewest number of groups but the greatest gain in A-statistics [22].
Modularity and nestedness
Modularity of the network was calculated using the LPAb + algorithm [23] which uses label propagation coupled with greedy multistep agglomeration to identify the communities (made of members of both types of nodes (bacteria and phage)) that maximise modularity in bipartite networks. As LPAb + is stochastic we choose the best modularity score, QB, returned from 1,000 trials each time we use the algorithm. Code for performing the modularity analysis is supplied [24].
Nestedness statistics were calculated using FALCON [25]. The nestedness measures used were NODF [26], NTC [27,28] and BR, the discrepancy score of Brualdi and Sanderson, 1999 [29]. NODF and NTC scores take values in the range [0,100], whilst BR is the absolute number of differences between the input and a maximally packed matrix. NODF has been recalculated here as NODF = 100-NODF, so that lower measure scores show greater nestedness with 0 representing perfect nestedness for each of the measures.
We tested for significance of both modularity and the nestedness found in our phage-bacteria infection network using two null models based on properties of our network. Null model one is a Bernoulli random null model where connections between phage j and bacteria i are made with probability pij = F/M, where F is the total number of edges in our network (number of infecting interactions) and M is the maximum number of potential interactions (number of TP’s × number of PT’s). Null model two is based on the information in the rows and columns in the network [30]; where a connection between phage j and bacteria i is made with probability pij = 0.5 (dj/r + ki/c) where dj is the number of infections caused by phage j, r is the number of PTs, ki is the number of phage that can infect bacteria i and c is the number of TPs. We tested 1,000 null matrices against our network for each null model in the modularity analysis, whilst we used the adaptive ensemble of FALCON for nestedness analysis and report the ensemble size used (N), p-values (probability of finding a more modular/nested network from the null model) and z-scores (effect size; the number of standard deviations our network was away from the mean average found in each null model).
BRIG plot
BRIG (Blast Ring Image Generator), a genome comparison tool [31], was used to compare similarities between the 12 T4 like typing phages by inputting all of the GenBank files for the assembled genomes and plotting blast hits against a MultiFASTA file of all of the phages. The image was displayed as a series of concentric rings with the central ring being the MultiFASTA reference; each outer ring displays hits (i.e. genomic regions that show a high percentage similarity to the central reference genome) for each phage. BRIG was also used to show the comparison of phages 9 and 10 (the two T7 like typing phages) against phage 9 as a reference.
SeqFindR and Easyfig plots
SeqFindR, a bioinformatics tool developed by the Beatson Laboratory at the University of Queensland, was used to identify gene presence and absence in the phage genomes. Easyfig [32] was used to visualise the coding regions and colour the accessory genes in red for each phage group.
Tail fiber analysis
Tail fiber encoding genes were extracted from the GenBank files of the typing phages and the protein sequences aligned using MEGA 5.2. The alignment told us how many changes in protein sequence there were within the groups.
Results
In the phage typing scheme there are 14 T4-like bacteriophages (TP1-8 and TP11-16) and two T7-like bacteriophages (TP9 and TP10). The reactivity of each of the typing phages with respect to the STEC O157 phage typing scheme was analysed. The two-way Euclidian cluster analysis combined the independent clustering of 66 STEC O157 bacterial phage types and the 16 typing phages into a single diagram and highlighted the associations between groups of phage types and typing phages (Figure 1). The analysis showed that the STEC O157 phage typing scheme formed a weak (Qb = 0.1575 (Table 1)) but significantly modular network where the TP groups were each specialised to infect a subset of PTs (Figure 2). There also exists a large number of between module interactions. Furthermore, the majority of PTs of STEC O157 react with at least one member of each group of typing phages. These groups can be regarded as universally infective against STEC O157. Using statistical tests we also found that the nestedness of our interaction network was statistically significantly different from that found under randomly formed networks (Table 1). This indicates a correlation between phage infectivity range and the resistance range of the host. These phages have been designed and chosen to infect STEC O157 and create a typing scheme with the simplest and minimum selection of phages so it makes sense that the system is nested.
Table 1.
Summary statistics for nestedness and modularity analysis
| Modularity | Nestedness | ||||
|---|---|---|---|---|---|
| Measure | QB | NODF | NTC | BR | |
| Measure score | x | 0.1575 | 27.9199 | 30.2532 | 130 |
| Null model 1 | N | 1000 | 1300 | 1300 | 1300 |
| p-value | <1/N | <1/N | <1/N | <1/N | |
| z-score | 4.8602 | -7.5382 | -11.9831 | -11.7632 | |
| Null model 2 | N | 1000 | 1000 | 1000 | 1000 |
| p-value | <1/N | <1/N | <1/N | <1/N | |
| z-score | 5.7693 | -4.6740 | -6.7842 | -7.1554 | |
Barber’s modularity (Qb) and three nestedness measures (NODF, NTC and BR) were calculated. Two null models were used to generate ensembles of networks (of size N) to evaluate the strength of the modularity and nestedness observed in the classified Escherichia coli O157:H7 phage-bacteria infection network. This is done by reporting the significance (as a p-value) and effect size (as a z-score) of the phage-bacteria infection network relative to the networks found in each null model ensemble. Note that, due to differences in how these measures are calculated, for modularity a positive z-score indicates that modularity is greater in the observed network than the mean average of the ensemble; whilst in the nestedness analysis a negative z-score indicates the observed network is more nested than the mean nestedness found within the null ensemble. The classified Escherichia coli O157:H7 phage-bacteria infection network was found to be both more nested and more modular than any of the networks generated by the tested null models.
Figure 2.

A visual representation of the modularity seen within the system with modules coloured. Phage type (PT) is represented on the y axis and Typing phage (TP) is represented on the x axis and the matrix showing presence of a reaction with that phage as a white or coloured block. The 4 observed modules are coloured as yellow, pink, green and black.
Fourteen of the 16 phages in the typing scheme were sequenced and successfully assembled. Despite several attempts, sequencing of typing phages 2 and 16 failed due to insufficient quantities of DNA extracted from the phage propagation preparations.
The BRIG plot showed that the 12 sequenced T4-like bacteriophages formed three distinct groups of similar genomic sequences (Figure 3). Group 1 included typing phages 1, 8, 11, 12 and 15; Group 2 comprised typing phages 3, 6, 7 and 13 and typing phages 4, 5 and 14 were in Group 3. Although the sequencing for TP2 and TP16 failed, the modularity analysis indicates that TP16 belonged to Group 1 and TP2 belonged to Group 3 (Figure 2). The TPs varied significantly in size between the three groups: the members of Group 1 were 93,000–95,000 bp, Group 2 members were 165,000–175,000 bp and those in Group 3 were 135,000–140,000 bp.
Figure 3.
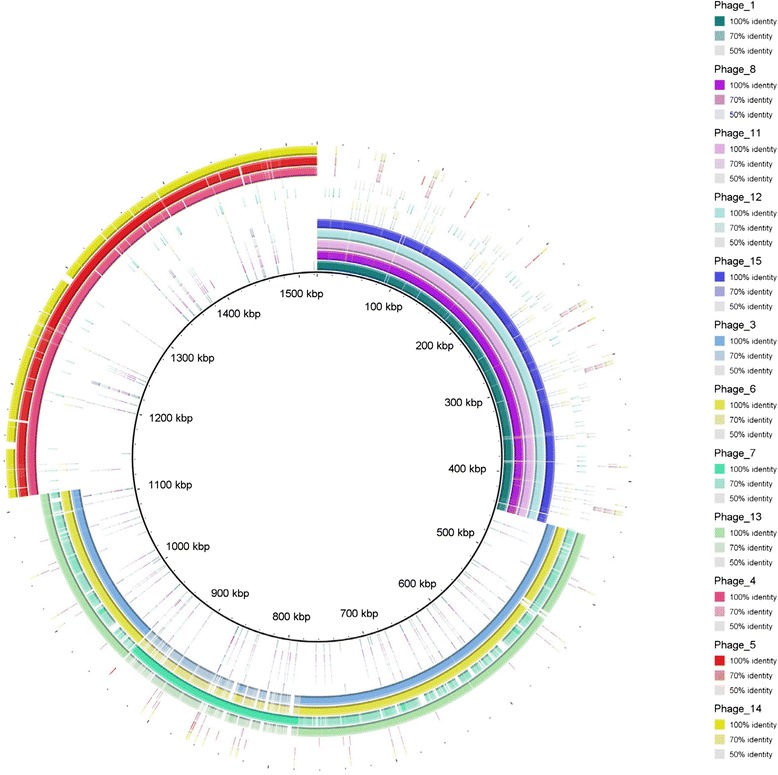
A genomic representative diagram drawn with BRIG of T4-like phage similarities, the coloured regions indicate high pairwise genomic sequence similarity according to blastn. Legend indicates which colours correspond with which phages and the shade of that colour indicates what level of similarity is observed. Central ring is multifasta of all T4-like phage genomes and each consecutive ring represents the similarity with a single phage. The multifasta and rings are in the same phage order.
The Group 1 phages (TP1, 8, 11, 12 and 15) were approximately 90,000 bp in length. These five phages were highly similar in genetic sequence content. The location, annotation and presence of accessory genes within Group 1 are shown in Figure 4, Additional file 1: Table S3. Figure 4 shows that there were 6 genes found in TP1 but absent in TP8, 11, 12 and 15 (five were annotated as hypothetical proteins and one tRNA). There were also five genes present in TP8, 11, 12 and 15 but not in TP1 (three were annotated as hypothetical proteins, one as AP2 domain protein and one was a tRNA gene) (Figure 4, Additional file 1: Table S3). TP8 was missing a region annotated as a putative prophage that was present in the other members of the group. With the exception of TP11, the Group 1 TPs are most closely related to each other by the two-way Euclidian cluster analysis demonstrating the link between gene content and phage typing profile.
Figure 4.
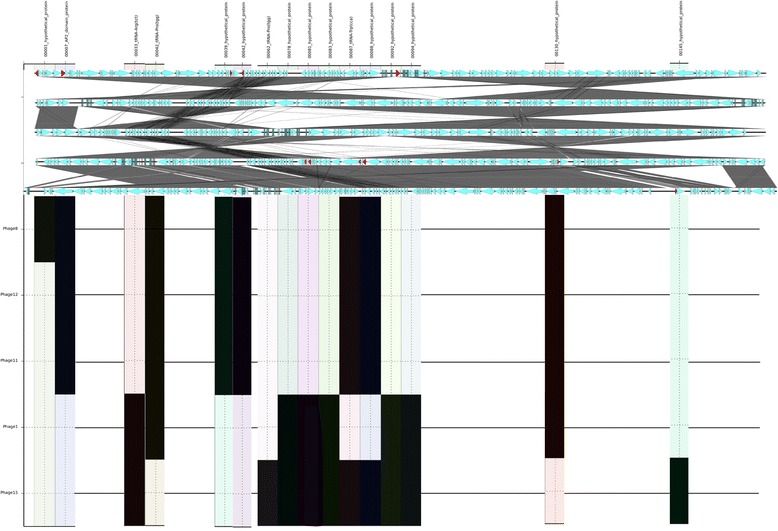
SeqFindR and Easyfig image combined representing the accessory gene content of group 1. Genomes of each phage in group 1 are represented by the Easyfig image showing linear visualisation of the genome and coding regions represented by arrows, accessory genes are coloured red. The order of phage genomes in the linear visualisation and the accessory content blocking is 8, 12, 11, 1 and 15 and was chosen based on similarity clustering in SeqFindR. Hits for the accessory genes in each genome are represented in labelled columns in the SeqFindR image underneath each accessory gene.
The typing phages in Group 2 (TP 3, 6, 7, and 13) were between 160–170,000 bp in length. The genomes were almost twice the size of the phages in Group 1 and exhibited less similarity. The accessory genes found in Group 2 were mostly annotated as encoding hypothetical proteins (Figure 5, Additional file 1: Table S4). The two-way Euclidian cluster analysis highlighted a close relationship between TP6 and TP13 and this corresponded with the level of sequence similarity of these two typing phages illustrated in Figure 5.
Figure 5.

SeqFindR and Easyfig image combined representing the accessory gene content of group 2. Genomes of each phage in group 2 are represented by the Easyfig image showing linear visualisation of the genome and coding regions represented by arrows, accessory genes are coloured red. The order of phage genomes in the linear visualisation and the accessory content blocking is 7, 3, 6 and 13 and was chosen based on similarity clustering in SeqFindR. Hits for the accessory genes in each genome are represented in labelled columns in the SeqFindR image underneath each accessory gene.
Typing phages 4, 5 and 14 were designated Group 3 and were 130–140,000 bp in length. Figure 6 shows the location, annotation and presence of accessory genes within Group 3. Figure 6 demonstrates that there were 29 gene differences within the group and the majorities (19) were annotated as hypothetical proteins. In addition, three genes encoded putative endonucleases and there were three genes designated tRNAs (Figure 6, Additional file 1: Table S5). The typing phages in Group 3 were most closely related to each other by the two-way Euclidian cluster analysis (Figure 1).
Figure 6.
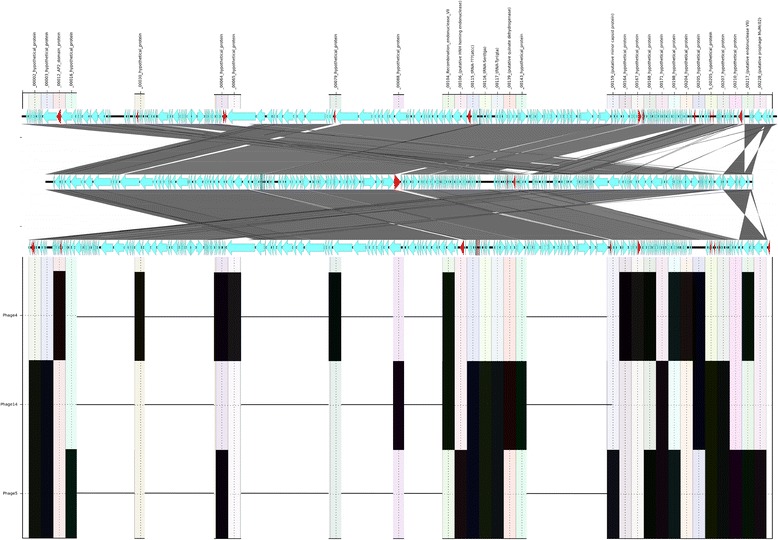
SeqFindR and Easyfig image combined representing the accessory gene content of group 3. Genomes of each phage in group 3 are represented by the Easyfig image showing linear visualisation of the genome and coding regions represented by arrows, accessory genes are coloured red. The order of phage genomes in the linear visualisation and the accessory content blocking is 4, 14 and 5 and was chosen based on similarity clustering in SeqFindR . Hits for the accessory genes in each genome are represented in labelled columns in the SeqFindR image underneath each accessory gene.
Phages 9 and 10, the two Podoviridae or T7 like phages that are found in the typing scheme, were successfully sequenced, assembled and annotated and revealed 40–45000 bp genomes consistent with the published sequences of T7 bacteriophages (Figure 7). Phages 9 and 10 differed by only three genes (annotated as encoded hypothetical proteins) that were found in Phage 9 but not in phage 10. The two-way Euclidian cluster analysis confirmed the close relationship between TP9 and TP 10 in terms of phage type profile. It also showed that there were six STEC O157 phage types (PT 2, 11, 17, 24, 50, and 51) that react with TP9 but not TP10 and none of the phage types react with TP10 but not TP9 (Figure 1). These three hypothetical proteins could be the key to the differences in the reactivity profiles of TP9 and 10.
Figure 7.
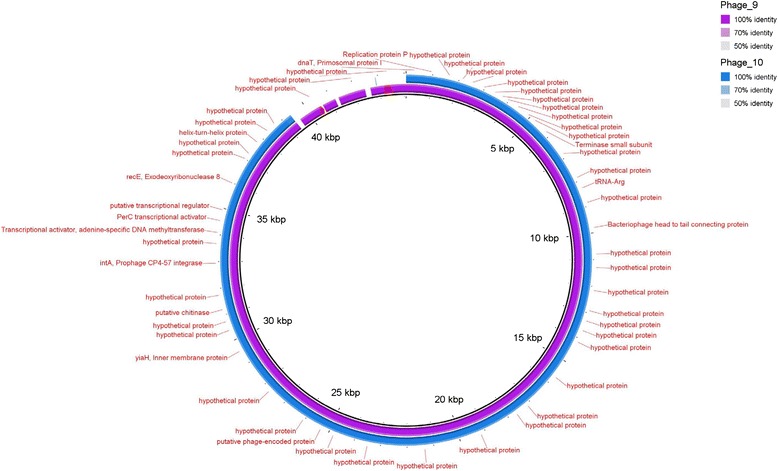
A genomic representative diagram drawn with BRIG, the coloured regions indicate high pairwise genomic sequence similarity according to blastn. The legend indicates which colours correspond with which phages. The central ring is a genbank file of Phage 9 as a reference and annotations of genes in red. The first ring is representative of Phage 9 and the second ring is representative of Phage 10 and the shade indicates the level of genomic similarity observed.
Tail-fiber encoding genes were analysed within each group and it was found that there were changes in the amino acid sequence for certain members of each group. Within the group 1 typing phages, phages 1 and 15 had 3 changes in amino acid sequence in their tail fibers, 2 of which were shared and 1 each unique to each phage. Within the group 2 typing phages, phage 7 has 47 changes in its amino acid sequence and 3 amino acid deletions. Within the group 3 typing phages, the same single position in all 3 members of the group has a different amino acid present and additionally there was another single position in typing phage 14 that had a different amino acid. The T7-like phages had identical tail fiber genes. There was no genetic similarity between tail fiber genes found in different groups.
Discussion
Phage-host interactions are key to understanding the virulence and success of E. coli O157 but little is known about the typing phages used in the O157 typing scheme. Sequencing these phages has enabled us to group the T4-like Myoviridae and the two Podoviridae or T7-like phages members of the typing phage scheme into four groups based on their sequence similarity. The two-way Euclidian cluster analysis demonstrated that similar phage groups react with STEC O157 in a similar way with closely related reaction profiles. The sequencing data also highlighted that a small number of gene differences may be responsible for the subtle differences in reaction profiles within the groups.
The large proportion of genes annotated as encoding hypothetical proteins hindered our investigations into the mechanisms of host-phage interactions. Attempts were made to annotate these genes further using protein BLAST and HMMER but only uncharacterised proteins were hit. However, the determination of which genes vary within each group will enable us to focus on the genes that may play a key role in the mechanisms of interactions between specific typing phages and strains belonging to specific phage types. For example, in Group 1, there were five genes that were found only in TP8, 11 and 12 and three PTs (PT21/28, 59 and 82) that only react with these TPs. The proteins encoded by these five genes may play a key role in the host-phage mechanisms between TP8, 11 and 12 and strains of STEC O157 belonging to PT21/28, 59 and 82. PT21/28 is the most common PT in the UK and is significantly associated with HUS [33]. Further details of unique host-phage interactions are described in Additional file 1: Table S6 and the genes referred to within can be found in Figures 4, 5 and 6.
Analysis of tail fibers genes showed that typing phages 1, 15, 7 and each individual member of Group 3 had different protein sequences encoded to the other members of their group. The changes that were found could partially account for infectivity differences [34]. These could explain a few of the differences in host specificity seen within those groups, although this will not apply to the T7-like typing phages that have had identical predicted tail fiber proteins.
Certain typing phages had almost identical genomes but different host susceptibility profiles, for example, TP11 belonged to the Group 1 phages but had a similar host susceptibility profile to the Group 2 phages. Each phage in the typing scheme has its own propagating strain (see Additional file 1: Figure S1, Table 1) so it is also possible that host-induced modification occurs [35]. For example, the propagating strains for the closely related typing phages TP9 and TP10 are STEC O157 PT2 and PT32, respectively. Modifications may be a result of methylation or other phenotypic changes that are not evident in the genome but may affect the host range of the virus.
Phenotypic differences in susceptibility patterns in genetically similar phages could be explained by the transcription order of genetic loci in the phage genome. It has been suggested that gene synteny constrains adaptation and is important for fitness and, therefore, infectivity of bacteriophages [36]. The order of transcription may be important in overcoming the host response to infection. The phages that transcribe their genetic loci in a different order may be killed and degraded by the host response, for example, TP 8, 11 and 12 are almost identical but have a different gene order and this may be key to their different infection profiles.
Our analysis showed that the significantly modular network exhibited by the STEC O157 phage typing scheme was linked to the genetic similarity groups mentioned above showing that these groups are specialised to infect a subset of PTs. However, the typing scheme as a whole is also significantly nested; more generalised phages minimise the number of phages needed in the scheme. Both of these network structures have also been found in other phage-bacteria infection networks [37,38]. The most common PTs in the UK: 2, 8, 21/28 and 32 are all found in different modules, meaning there is an abundant PT in each module. When looking at these PTs with nestedness, PT 8 and 2 both have a phage susceptibility range of 14 and 13 respectively so are quite generalised but PT 21/28 and 32 both have a host range of 7, and lie more towards the specialised end of the spectrum. It is interesting that the more abundant strains seem to appear at two levels of host range – perhaps suggestive of a trade-off between host range and phage productivity. It would be interesting to see, in conditions where the phages are allowed to evolve with their hosts, if a more modular network arises with further specialisation of the phages to maintain a kill-the-winner dynamic and less broad range infectivity [39]. This is an artificial system that we are observing and it is likely that we would see a different network arising in nature’s ecological systems.
Phage-typing has been used for epidemiological and surveillance studies by a number of groups [40,41] for different organisms. Phage-type association with increased strain virulence is of high interest to public health workers dealing with STEC O157, the replacement of phage-typing with whole genome sequencing should still incorporate our knowledge of phage type and associated virulence. For this reason it is valuable to find the molecular markers associated with high frequency and highly pathogenic phage types; elucidating the determinants underpinning differences in phage typing should contribute to this.
Phage-mediated therapies will continue to be an area of interest as we struggle with resistance to conventional antibiotics. It makes sense that moving forward there will be considerable interest in being able to predict bacterial susceptibility to ‘treatment’ phages based on sequence information alone. Furthermore, the next step would be modification of specific phages to improve their targeting/activity. This will rely on understanding of the phage genes that govern the specificity of infection in different backgrounds. The place to start is with certain key bacterial pathogens and a bank of phages.
Conclusions
In this study, the STEC O157 typing phages we clustered into four distinct groups of similar genomic sequences, that broadly correlated with phage typing profile groups determined by two-way Euclidian clustering. Genetic variation within the TP groups may explain the subtle differences between the phage typing profiles exhibited by the E. coli O157 typing phages. This analysis was hindered by the lack of detailed annotation of protein encoding genes in T4 and T7-like phages. The impact of the order of transcription of the blocks of genetic loci and the role of host-induced modification further confound the analysis. However, sequencing the typing phage has enabled us to identify the variable genes within each group and to determine how these correspond to changes in phage type. Future studies will focus on the genes that appear to alter host-phage interactions and we aim to identify bacterial genes that influence typing phage resistance and susceptibility using random mutagenesis approaches. In order to understand the best combination of strains and individual phages to work with, the network of interactions needs to be analysed. This information can also provide insight on how phage typing can potentially be simplified in the future. A better understanding of the genetic differences between bacterial phage types, and the possible differences in virulence factors, could help elucidate why different phage types occupy specific niches and are associated with different patient age groups and disease severity.
Availability of supporting data
The raw sequencing reads have been deposited in the short read archive under project alias PRJNA252693. The assembled sequences and annotations of the typing phages have been deposited in Genbank under the following accessions;
Phage 1: KP869100
Phage 3: KP869101
Phage 4: KP869102
Phage 5: KP869103
Phage 6: KP869104
Phage 7: KP869105
Phage 8: KP869106
Phage 9: KP869107
Phage 10: KP869108
Phage 11: KP869109
Phage 12: KP869110
Phage 13: KP869111
Phage 14: KP869112
Phage 15: KP869113
All other supporting data is included as additional files.
Acknowledgements
We would like to thank Matthew Hannah for performing the electron microscopy needed to take the pictures of the bacteriophages, Richard Ellis for performing sequencing of the bacteriophages at AHVLA, Vivienne do Nascimento for propogating the bacteriophage and Phillip Ashton for bioinformatics support. LAC acknowledges PhD funding from Public Health England and the National Institute for Health Research scientific research development fund (108601). DLG is supported by BBSRC institute strategic grant funding from the BBSRC.
Abbreviations
- STEC
Shiga Toxin producing Escherichia coli
- HUS
Haemolytic uraemic syndrome
- PT
Phage-type
- TP
Typing phage
- TGAC
The genome analysis centre
- AHVLA
Animal health and veterinary laboratories agency
- PHE
Public health England
- GBRU
Gastrointestinal bacterial reference unit
- BRIG
Blast ring image generator
Additional file
Propagating strain table. Table showing propagating strain and corresponding typing phage number that the strain propagates. Table S2. E. coli O157 phage typing scheme. Table showing reactions of the E. coli O157 type strains with the typing phages. Table S3. Table of the accessory variation of the Group 1 typing phages. Table detailing the accessory variation of Group 1 as depicted in figure 4. Table S4. Table of the accessory variation of the Group 2 typing phages. Table detailing the accessory variation of Group 2 as depicted in figure 5. Table S5. Table of the accessory variation of the Group 3 typing phages. Table detailing the accessory variation of Group 3 as depicted in figure 6. Table S6. Table of unique reactions. Table representing unique reaction that only occur within a subset of groups 1, 2 and 3 with specific PTs and number of genes found only in that subset. Figure S1. Phylogenetic tree of propagating strains. Phylogenetic tree of propagating strains for each typing phage and sakai as a reference. Figure S2. Visual representation of nestedness. A visual representation of the degree of nestedness found within the classified E. coli O157 phage-bacteria infection network. Figure S3. Electron microscopy image of typing phage 7 A representation of the the T4-like long-tailed phage morphology within the typing phages. Figure S4. Electron microscopy image of typing phage 9 A representation of the T7-like short-tailed phage morphology within the typing phages.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LC carried out the genetic analysis of the typing phages and drafted the manuscript. SB carried out the modularity/nestedness analysis. MCT performed the statistical analysis necessary to draw the two-way Eculidian tree. NP propagated the typing phages. TJD helped to draft the manuscript. DLG helped to draft the manuscript. CJ conceived the wider study and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Lauren A Cowley, Email: lauren.cowley@phe.gov.uk.
Stephen J Beckett, Email: S.J.Beckett@exeter.ac.uk.
Margo Chase-Topping, Email: margo.chase@ed.ac.uk.
Neil Perry, Email: neil.perry@phe.gov.uk.
Tim J Dallman, Email: tim.dallman@phe.gov.uk.
David L Gally, Email: david.gally@roslin.ed.ac.uk.
Claire Jenkins, Email: claire.jenkins1@phe.gov.uk.
References
- 1.Adak GK, Long SM, O’Brien SJ. Trends in indigenous foodborne disease and deaths, England and Wales: 1992 to 2000. Gut. 2002;51:832–841. doi: 10.1136/gut.51.6.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Griffin PM, Ostroff SM, Tauxe RV, Greene KD, Wells JG, Lewis JH, et al. Illnesses associated with Escherichia coli O157:H7 infections. A broad clinical spectrum. AnnInternMed. 1988;109:705–712. doi: 10.7326/0003-4819-109-9-705. [DOI] [PubMed] [Google Scholar]
- 3.Underwood AP, Dallman T, Thomson NR, Williams M, Harker K, Perry N, et al. Public health value of next-generation DNA sequencing of enterohemorrhagic Escherichia coli isolates from an outbreak. JClinMicrobiol. 2013;51:232–237. doi: 10.1128/JCM.01696-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perry N, Cheasty T, Dallman T, Launders N, Willshaw G. Application of multilocus variable number tandem repeat analysis to monitor Verocytotoxin-producing Escherichia coli O157 phage type 8 in England and Wales: emergence of a profile associated with a national outbreak. JApplMicrobiol. 2013;115:1052–1058. doi: 10.1111/jam.12303. [DOI] [PubMed] [Google Scholar]
- 5.Ihekweazu C, Carroll K, Adak B, Smith G, Pritchard GC, Gillespie IA, et al. Large outbreak of verocytotoxin-producing Escherichia coli O157 infection in visitors to a petting farm in South East England, 2009. EpidemiolInfect. 2012;140:1400–1413. doi: 10.1017/S0950268811002111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrne L, Elson R, Dallman TJ, Perry N, Ashton P, Wain J, et al. Evaluating the use of multilocus variable number tandem repeat analysis of Shiga toxin-producing Escherichia coli O157 as a routine public health tool in England. PLoSOne. 2014;9:e85901. doi: 10.1371/journal.pone.0085901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brockhurst MA, Koskella B. Experimental coevolution of species interactions. Trends EcolEvol. 2013;28:367–375. doi: 10.1016/j.tree.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 8.Hoey DE, Currie C, Else RW, Nutikka A, Lingwood CA, Gally DL, et al. Expression of receptors for verotoxin 1 from Escherichia coli O157 on bovine intestinal epithelium. J Med Microbiol. 2002;51:143–149. doi: 10.1099/0022-1317-51-2-143. [DOI] [PubMed] [Google Scholar]
- 9.Ohnishi M, Kurokawa K, Hayashi T. Diversification of Escherichia coli genomes: are bacteriophages the major contributors? Trends Microbiol. 2001;9:481–485. doi: 10.1016/S0966-842X(01)02173-4. [DOI] [PubMed] [Google Scholar]
- 10.Scholl D, Rogers S, Adhya S, Merril CR. Bacteriophage K1-5 encodes two different tail fiber proteins, allowing it to infect and replicate on both K1 and K5 strains of Escherichia coli. JVirol. 2001;75:2509–2515. doi: 10.1128/JVI.75.6.2509-2515.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahmed R, Bopp C, Borczyk A, Kasatiya S. Phage-typing scheme for Escherichia coli O157:H7. J Infect Dis. 1987;155:806–809. doi: 10.1093/infdis/155.4.806. [DOI] [PubMed] [Google Scholar]
- 12.Weitz JS, Poisot T, Meyer JR, Flores CO, Valverde S, Sullivan MB, et al. Phage-bacteria infection networks. Trends Microbiol. 2013;21:82–91. doi: 10.1016/j.tim.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Hausmann R. Bacteriophage T7 genetics. Curr Top Microbiol Immunol. 1976;75:77–110. doi: 10.1007/978-3-642-66530-1_3. [DOI] [PubMed] [Google Scholar]
- 14.Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, Ruger W. Bacteriophage T4 genome. MicrobiolMolBiol Rev. 2003;67:86–156. doi: 10.1128/MMBR.67.1.86-156.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kropinski AM, Lingohr EJ, Moyles DM, Chibeu A, Mazzocco A, Franklin K, et al. Escherichia coli O157:H7 typing phage V7 is a T4-like virus. JVirol. 2012;86:10246. doi: 10.1128/JVI.01642-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kropinski AM, Waddell T, Meng J, Franklin K, Ackermann HW, Ahmed R, et al. The host-range, genomics and proteomics of Escherichia coli O157:H7 bacteriophage rV5. VirolJ. 2013;10:76. doi: 10.1186/1743-422X-10-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perry LL, SanMiguel P, Minocha U, Terekhov AI, Shroyer ML, Farris LA, et al. Sequence analysis of Escherichia coli O157:H7 bacteriophage PhiV10 and identification of a phage-encoded immunity protein that modifies the O157 antigen. FEMS MicrobiolLett. 2009;292:182–186. doi: 10.1111/j.1574-6968.2009.01511.x. [DOI] [PubMed] [Google Scholar]
- 18.Santos MA. An improved method for the small scale preparation of bacteriophage DNA based on phage precipitation by zinc chloride. Nucleic Acids Res. 1991;19:5442. doi: 10.1093/nar/19.19.5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014. doi:10.1093/bioinformatics/btu153First published online: March 18, 2014. [DOI] [PubMed]
- 21.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. JComputBiol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCune B, Grace JB. Analysis of ecological communities. Gleneden Beach, Ore: MjM Software; 2002. [Google Scholar]
- 23.Liu X, Murata T. An efficient algortihm for optimizing bipartite modularity in bipartite networks. JACIII. 2010;14:408–415. [Google Scholar]
- 24.Beckett, SJ. A weighted modularity algorithm for bipartite networks. figshare. 2014. http://dx.doi.org/10.6084/m9.figshare.999114.
- 25.Beckett SJ, Boulton CA, Williams HT. FALCON: a software package for analysis of nestedness in bipartite networks. F1000Res. 2014;3:185. doi: 10.12688/f1000research.4831.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Almeida-Neto M, Guimaraes P, Guimaraes PR, Jr, Loyola RD, Ulrich W. A consistent metric for nestedness analysis in ecological systems: reconciling concept and measurement. Oikos. 2008;117:1227–1239. doi: 10.1111/j.0030-1299.2008.16644.x. [DOI] [Google Scholar]
- 27.Atmar W, Patterson BD. The measure of order and disorder in the distribution of species in fragmented habitat. Oecologia. 1993;96:373–382. doi: 10.1007/BF00317508. [DOI] [PubMed] [Google Scholar]
- 28.Oksanen, J, Blanchet, FG, Kindt, R, Legendre, P, Minchin, PR, O’Hara, RB, Simpson, GL, Solymos, P, Stevens, MHH, Wagner H. vegan: community ecology package. R package version 2.0-10. 2013. http://CRAN.R-project.org/package=vegan.
- 29.Brualdi RA, Sanderson JG. Nested species subsets, gaps, and discrepancy. Oecologia. 1999;119:256–264. doi: 10.1007/s004420050784. [DOI] [PubMed] [Google Scholar]
- 30.Bascompte J, Patterson BD. The nested assembly of plant-animal mutualistic networks. Proc Natl Acad Sci U S A. 1993;100:9383–0387. doi: 10.1073/pnas.1633576100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMCGenomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sullivan MJ, Petty NK, Beatson SA. Easyfig: a genome comparison visualizer. Bioinformatics. 2011;27:1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matthews L, Reeve R, Gally DL, Low JC, Woolhouse ME, McAteer SP, et al. Predicting the public health benefit of vaccinating cattle against Escherichia coli O157. Proc Natl Acad Sci U S A. 2013;110:16265–16270. doi: 10.1073/pnas.1304978110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leiman PG, Battisti AJ, Bowman VD, Stummeyer K, Muhlenhoff M, Gerardy-Schahn R, et al. The structures of bacteriophages K1E and K1-5 explain processive degradation of polysaccharide capsules and evolution of new host specificities. JMolBiol. 2007;371:836–849. doi: 10.1016/j.jmb.2007.05.083. [DOI] [PubMed] [Google Scholar]
- 35.Hattman S, Fukasawa T. Host-induced modification of t-even phages due to defective glucosylation of their DNA. Proc Natl Acad Sci U S A. 1963;50:297–300. doi: 10.1073/pnas.50.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Springman R, Badgett MR, Molineux IJ, Bull JJ. Gene order constrains adaptation in bacteriophage T7. Virology. 2005;341:141–152. doi: 10.1016/j.virol.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 37.Beckett SJ, Williams HT. Coevolutionary diversification creates nested-modular structure in phage-bacteria interaction networks. Interface Focus. 2013;3:20130033. doi: 10.1098/rsfs.2013.0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flores CO, Meyer JR, Valverde S, Farr L, Weitz JS. Statistical structure of host-phage interactions. Proc Natl Acad Sci U S A. 2011;108:E288–E297. doi: 10.1073/pnas.1101595108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodriguez-Valera F, Martin-Cuadrado AB, Rodriguez-Brito B, Pasic L, Thingstad TF, Rohwer F, et al. Explaining microbial population genomics through phage predation. NatRevMicrobiol. 2009;7:828–836. doi: 10.1038/nrmicro2235. [DOI] [PubMed] [Google Scholar]
- 40.Mora A, Blanco M, Blanco JE, Alonso MP, Dhabi G, Thomson-Carter F, et al. Phage types and genotypes of shiga toxin-producing Escherichia coli O157:H7 isolates from humans and animals in spain: identification and characterization of two predominating phage types (PT2 and PT8) JClinMicrobiol. 2004;42:4007–4015. doi: 10.1128/JCM.42.9.4007-4015.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baggesen DL, Sorensen G, Nielsen EM, Wegener HC. Phage typing of Salmonella Typhimurium - is it still a useful tool for surveillance and outbreak investigation? Euro Surveill. 2010;15:19471. [PubMed] [Google Scholar]