

Abstract
Under the remit of the Ministerial Industry Strategy Group (MISG), the Association of the British Pharmaceutical Industry (ABPI) and Medicines and Healthcare products Regulatory Agency (MHRA) hosted a meeting to explore physiologically based pharmacokinetic modeling and simulation, focusing on the clinical component of regulatory applications. The meeting took place on 30 June 2014 with international representatives from industry, academia, and regulatory agencies. Discussion topics were selected to be complementary to those discussed at an earlier US Food and Drug Administration (FDA) meeting. This report summarizes the meeting outcomes, focusing on the European regulatory perspective.
INDUSTRY VIEWPOINTS
The current application of physiologically based pharmacokinetic (PBPK) modeling in drug development was summarized by three industry representatives (links to all presentations are provided in Supplementary Table 1 and a comprehensive meeting report is available online1).
Jan Snoeys (Janssen) provided an overview of PBPK in drug development and two additional presenters gave specific examples of the use of PBPK in industry. In the first presentation two approaches to PBPK modeling were described: models using only in vitro/in silico data (bottom up) and those combining in vitro/in silico data with observed PK data. The importance of validated in vitro assays was stressed, ideally with adoption of common assay methodology across different laboratories. Also highlighted was that the process of model optimization using sensitivity analysis and parameter optimization is integral to development of robust PBPK models and must be conducted recognizing information gaps and consequences on model utilization. Often optimization of several independent factors may lead to improved simulated PK profiles. In such circumstances it is of value to test such hypotheses with experimental data. In system model verification, use of prospective simulations and large compound datasets could help identify model limitations. Drug-independent system components and virtual populations may be verified with compounds representing a range of physicochemical properties.
Hannah Jones (Pfizer) provided two examples of PBPK modeling. Differences in UGT-mediated clearance and UGT2B15 polymorphism between Japanese and Caucasian subjects were considered in the model of an investigational drug. Resulting simulations supported regulatory interactions aimed at avoiding additional clinical studies in Japanese subjects. In the second example, a PBPK model included drug disposition mediated by organic anion-transporting polypeptides (OATP). Literature data of seven compounds and a scaling method were employed to develop the model. The model was valuable in prospectively predicting human PK for four investigational drugs tested to be OATP substrates in vitro.
Patrice Larger (Novartis) described PK predictions using two PBPK pregnancy models: one with limited pregnancy factors and a second expanded with additional factors such as fetal–placental volume and blood flow. Model performance was assessed using four reference compounds. Specific pregnancy factors in the second model did not significantly impact the predicted pharmacokinetic profiles for the reference compounds. A PBPK model was also described to facilitate dose selection in a pediatric trial. The model incorporated PK data from adults, child physiology, and UGT ontogeny. The model was used to explore dosing options for an investigational drug in pediatrics.
EUROPEAN REGULATORY VIEWPOINT
PBPK is viewed as of great potential value to support benefit–risk evaluations, providing a mechanistic basis for extrapolation beyond the clinical trial population, reducing uncertainty, and enabling better labeling around drug–drug interactions (DDIs) and in special populations (e.g., elderly, pediatric, etc). PBPK is increasingly submitted as part of marketing authorization applications (MAAs). To date these have comprised mainly DDI applications and have been included in a number of European Summaries of Product Characteristics (SmPC) and/or European Public Assessment Reports (e.g., Halavan,2 Jakavi,3 and Olysio4). So far, few submissions with application to pediatric dose selection have been received.
“PBPK-thinking” in drug development is encouraged, as it leads to a mechanistic understanding of the processes involved in drug disposition. The modeling approach helps to identify gaps in understanding of absorption, distribution, metabolism, and excretion (ADME), informs improved study designs, complements other modeling and simulation (M&S) approaches and builds confidence for extrapolation. When systematically applied over many drugs, this will support continued development and validation of system models. The continued evolution of PBPK is key to facilitating greater confidence in extrapolation (e.g., pediatric, elderly, DDI, renal impairment, etc.), thereby reducing experimental data requirements.
This meeting was viewed as the beginning of an important dialog towards defining the standards to facilitate a greater role of PBPK in European regulatory decision making.
There is now sufficient experience within the European regulatory system to support development of regulatory standards, guidelines, and practice. A PBPK concept paper has been published5 which will lead to a specific European guideline on qualification and reporting of PBPK modeling and analysis. The guideline will be informed by comments received during the consultation period and discussions reported here and the earlier US Food and Drug Administration (FDA) workshop.6
DISCUSSION TOPICS
The steps involved in PBPK model development relevant for regulatory applications within clinical pharmacology are illustrated in Figure 1a, which formed the basis for selection of the four discussion topics:
Drug-specific input parameters
Clinical data for verification of drug-specific input parameters
Qualification of system models
Reporting
Figure 1.
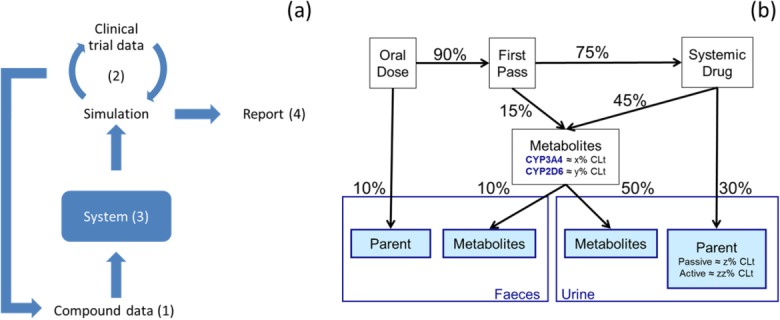
(a) Schematic illustrating overlay of discussion topics implicit in best practice during PBPK model development and reporting. (b) Example quantitative mass balance diagram following oral and intravenous dosing.
Prior to the meeting participants were involved in defining key questions for discussion. The recommendations and actions from each session are shown in Table 1.
Table 1.
Key points and recommendations from discussion sessions
| Key points | Recommendations |
|---|---|
| Topic 1: What are appropriate data standards for drug input data for PBPK models? | |
| Not all parameters are equal: critical values depend on the physicochemical properties of the molecule and on the application or interest (DDI, specific populations such as pediatrics, biopharmaceutics). Critical parameters are those that have an impact on addressing clinically relevant questions. |
|
| Methodologies are not consistent across companies or even within some companies. While considered highly desirable, the ideal of a standard in vitro methodology across the industry was not thought to be a realistic aim. Rather, a full understanding and description of methodology with adoption of common reference standards to be utilized across companies should be encouraged. |
|
| There is mixed acceptance around scaling factors. Physiological scaling factors can be easily accepted, e.g., MPPGL (mg of protein per gram of liver), and where a consensus exists would not be expected to be altered. Empirical scaling factors, derived to account for a lack of direct extrapolation from in vitro to in vivo, are likely to be dependent on the in vitro methodology utilized and on the compound, and will be company- or lab-specific. |
|
| Depending on the stage of development, in silico values may be useful, but measured values are often preferred. The exception to this is where there is evidence that in silico values are more accurate (e.g., log D for highly lipophilic compounds). | |
| It is important to try to understand when poor simulations result from inadequate in vitro data or are due to incomplete understanding of in vivo drug disposition. | |
| Suggested decision metric should be “Does it modulate dose requirements?” | |
| Topic 2: Verification of drug specific input parameters | |
| During drug development, it is best practice to have a quantitative understanding of the contribution of the various pathways involved in a drug's ADME. |
|
| Given the discussion around sensitivity analysis, it would appear that a general guidance could be developed around the choice of parameters and range of values included in sensitivity analysis. |
|
| Although the focus of the discussion was on sensitivity analysis to address input parameter uncertainty, attention should also be paid to the experimental systems themselves and the possibility of improving confidence in key input parameters. |
|
| There are a number of gaps to be filled before PBPK models of enzyme induction will be viewed as sufficiently reliable to support waiver of in vivo studies for a potential perpetrator within the European regulatory system. Further development in this area would be welcomed. | |
| Topic 3: Best practice for qualification of system models | |
| It was acknowledged that system model qualification is an area that has not been extensively discussed within the PBPK community and that standards are not currently agreed. Further progress to establish best practice is needed. |
|
| There is some mismatch between the terminology used within PBPK and computational science communities: “qualification” or “verification” vs. “validation.” |
|
| The meeting identified that each software provider has developed internal systems to evaluate and track the reliability of their system models and associated libraries. | |
| In the field of PBPK, open source software published with the training datasets utilized presents a challenge for software providers wishing to protect their intellectual property. Other solutions, such as open source validation datasets against which commercially available software are validated (for a specific condition of use), could potentially serve the same purpose. | |
| Topic 4: What should a PBPK report look like? | |
| The model development “story” should be presented but ought to be fit for purpose and sufficiently detailed to facilitate regulatory review without being overly detailed. |
|
| For regulatory submissions it is important to contextualize the purpose of the PBPK model. |
|
| The acceptability of the PBPK model in terms of targets for successful prediction of clinical data should be interpreted within the context of therapeutic index. |
|
| Development of a PBPK model during a drug development program can be helpful in promoting a full and integrated understanding of a drug's quantitative disposition. Alongside this overall objective, it is helpful to develop specific plans for the application of PBPK modeling to clinical pharmacology programs. | |
| Whether PBPK modeling is used or not, dose adjustment for DDIs or other extrinsic and intrinsic factors should be framed within the context of the therapeutic index. | |
The objective of Session 1 was to help establish best practice for defining drug-specific input parameters. Key parameters were identified prior to the meeting (see link in Supplementary Table 1). Questions discussed were: (1) model improvement (optimization of parameters and use of scaling factors), (2) sensitivity testing of parameters, and (3) methodology to define inputs. It was agreed that optimization is routinely performed but should be well justified and that scaling factors vary in plausibility; some being physiologically based but others more empirical, and may vary in size between analyses. Sensitivity testing of model predictions to changes in model parameters was considered to be essential, with the suggestion that the importance of a parameter and the range of sensitivity analysis performed be defined depending on the physiochemical properties of the drug and/or the experimental systems under consideration. It was noted that in vitro methodology to determine input values often varies across and sometimes within companies. Important points identified for follow up were the need for a full understanding and description of methodology to measure input parameters, preferably with adoption of common reference standards across companies, and a need for greater clarification and justification of empirical scaling factors.
In Session 2, verification of PBPK models was discussed and best practice in the use of ADME data (in vitro and in vivo) in the verification of drug-specific input parameters was explored. An example of a useful quantitative mass balance diagram for a compound administered both orally and intravenously is illustrated in Figure 1b. Based on the feedback, the following points were identified as important to support a quantitative mass balance diagram: (1) What are the clearance pathways and their quantitative contributions? (2) What is the extent of absorption of the drug and is parent drug in feces a result of lack of absorption (fa) or biliary/intestinal excretion? (3) When oral bioavailability is low, what are the contributions of intestinal and hepatic first pass loss? (4) What is the rate-limiting step in hepatic drug clearance (basolateral uptake or metabolism/biliary secretion)? In general, attendees agreed that intravenous data can be important in PBPK model development (depending on the pathways). Sensitivity analysis for inhibition constant (Ki), fraction unbound in plasma, microsomes and gut (fup, fumic, fugut, depending on the gastrointestinal model utilized), and permeability is considered crucial when simulating the impact of a possible CYP3A perpetrator on the PK of a victim drug. Further simulations for sensitivity evaluation should address confidence in liver or intrahepatic concentration and intraenterocyte concentration when inhibition occurs intracellularly. PBPK modeling of enzyme induction was also discussed: it was highlighted that currently there is insufficient confidence within the European regulatory system to support waiver of studies based on PBPK. An important point identified for follow up was development of a statement, with a supporting rationale, which explains the expectation of intravenous data (unless its absence can be justified) as a key element in the quantitative mechanistic understanding of drug disposition.
The system qualification discussion (Session 3) focused on commercially available software, which is utilized in the vast majority of regulatory applications of PBPK. The three main companies providing PBPK software used in clinical pharmacology regulatory submissions to date (GastroPlus, PK-Sim, and SimCYP) provided short summaries of their approach to system model qualification. The companies generally appeared to take similar approaches. The system validation consists of tracking and documenting (user manual, publication) changes and (re)validating the software using former or new test cases. The libraries' qualification consists of continuously updating the databases with emerging knowledge from the literature or provided by (pharmaceutical) companies. Additional elements presented by the companies included: version stamp for model output, training datasets with tutorials available to users, validation datasets that remain with the software to allow verification of continued system model reliability, and transparency regarding source of data and assumptions within the system. Overall it was concluded that each software provider has developed internal systems to evaluate and track the reliability of their system models and associated libraries. Further discussion focused on the framework proposed for assessment of systems pharmacology models7 and referred to the US National Academies framework on Validation, Verification, and Uncertainty Quantitation.8 In summary, the framework recommends that the software should be open access, “bug-free,” with no copying errors. In addition, two types of data should be clearly differentiated: training data published with the model and novel and varied validation data. It was acknowledged that in the field of PBPK, open source software published with training datasets presents a challenge for software providers wishing to protect their intellectual property. Other solutions, such as open source validation datasets (for a specific condition of use), could potentially serve the same purpose. Importantly, there is some mismatch between the terminology used within PBPK and computational science communities: “qualification” or “verification” vs. “validation.” Also acknowledged was that system model qualification is an area that has not been extensively discussed within the PBPK community and that standards are not currently agreed, and that progress to establish best practice is needed.
Session 4 discussed best practice in PBPK reporting focusing on what information is needed in reports for regulatory review. It was considered that reports should clearly address the regulatory question, the clinical context, and whether the simulations will lead to changes in dose recommendations. There was general agreement that including background information (clinical pharmacology, purpose of the modeling effort, history of PBPK model development, etc.) is helpful and that the level of detail should be fit for purpose. It was also pointed out that background information on clinical pharmacology is of limited value without integration across studies. Uncertainty, sensitivity analysis, and plausibility of the assumptions should always be presented and discussed. Reports of simulations used in support of a waiver of an in vivo study (such as a DDI study) require particular care to integrate the simulations with the rest of the clinical data, including a discussion of the implications for drug dosing, taking into account the PKPD profile of the drug.
Acknowledgments
The authors thank the organizers and presenters: Eva Gil Berglund (MPA), Michael Bolger (Simulations Plus), Liberty Dixon (ABPI), Rob Hemmings (MHRA), Hannah Jones (Pfizer), Patrice Larger (Novartis), Louise Leong (ABPI), Christoph Niederalt (Bayer Technology Services), Munir Pirmohamed (meeting chair), Malcolm Rowland (University of Manchester), Masanobu Sato (PMDA), Vikram Sinha (FDA), Jan Snoeys (Janssen), and Karen Yeo (Certara). The views expressed in this article are the personal views of the authors (SC, AN, and TS) and may not be understood or quoted as being made on behalf of or reflecting the position of the European Medicines Agency or any National Competent Authority.
Conflict of Interest
The authors declare no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
Supporting Information
References
- Shepard T, Scott G, Cole S, Nordmark A, Bouzom F, Rowland M. Zhao P , & Meeting report. MISG New Technologies Forum on Physiologically-based Pharmacokinetic (PBPK) Modeling and Simulation. < http://www.mhra.gov.uk/home/groups/comms-ic/documents/websiteresources/con457608.pdf >.
- Halavan EPAR. < http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/002084/WC500170478.pdf >.
- Jakavi EPAR and SmPC < http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002464/WC500133226.pdf http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002464/WC500133223.pdf >.
- Olysio EPAR < http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002777/WC500167870.pdf >.
- Concept paper on qualification and reporting of physiologically based pharmacokinetic (PBPK) modeling and analyses. EMA/CHMP/211243/2014.
- Reference to FDA meeting report in same CPT edition.
- Understanding the Potential of Systems Pharmacology. Unpublished paper written by G Mirams (University of Oxford, UK), D Gavaghan (University of Oxford, UK), M Davies (AstraZeneca, UK) for the Medical Research Council.
- National Research Council. Assessing the Reliability of Complex Models: Mathematical and Statistical Foundations of Verification, Validation, and Uncertainty Quantification. . (The National Academies Press, Washington, DC, 2012) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information