

Abstract
Novel allosteric modulators of the dopamine transporter (DAT) have been identified. We have shown previously that SRI-9804 [N-(diphenylmethyl)-2-phenyl-4-quinazolinamine], SRI-20040 [N-(2,2-diphenylethyl)-2-phenyl-4-quinazolinamine], and SRI-20041 [N-(3,3-diphenylpropyl)-2-phenyl-4-quinazolinamine] partially inhibit [125I]RTI-55 ([125I]3β-(4′-iodophenyl)tropan-2β-carboxylic acid methyl ester) binding and [3H]dopamine ([3H]DA) uptake, slow the dissociation rate of [125I]RTI-55 from the DAT, and allosterically modulate d-amphetamine–induced, DAT-mediated DA release. We synthesized and evaluated the activity of >500 analogs of these ligands and report here on 36 selected compounds. Using synaptosomes prepared from rat caudate, we conducted [3H]DA uptake inhibition assays, DAT binding assays with [3H]WIN35428 ([3H]2β-carbomethoxy-3β-(4-fluorophenyl)tropane), and DAT-mediated release assays with either [3H]MPP+ ([3H]1-methyl-4-phenylpyridinium) or [3H]DA. We observed three groups of [3H]DA uptake inhibitors: 1) full-efficacy agents with a one-site fit, 2) full-efficacy agents with a two-site fit, and 3) partial-efficacy agents with a one-site fit—the focus of further studies. These agents partially inhibited DA, serotonin, and norepinephrine uptake, yet were much less potent at inhibiting [3H]WIN35428 binding to the DAT. For example, SRI-29574 [N-(2,2-diphenylethyl)-2-(imidazo[1,2-a]pyridin-6-yl)quinazolin-4-amine] partially inhibited DAT uptake, with an IC50 = 2.3 ± 0.4 nM, without affecting binding to the DAT. These agents did not alter DAT-mediated release of [3H]MPP+ in the absence or presence of 100 nM d-amphetamine. SRI-29574 had no significant effect on the d-amphetamine EC50 or Emax value for DAT-mediated release of [3H]MPP+. These studies demonstrate the existence of potent DAT ligands that partially block [3H]DA uptake, without affecting DAT binding or d-amphetamine–induced [3H]MPP+ release. These compounds may prove to be useful probes of biogenic amine transporter function as well as novel therapeutics.
Introduction
The biogenic amine transporters (BATs) are members of the neurotransmitter/sodium symporter (NSS) protein superfamily. These membrane-spanning proteins cotransport neurotransmitters and Na+ ions from the extracellular space into the cytoplasm, utilizing the potential energy inherent to the inwardly directed transmembrane Na+ gradient (Gether et al., 2006; Forrest et al., 2011). Under normal circumstances, BATs tightly control the extracellular concentrations of previously released biogenic amine transmitters [dopamine (DA), norepinephrine (NE), and serotonin (5-HT)] by translocating these molecules back into the nerve terminal, a process termed “uptake.” Dopaminergic signaling is involved in several aspects of brain function such as cognition, movement, motivation, affect, behavioral reinforcement, and economic analysis (reward prediction and valuation) (Greengard, 2001; Montague and Berns, 2002; Salamone et al., 2009). Perturbation of dopamine transporter (DAT) function is implicated in a number of neuropsychiatric disorders: attention-deficit/hyperactivity disorder, Parkinson’s disease, depression, anhedonia, and addictive/compulsive disorders (Gainetdinov and Caron, 2003; Felten et al., 2011; Kurian et al., 2011). Moreover, the DAT is a target of several important medications and a number of recreational drugs (Reith et al., 2015; Sitte and Freissmuth, 2015). For example, clinically used DAT ligands include psychostimulants (e.g., d-amphetamine, methylphenidate, and modafinil), antidepressants (e.g., bupropion), and certain anorectics [e.g., phendimetrazine, a prodrug that is converted to the DAT ligand phenmetrazine in vivo (Rothman et al., 2002)]. Interaction with the DAT also contributes to the powerful reinforcing and locomotor stimulant effects of cocaine, one of the most prominent drugs of addiction (reviewed in Gainetdinov and Caron, 2003; and Schmitt and Reith, 2010).
Drugs that interact with the BATs are typically classified into two categories: 1) ligands that bind to the BAT but are not transported (i.e., uptake inhibitors), and 2) ligands that bind to the BAT and are translocated through the transporter into the intracellular medium (i.e., substrates). Cocaine and the widely used antidepressant fluoxetine are examples of uptake inhibitors. A variety of psychoactive drugs are substrates for the BATs, and these compounds are often called “releasers” because they induce the release of neurotransmitters by reversing the normal direction of flux through the transporter. Using the DAT as an example, the DA release process occurs via a mechanism classically called “carrier-mediated exchange.” According to this mechanism, the inward transport of an exogenous substrate, like amphetamine, releases cytoplasmic DA via reverse transport. Reverse transport by the DAT depends upon increased concentration of intracellular Na+ (Khoshbouei et al., 2003), which accompanies translocation of amphetamine-like substrates, thereby promoting DA efflux (Sitte et al., 1998). The molecular basis for this reverse transport process is complex and still under investigation (see Schmitt et al., 2013, for a review).
With regard to DAT ligands, one simple hypothesis is that all uptake inhibitors will interact with the DAT in a similar manner, and therefore produce similar in vitro and behavioral effects. By extension, all DAT substrates will also interact with the DAT in a similar manner to produce similar in vitro and behavioral effects. Recent studies, reviewed by Schmitt et al. (2013), are not compatible with this hypothesis. For example, atypical DA uptake inhibitors, based on the benztropine structure and developed primarily by the Katz group (Tanda et al., 2009), block DA uptake but do not produce the expected cocaine-like behavioral effects. The efforts of our laboratory have entailed the screening of an extensive chemical library to identify potential BAT ligands. These efforts identified three quinazolinamine DAT allosteric modulators: SRI-20040 [N-(2,2-diphenylethyl)-2-phenyl-4-quinazolinamine], SRI-20041 [N-(3,3-diphenylpropyl)-2-phenyl-4-quinazolinamine], and SRI-9804 [N-(diphenylmethyl)-2-phenyl-4-quinazolinamine] (see Fig. 1, A and B; and Supplemental Fig. 1 for structures) (Pariser et al., 2008). A key finding with these compounds is that they are partial inhibitors of both DA uptake and [125I]RTI-55 ([125I]3β-(4′-iodophenyl)tropan-2β-carboxylic acid methyl ester) binding to the DAT. Unlike cocaine, which can inhibit DA uptake and DAT binding with 100% efficacy, these compounds display maximal response (Emax) values ranging from 40–60%. These allosteric modulators increase the KD value and decrease the Bmax value for [125I]RTI-55 binding to DAT, and also slow the dissociation rate of bound [125I]RTI-55, further suggesting that these ligands do not compete for the same binding site as phenyltropane ligands (Pariser et al., 2008). Perhaps most importantly, while two of the quinazolinamine modulators (SRI-9804 and SRI-20040) partially inhibit both uptake of [3H]DA (forward transport) and DAT-mediated release of preloaded [3H]DA (reverse transport), a third compound (SRI-20041) inhibits substrate uptake, without appreciable effects on efflux (Rothman et al., 2009). This latter compound appeared to be the first DAT ligand to differentially affect substrate uptake versus transmitter release, suggesting that it may be possible to design compounds that selectively influence a single component of the NSS translocation cycle.
Fig. 1.
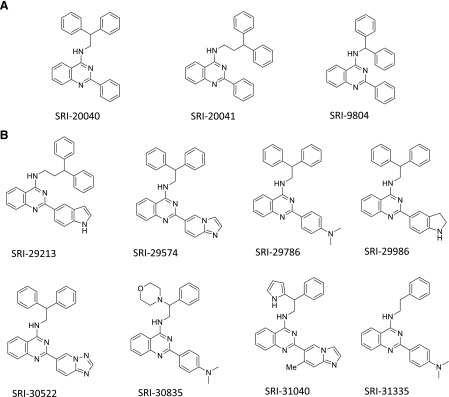
Structure of test compounds (A and B). Note that SRI-20040, SRI-20041, and SRI-9804 were formerly designated SoRI-20040, SoRI-20041, SoRI-9804, respectively.
These aforementioned first-generation allosteric modulators were limited by their weak potency (low-micromolar range) in affecting DAT function. This low potency made in vivo study of their possible behavioral effects difficult. We therefore continued this effort by additional structure-activity studies, which are still ongoing, to develop allosteric ligands with greater potency. At the current time, >500 analogs have been synthesized and evaluated in various in vitro assays (see Materials and Methods) for possible allosteric modulation of the DAT. We report here the initial results with 36 second-generation compounds, some of which allosterically modulate the DAT with nanomolar potency.
Materials and Methods
Animals.
Male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) weighing 300–400 g were used as subjects in these experiments. Rats were housed in standard conditions (12-hour light/dark cycle) with food and water freely available. Animals were maintained in facilities fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, and experiments were performed in accordance with the Institutional Animal Care and Use Committee of the National Institute on Drug Abuse Intramural Research Program.
Neurotransmitter Uptake Assays.
Uptake inhibition assays for the DAT and transporters for norepinephrine (NET) and serotonin (SERT) were conducted in rat brain synaptosomes as described elsewhere with minor modifications (Rothman et al., 2001). Freshly removed caudate (DAT), or whole brain minus cerebellum and caudate (NET and SERT), was homogenized in 10% ice-cold sucrose with 12 strokes of a handheld Potter-Elvehjem homogenizer (Thomas Scientific, Swedesboro, NJ) followed by centrifugation at 1000g for 10 minutes. The supernatants were saved on ice and used immediately. Transporter activity at the DAT, NET, and SERT was assessed using 5 nM [3H]DA, 10 nM [3H]NE, and 5 nM [3H]5-HT, respectively. The assay buffer was Krebs phosphate buffer (pH 7.4) containing 126 mM NaCl, 2.4 mM KCl, 0.5 mM KH2PO4, 1.1 mM CaCl2, 0.83 mM MgCl2, 0.5 mM Na2SO4, 11.1 mM glucose, 13.7 mM Na2HPO4, 1 mg/ml ascorbic acid, and 50 μM pargyline. For NET uptake assays, 50 nM GBR12935 [1-(2-diphenylmethoxyethyl)-4-(3-phenylpropyl)piperazine] was added to the sucrose solution and assay buffer to prevent uptake of [3H]NE by DAT. For SERT uptake assays, the sucrose solution and assay buffer contained 100 nM nomifensine and 50 nM GBR12935 to prevent uptake of [3H]5-HT by NET and DAT, respectively. Uptake inhibition assays were conducted at 25°C (DAT and SERT) or 37°C (NET), and were initiated by adding 100 μl of tissue to 900 μl of assay buffer containing test drug and 3H-neurotransmitter. Test drugs were diluted in assay buffer containing 1 mg/ml of bovine serum albumin (BSA) prior to addition. Nonspecific uptake was measured by incubating in the presence of 1 μM indatraline. The reactions were stopped after 15 minutes (DAT), 10 minutes (NET), or 30 minutes (SERT) by rapid vacuum filtration with a Brandel cell harvester (Brandel, Gaithersburg, MD) over Whatman GF/B filters presoaked in wash buffer maintained at 25°C [10 mM Tris-HCl (pH 7.4), 150 mM NaCl]. Filters were rinsed with 6 ml of wash buffer, and retained tritium was measured with a MicroBeta liquid scintillation counter (PerkinElmer, Waltham, MA) after overnight extraction in 0.6 ml of liquid scintillation cocktail (Cytoscint; ICN Biomedicals, Santa Ana, CA).
Neurotransmitter Release Assays.
DAT-mediated release assays were carried out as previously described with minor modifications (Rothman et al., 2003). Synaptosomes were prepared from rat caudate tissue as described for uptake inhibition assays, except that the sucrose solution contained 1 μM reserpine to block vesicular uptake of substrates. Synaptosomal preparations were incubated to steady state with 9 nM [3H]MPP+ ([3H]1-methyl-4-phenylpyridinium) (60 minutes, 25°C) in uptake assay buffer containing 1 μM reserpine to block vesicular uptake of substrates and 100 nM citalopram and 100 nM desipramine to block uptake of [3H]MPP+ by SERT and NET. Subsequently, 850 μl of synaptosomes preloaded with [3H]MPP+ were added to polystyrene test tubes that contained 150 μl of test drug in assay buffer plus 1 mg/ml BSA. After 30 minutes at 25°C, the release reaction was terminated by rapid vacuum filtration as described for uptake assays. Nonspecific values were measured by incubations in the presence of 10 μM tyramine. The retained tritium was measured as described for uptake assays.
[3H]WIN35428 Binding Assays.
The ability of test drugs to inhibit [3H]WIN35428 ([3H]2β-carbomethoxy-3β-(4-fluorophenyl)tropane) binding to DAT in rat caudate membranes was assessed as follows. For each experiment, caudates from four rat brains were suspended in 15 ml of ice-cold assay buffer (50 mM sodium phosphate, pH 7.4) and homogenized using a polytron (setting 6, 20 seconds). The homogenate was centrifuged at 30,000g for 10 minutes at 4°C. The pellet was resuspended with vigorous vortexing in 15 ml of fresh ice-cold assay buffer, and the centrifugation was repeated. The pellet was resuspended in 15 ml of fresh ice-cold assay buffer with vigorous vortexing followed by six strokes with a glass-on-glass handheld homogenizer and was diluted to a final volume of 235 ml in ice-cold assay buffer. [3H]WIN35428 was diluted to 10 nM in assay buffer that contained 25 μg/ml chymostatin, 25 μg/ml leupeptin, 0.1 mM EDTA, and 0.1 mM EGTA. Each assay tube contained 0.75 ml of membrane preparation, 0.15 ml of test drug diluted in assay buffer containing 1 mg/ml BSA, and 0.1 ml of [3H]WIN35428 preparation (final concentration of 1 nM). Assays were initiated by the addition of membranes and were terminated after 2 hours at 25°C by rapid vacuum filtration as described above. Retained tritium was measured as described above.
Screening Methods.
All of the test compounds were synthesized at Southern Research Institute. Details of the chemical synthesis will be published elsewhere. Test compounds were evaluated in a stepwise manner. First, 8-point dose-response curves were generated for each compound (1–20,000 nM) in the [3H]DA uptake assay. The data from three experiments were pooled and fit to a dose-response curve equation (using KaleidaGraph; Synergy Software, Reading, PA) to yield an Emax and IC50 value. Next, because we were interested in characterizing potent partial DAT inhibitors, compounds were selected for further evaluation only if they had an IC50 ≤ 20 nM (high potency) and an Emax ≤ 70% (partial efficacy). Compounds that displayed properties of full-efficacy inhibitors of [3H]DA uptake were not tested further. A subset of potent partial inhibitors was then tested for dose-response effects in the [3H]NE and [3H]5-HT uptake inhibition assays. In addition, these same compounds were tested for their ability to alter DAT-mediated release of [3H]MPP+ in the absence and presence of 100 nM d-amphetamine. Selected compounds were also tested for their ability to inhibit [3H]WIN35428 binding to rat caudate DAT.
Data Analysis and Statistics.
For release experiments, dose-response curves were generated using eight concentrations of test drug. To describe the method for calculating the release dose-response curves, the following definitions are necessary: total binding (TB) = cpm in the absence of any drug; nonspecific binding (NS) = cpm in the presence of 10 μM tyramine; maximal release (MR) = TB−NS; specific release (SR) = (cpm in the presence of drug)−NS; and % MAX release = 100−SR/MR*100.
The data from three experiments, expressed as % MAX Release, were then fit to a dose-response curve equation,
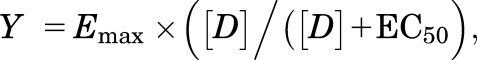 |
for the best-fit estimates of the Emax and EC50 using either KaleidaGraph version 3.6.4 or MLAB-PC (Nightingale et al., 2005). In some cases, dose-response curves were fit to a two-component equation:
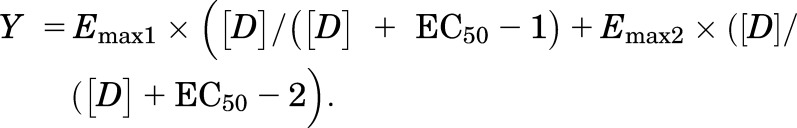 |
Statistical significance of the one-site versus two-site fits was based on F test results. In “shift” experiments, a substrate dose-response curve was generated in the absence and presence of a test drug. Apparent Ke values were calculated according to the equation
where EC50-2 is the EC50 value in the presence of the test drug and EC50-1 is the value in the absence of the uptake inhibitor.
Results
Initial Screen of Compounds.
As noted in Materials and Methods, compounds were first evaluated in the [3H]DA uptake inhibition assay. Some agents acted as full-efficacy [3H]DA uptake inhibitors [for example, see SRI-31335 [2-(4-(dimethylamino)phenyl)-N-phenethylquinazolin-4-amine] (Fig. 2A)]. A large set of agents also acted as partial inhibitors when the dose-response curves were fit to the one-component equation (for example, see SRI-29986 [N-(2,2-diphenylethyl)-2-(indolin-5-yl)quinazolin-4-amine] and SRI-30835 [2-(4-(dimethylamino)phenyl)-N-(2-morpholino-2-phenylethyl)quinazolin-4-amine] in Fig. 2A). However, upon visual inspection, it is clear that whereas the SRI-29986 dose-response curve is well described by a one-component equation, the SRI-30835 dose-response curve is not. Fitting the same three dose-response curves to a two-component equation led to a highly significant improvement in the goodness-of-fit for SRI-30835, but not the other two agents (Fig. 2B; Table 1). These data illustrate that the initial set of compounds binned into three groups of [3H]DA uptake inhibitors: 1) apparent full-efficacy one-component agents, 2) apparent full-efficacy two-component agents, and 3) partial-efficacy agents. We focused further studies on the initial set of 36 partial-efficacy agents. Of interest, preliminary experiments suggest that some of the apparent full-efficacy one-component agents are also allosteric modulators (data not shown).
Fig. 2.

Initial screening of compounds for inhibition of [3H]DA uptake using a one-component (A) or two-component (B) fit. Panel (A) shows that some agents acted as full-efficacy [3H]DA uptake inhibitors (e.g., SRI-31335), whereas others acted as partial inhibitors (e.g., SRI-29986 and SRI-30835), when the dose-response curves were fit to the one-component equation. Panel (B) shows that the SRI-30835 dose-response curve was better described by a two-site fit (also see Table 1) and that SRI-30835 was a full-efficacy agent when fit to a two-component model. We focused further studies on the initial set of 36 partial-efficacy agents well described by a one-component model.
TABLE 1.
[3H]DA uptake inhibition by select SRI compounds: one-component versus two-component fits
Dose-response curves (eight points) reported in Fig. 2 were fit to one- and two-component equations for the best-fit estimates of the IC50 and Emax values (± S.D.).
| Drug | One-Site IC50 | One-Site Emax | Two-Site IC50-1 | Two-Site Emax1 | Two-Site IC50-2 | Two-Site Emax2 | Sum-of-Squares One-Site Fit | Sum-of-Squares Two-Site Fit | F Test |
|---|---|---|---|---|---|---|---|---|---|
| nM | %I | nM | %I | nM | %I | ||||
| SRI-29986 | 18 ± 4 | 75 ± 3 | 18 × 105 ± 1.7 × 105 | 37 × 108 ± 2.7 × 108 | 18 × 105 ± 1 × 105 | 37 × 108 ± 0.7 × 108 | 175 | 175 | 0 |
| SRI-31335 | 156 ± 12 | 100 ± 1.5 | 99 ± 63 | 76 ± 54 | 564 ± 1177 | 27 ± 53 | 28.9 | 19.0 | 1.56 |
| SRI-30835 | 15 ± 7 | 75 ± 5 | 6 ± 0.9 | 57 ± 2 | 4000 ± 1330 | 42 ± 4 | 677 | 14.2 | 140* |
P < 0.001 vs. one-component fit (F test).
Evaluation of Test Agents for Inhibition of DAT, SERT, and NET Uptake and DAT Binding.
Figure 3 illustrates that SRI-29574 [N-(2,2-diphenylethyl)-2-(imidazo[1,2-a]pyridin-6-yl)quinazolin-4-amine] was a partial inhibitor not only of DAT uptake but also of SERT and NET uptake. All agents tested (Table 2) were partial inhibitors of DAT, SERT, and NET uptake, though in general the efficacy was lower at SERT than at NET and DAT. Although many of the test agents had similar IC50 values for BAT uptake inhibition, in general the order of potency was DAT > SERT > NET. Another striking aspect of the data set was that most compounds were ∼3 orders of magnitude less potent in inhibiting [3H]WIN35428 binding to DAT than in blocking uptake of [3H]DA. This is illustrated in Fig. 4A for two compounds. SRI-29574 partially inhibited DAT uptake (IC50 = 2.3 ± 0.4 nM) while being inactive in inhibiting DAT binding. In contrast, SRI-29786 [2-(4-(dimethylamino)phenyl-N-(2,2-diphenylethyl)quinazolin-4-amine] partially inhibited DAT uptake (IC50 = 7.1 ± 2.2 nM) but also inhibited DAT binding, with full efficacy and an IC50 value (1100 ± 10 nM) 155-fold weaker than the IC50 for inhibition of DAT uptake. Overall, only 5 of the 36 compounds were full-efficacy inhibitors of DAT binding, and in most cases the agents were much less potent at DAT binding inhibition than at DAT uptake inhibition. In contrast, the prototypical DAT blockers GBR12935 and cocaine displayed similar potency and efficacy in both assays. There was no significant correlation between the Emax values observed in the DAT uptake and binding assays (Fig. 4B).
Fig. 3.

Inhibition of [3H]DA, [3H]5-HT, and [3H]NE uptake by SRI-29574 in rat brain synaptosomes. SRI-29574 was a partial inhibitor not only of DAT uptake but also of SERT and NET uptake. All agents tested (see Table 2) were partial inhibitors of DAT, SERT, and NET uptake, though in general the efficacy was lower at SERT than at NET and DAT. The data were fit to a one-component dose-response curve equation for the best-fit estimates of the IC50 (± S.D.) and Emax (± S.D.), respectively: [3H]DA uptake (2.3 ± 0.4 nM; 68 ± 2%), [3H]5-HT uptake (23 ± 5 nM; 52 ± 2%), and [3H]NE uptake (52 ± 15 nM; 72 ± 4%). Each data point is the mean ± S.D. of three separate experiments.
TABLE 2.
Summary of results obtained for the 36 partial-efficacy DAT uptake blockers
Dose-response curves for each indicated agent were generated as described in Materials and Methods for DAT, NET, and SERT uptake inhibition and DAT binding. Each value is the mean ± S.D.; n = 3.
| Drug | DAT Uptake IC50 | DAT Uptake Emax | NET Uptake IC50 | NET Uptake Emax | SERT Uptake IC50 | SERT Uptake Emax | DAT Binding IC50 | DAT Binding Emax | 5-HT/DA IC50 (Uptake) | NE/DA IC50 (Uptake) |
|---|---|---|---|---|---|---|---|---|---|---|
| nM | % | nM | % | nM | % | μM | % | |||
| SRI-29070 | 174 ± 58 | 66 ± 4 | 1740 ± 1150 | 64 ± 10 | 699 ± 164 | 48 ± 3 | 1.8 ± 0.4 | 71 ± 4 | 4 | 10 |
| SRI-29072 | 212 ± 49 | 71 ± 3 | 5850 ± 1746 | 62 ± 5 | 6382 ± 2636 | 56 ± 9 | 2.7 ± 0.3 | 77 ± 2 | 30 | 28 |
| SRI-29153 | 20 ± 1 | 73 ± 1 | 181 ± 46 | 73 ± 4 | 37 ± 18 | 55 ± 5 | 1.7 ± 0.2 | 76 ± 2 | 1.9 | 9 |
| SRI-29155 | 10 ± 1.0 | 74 ± 1 | 290 ± 52 | 70 ± 3 | 68 ± 13 | 54 ± 2 | 0.9 ± 0.2 | 87 ± 4 | 6.8 | 29 |
| SRI-29212 | 672 ± 204 | 67 ± 4 | 8126 ± 2273 | 64 ± 5 | 3805 ± 1645 | 50 ± 7 | 2.2 ± 0.5 | 72 ± 4 | 5.7 | 12 |
| SRI-29213 | 16 ± 4 | 81 ± 3 | 346 ± 59 | 77 ± 3 | 89 ± 51 | 43 ± 4 | 0.2 ± 0.0 | 94 ± 2 | 5.6 | 22 |
| SRI-29338 | 9.0 ± 1.5 | 71 ± 2 | 204 ± 47 | 62 ± 3 | 56 ± 21 | 52 ± 3 | 1.18 ± 0.33 | 63 ± 4 | 6 | 23 |
| SRI-29554 | 11 ± 1 | 71 ± 1 | 179 ± 27 | 78 ± 2 | 57 ± 18 | 60 ± 3 | 0.98 ± 0.46 | 59 ± 6 | 5 | 16 |
| SRI-29574 | 2.3 ± 0.4 | 68 ± 2 | 52 ± 15 | 72 ± 4 | 23 ± 5 | 52 ± 2 | Inactive | Inactive | 10 | 23 |
| SRI-29577 | 4.4 ± 0.8 | 70 ± 2 | 90 ± 15 | 71 ± 2 | 20 ± 5 | 56 ± 2 | 3.4 ± 1.2 | 65 ± 6 | 4.6 | 20 |
| SRI-29776 | 19 ± 4 | 69 ± 2 | 229 ± 43 | 71 ± 3 | 106 ± 19 | 61 ± 2 | 6.09 ± 0.97 | 58 ± 3 | 6 | 12 |
| SRI-29779 | 7.3 ± 2.2 | 63 ± 3 | 147 ± 63 | 71 ± 6 | 42 ± 12 | 51 ± 2 | 1.2 ± 0.2 | 83 ± 3 | 5.8 | 20 |
| SRI-29786 | 7.1 ± 2.2 | 70 ± 3 | 143 ± 61 | 68 ± 7 | 49 ± 27 | 44 ± 4 | 1.1 ± 0.1 | 100 ± 3 | 6.9 | 20 |
| SRI-29982 | 13 ± 2 | 73 ± 2 | 259 ± 41 | 71 ± 2 | 54 ± 7 | 51 ± 1 | 2.46 ± 1.21 | 47 ± 6 | 4 | 20 |
| SRI-29983 | 11 ± 2 | 70 ± 2 | 203 ± 79 | 73 ± 6 | 35 ± 9 | 57 ± 2 | 1.29 ± 0.34 | 55 ± 3 | 3 | 19 |
| SRI-29991 | 2.1 ± 0.3 | 68 ± 1 | 63 ± 9 | 69 ± 2 | 4.7 ± 0.6 | 51 ± 1 | 1.22 ± 1.12 | 18 ± 4 | 2 | 30 |
| SRI-30503 | 9.2 ± 1.2 | 70 ± 1 | 98 ± 23 | 65 ± 3 | 16 ± 5 | 55 ± 2 | 0.67 ± 0.37 | 34 ± 4 | 2 | 11 |
| SRI-30504 | 11 ± 2 | 70 ± 2 | 426 ± 49 | 70 ± 2 | 50 ± 14 | 55 ± 3 | 0.97 ± 0.65 | 47 ± 7 | 5 | 39 |
| SRI-30507 | 18 ± 3 | 71 ± 2 | 132 ± 16 | 77 ± 2 | 28 ± 6 | 54 ± 2 | 4.80 ± 1.81 | 62 ± 7 | 2 | 7 |
| SRI-30508 | 9.3 ± 1.1 | 65 ± 1 | 69 ± 18 | 76 ± 3 | 3.9 ± 0.4 | 56 ± 1 | 0.15 ± 0.09 | 34 ± 3 | 0.4 | 8 |
| SRI-30513 | 12 ± 2 | 72 ± 2 | 153 ± 55 | 65 ± 4 | 83 ± 28 | 59 ± 4 | 2.97 ± 1.03 | 50 ± 4 | 7 | 13 |
| SRI-30517 | 6.0 ± 0.7 | 70 ± 1 | 95 ± 12 | 70 ± 2 | 23 ± 3 | 54 ± 1 | 0.16 ± 0.08 | 43 ± 4 | 4 | 16 |
| SRI-30522 | 8.8 ± 1.1 | 63 ± 1 | 86 ± 55 | 40 ± 5 | 13 ± 6 | 35 ± 3 | Inactive | Inactive | 1 | 10 |
| SRI-30524 | 4.8 ± 0.6 | 71 ± 1 | 44 ± 6 | 76 ± 2 | 8.7 ± 0.9 | 56 ± 1 | 2.36 ± 1.17 | 52 ± 6 | 2 | 9 |
| SRI-30810 | 5.6 ± 0.8 | 64 ± 1 | 60 ± 10 | 69 ± 2 | 11 ± 3 | 51 ± 2 | 0.82 ± 0.36 | 25 ± 2 | 2 | 11 |
| SRI-30826 | 6.1 ± 1 | 64 ± 2 | 88 ± 15 | 81 ± 2 | 18 ± 4 | 56 ± 2 | 1.28 ± 0.40 | 42 ± 3 | 3 | 14 |
| SRI-30827 | 0.5 ± 0.1 | 63 ± 2 | 21 ± 7 | 67 ± 3 | 3.2 ± 0.9 | 58 ± 3 | 1.99 ± 0.33 | 79 ± 3 | 6 | 42 |
| SRI-30828 | 8.9 ± 1.6 | 60 ± 2 | 144 ± 30 | 65 ± 3 | 20 ± 5 | 50 ± 2 | 2.61 ± 1.14 | 60 ± 7 | 2 | 16 |
| SRI-30837 | 11 ± 1 | 61 ± 1 | 300 ± 56 | 75 ± 3 | 67 ± 8 | 55 ± 1 | 1.70 ± 0.62 | 44 ± 4 | 6 | 27 |
| SRI-30946 | 21 ± 3 | 70 ± 2 | 78 ± 17 | 75 ± 3 | 35 ± 11 | 59 ± 3 | 1.17 ± 0.32 | 44 ± 3 | 2 | 4 |
| SRI-31034 | 7.4 ± 1.1 | 69 ± 1 | 94 ± 22 | 63 ± 3 | 25 ± 7 | 49 ± 2 | Inactive | Inactive | 3 | 13 |
| SRI-31039 | 7.4 ± 2 | 74 ± 4 | 31 ± 7 | 71 ± 3 | 8.0 ± 2.8 | 58 ± 3 | 3.15 ± 1.11 | 72 ± 7 | 1 | 4 |
| SRI-31040 | 1.2 ± 0.1 | 69 ± 1 | 11 ± 4 | 70 ± 4 | 3.1 ± 0.7 | 54 ± 2 | 3.74 ± 1.10 | 91 ± 7 | 3 | 9 |
| SRI-31043 | 11 ± 1 | 67 ± 1 | 47 ± 10 | 67 ± 2 | 22 ± 5 | 51 ± 2 | 2.16 ± 0.97 | 30 ± 3 | 2 | 4 |
| SRI-31142 | 1.9 ± 0.3 | 72 ± 2 | 17 ± 4 | 61 ± 2 | 2.4 ± 0.4 | 48 ± 1 | 2.34 ± 0.45 | 92 ± 4 | 1.3 | 9 |
| SRI-31143 | 1.7 ± 0.1 | 69 ± 1 | 16 ± 5 | 69 ± 4 | 3.0 ± 0.6 | 51 ± 2 | 3.39 ± 1.99 | 52 ± 8 | 1.8 | 9 |
| Cocaine | 200 ± 19 | 100 ± 2 | 329 ± 22 | 102 ± 2 | 273 ± 24 | 98 ± 2 | 0.28 ± 0.03 | 97 ± 3 | 1.4 | 1.7 |
| GBR12935 | 1.1 ± 0.1 | 104 ± 3 | N.D. | N.D. | N.D. | N.D. | 2.0 × 10−3 ± 0.08 × 10−3 | 101 ± 0.9 |
N.D., not determined.
Fig. 4.

Comparison of the inhibition of [3H]WIN35428 binding versus [3H]DA uptake by test agents. The test drugs shown here (A), and most of the drugs examined (see Table 2), were ∼3 orders of magnitude less potent in inhibiting [3H]WIN35428 binding to DAT than in blocking uptake of [3H]DA. For example, SRI-29574 partially inhibited DAT uptake (IC50 = 2.3 ± 0.4 nM) while being inactive in inhibiting DAT binding. In contrast, SRI-29786 partially inhibited DAT uptake (IC50 = 7.1 ± 2.2 nM) but also inhibited DAT binding, with full efficacy and an IC50 value (1100 ± 10 nM) 155-fold weaker than the IC50 for inhibition of DAT uptake. (B) The correlation plot of DAT uptake Emax versus DAT binding Emax for 36 DAT partial inhibitors shows that there was no significant correlation between the Emax values observed in the DAT uptake and binding assays for 36 DAT partial inhibitors. Each data point is the mean ± S.D. of three separate experiments. The data were fit to a one-component dose-response curve equation for the best-fit estimates of the IC50 (± S.D.) and Emax (± S.D.), which are reported in Table 2.
Effect of Test Agents on DAT-Mediated [3H]MPP+ Release.
The first set of release experiments determined the effect of test agents on DAT-mediated [3H]MPP+ release in the absence and presence of 100 nM d-amphetamine. Overall, at concentrations of <1 μM, none of the agents altered DAT-mediated [3H]MPP+ release in the absence or presence of 100 nM d-amphetamine (data not shown). The ability of these agents to shift d-amphetamine–induced DAT-mediated [3H]MPP+ release, using blocking concentrations ∼25 times greater than the corresponding IC50 for DAT uptake inhibition, were then determined. Figure 5A reports representative results. SRI-29574 had no significant effect on the d-amphetamine EC50 or Emax value. SRI-29213 [N-(3,3-diphenylpropyl)-2-(1H-indol-5-yl)quinazolin-4-amine], in contrast, significantly increased the EC50 value and also decreased the Emax value. Of the 23 agents tested in this manner (see Table 3), only SRI-29213 increased EC50 and decreased Emax. GBR12935, a competitive DAT uptake inhibitor, shifted the d-amphetamine release curve to the right in a parallel fashion without changing the Emax value. Similar results were obtained when [3H]DA was used instead of [3H]MPP+ (Fig. 5B).
Fig. 5.

Effect of test agents on DAT-mediated release of [3H]MPP+ (A) or [3H]DA (B). (A) SRI-29574 had no significant effect on the d-amphetamine EC50 or Emax value. SRI-29213, in contrast, significantly increased the EC50 value and also decreased the Emax value. GBR12935, a competitive DAT uptake inhibitor, shifted the d-amphetamine release curve to the right in a parallel fashion without changing the Emax value. (B) Similar results were observed for [3H]DA release. Each data point is the mean ± S.D. of three separate experiments. The data were fit to a one-component dose-response curve equation for the best-fit estimates of the EC50 (± S.D.) and Emax (± S.D.), which are reported in Table 2.
TABLE 3.
Effect of test agents on d-amphetamine–induced, DAT-mediated [3H]MPP+ or [3H]DA release
d-Amphetamine dose-response curves were generated in the absence and presence of each test agent as described in Materials and Methods and illustrated in Fig. 5A. Each value is the mean ± S.D.; n = 3. The apparent Ke was calculated according to the following equation: Apparent Ke = [Blocker]/((EC50-2/EC50-1) − 1), where EC50-1 is the EC50 in the absence of blocker and EC50-2 is the EC50 in the presence of blocker. A negative apparent Ke occurs when a shifted EC50 is less than the control EC50 value.
| Blocker |
IC50 for DAT Uptake Inhibition |
Emax for DAT Uptake Inhibition |
Blocker Concentration |
d-Amphetamine EC50 |
d-Amphetamine Emax |
Apparent Ke |
|---|---|---|---|---|---|---|
| nM | % | nM | % | nM | ||
| [3H]MPP+ Release | ||||||
| None | – | – | – | 6.4 ± 1.2 | 104 ± 4 | – |
| SRI-29574 | 2 | 68 | 50 | 5.4 ± 0.6 | 103 ± 3 | −388 |
| SRI-29577 | 4 | 70 | 125 | 4.8 ± 0.4 | 102 ± 2 | −553 |
| SRI-29786 | 7 | 70 | 250 | 7.0 ± 0.5 | 101 ± 2 | 1940 |
| SRI-29779 | 7 | 63 | 250 | 7.9 ± 0.6 | 99 ± 2 | 912 |
| SRI-29155 | 10 | 74 | 250 | 9.6 ± 1.0* | 94 ± 2* | 456 |
| SRI-29213 | 16 | 81 | 500 | 9.7 ± 1.0* | 78 ± 2*** | 886 |
| SRI-29153 | 20 | 73 | 500 | 7.9 ± 0.9 | 101 ± 3 | 1820 |
| SRI-29070 | 174 | 66 | 5000 | 10.6 ± 0.7** | 98 ± 1 | 7050 |
| SRI-29072 | 212 | 71 | 5000 | 7.4 ± 1.0 | 96 ± 3 | 25830 |
| SRI-29212 | 672 | 67 | 12,500 | 7.4 ± 0.8 | 103 ± 2 | 64580 |
| SRI-29991 | 2 | 68 | 50 | 6.1 ± 0.7 | 102 ± 2 | −1070 |
| SRI-30517 | 6 | 70 | 150 | 7.0 ± 1.3 | 104 ± 4 | 1600 |
| SRI-30522 | 9 | 63 | 250 | 6.4 ± 1.1 | 105 ± 4 | N.A. |
| SRI-30524 | 5 | 71 | 125 | 7.1 ± 0.9 | 103 ± 3 | 1140 |
| SRI-30810 | 6 | 64 | 150 | 7.2 ± 1.5 | 104 ± 5 | 1200 |
| SRI-30826 | 6 | 64 | 150 | 7.0 ± 1.3 | 105 ± 4 | 1600 |
| SRI-30827 | 0.5 | 63 | 12.5 | 6.7 ± 0.9 | 104 ± 3 | 267 |
| SRI-31034 | 7 | 69 | 200 | 6.8 ± 1.5 | 104 ± 5 | 3200 |
| SRI-31040 | 1 | 69 | 25 | 9.3 ± 1.3* | 105 ± 3 | 55 |
| SRI-31142 | 2 | 72 | 50 | 7.2 ± 1.0 | 103 ± 3 | 400 |
| SRI-31143 | 2 | 67 | 50 | 6.9 ± 1.3 | 104 ± 4 | 46 |
| GBR12935 | 2 | 100 | 5 | 150 ± 25*** | 122 ± 8* | 0.22 |
| [3H]DA Release | ||||||
| None | – | – | – | 67 ± 10 | 97 ± 3 | – |
| GBR12935 | 2 | 100 | 5 | 519 ± 95** | 91 ± 6 | 0.74 |
| SRI-29574 | 2 | 68 | 50 | 53 ± 6 | 95 ± 2 | −239 |
| SRI-29213 | 16 | 81 | 500 | 72 ± 12 | 71 ± 3*** | 6700 |
N.A., not applicable.
P < 0.05 vs. control (Student's t test); **P < 0.01 vs. control (Student's t test); ***P < 0.001 vs. control (Student's t test).
Effect of SRI-29574 and Cocaine on [3H]DA Uptake/Accumulation.
We next assessed the effect of SRI-29574, a potent partial [3H]DA uptake inhibitor, on the time course of [3H]DA uptake, in comparison with cocaine. We predicted that SRI-29574 would reduce the maximum level of [3H]DA accumulation, consistent with noncompetitive inhibition. As reported in Fig. 6A and Table 4, SRI-29574 had no significant effect on the time to half-maximal accumulation (t1/2) but decreased the Emax in a dose-dependent manner (Fig. 6B) (EC50 = 5.4 ± 0.2 nM; Emax = 69 ± 1%). In contrast, the most striking effect of cocaine (Fig. 7, A and B) was to increase the t1/2 in a dose-dependent linear manner. The effect of cocaine on the Emax was more complex. Post hoc Student’s t test showed that two of the four Emax values were not significantly different from control, indicating that cocaine did not have a consistent effect on the Emax. Viewed collectively, these results are consistent with SRI-29574 being a noncompetitive inhibitor of [3H]DA uptake.
Fig. 6.

Effect of SRI-29574 on [3H]DA uptake/accumulation. As shown in (A) and Table 4, SRI-29574 had no significant effect on the t1/2 but decreased the Emax in a dose-dependent manner (IC50 = 5.4 ± 0.2 nM; Emax = 69 ± 1%) (B). Each data point is the mean ± S.D. of three separate experiments. The data were fit to a one-component time-response curve equation for the best-fit estimates of the t1/2 (minutes ± S.D.) and Emax (% ± S.D.), which are reported in Table 4.
TABLE 4.
Effect of SRI-29574 on [3H]DA accumulation
[3H]DA uptake was assessed at various time points in the absence and presence of the indicated concentrations of SRI-29574. The combined data from all three experiments (Fig. 6A) (180 data points) were fit to the equation B = Emax × (T/(T + t1/2)), where B is the observed level of uptake, T is the time in minutes, and t1/2 is the time to half-maximal accumulation. The time course data for each drug condition were separately applied to the above equation, each of which differed only in the names of the corresponding parameters: Emax1, Emax2, Emax3, Emax4, and Emax5 and t1/2-1, t1/2-2, t1/2-3, t1/2-4, and t1/2-5. Thus, the unconstrained fit led to the reported parameter values. The data were then fit with the constraint that the Emax values were all equal. This resulted in a highly significant increase in the sum-of-squares (F = 18.9, P = 0). In contrast, fitting the data with the constraint that the t1/2 values were equal did not significantly increase the sum-of-squares (F = 0.46, P = 0.76).
| [SRI-29574] | t1/2 | Emax | Inhibition of the Emax Value |
|---|---|---|---|
| nM | min | % | |
| 0 | 32 ± 4 | 98 ± 4 | 0 |
| 1 | 28 ± 3 | 89 ± 4 | 11 |
| 4 | 27 ± 3 | 71 ± 2* | 29 |
| 8 | 34 ± 3 | 58 ± 2* | 42 |
| 25 | 31 ± 3 | 43 ± 3* | 57 |
P < 0.05 vs. control (unpaired Student's t test).
Fig. 7.

Effect of cocaine on [3H]DA uptake/accumulation. Cocaine increased the t1/2 for [3H]DA uptake/accumulation in a dose-dependent (A and Table 5) and linear (B) manner. The effect of cocaine on the Emax was more complex. Post hoc Student’s t test showed that two of the four Emax values were not significantly different from control, indicating that cocaine did not have a consistent effect on the Emax. Each data point is the mean ± S.D. of three separate experiments. The data were fit to a one-component time-response curve equation for the best-fit estimates of the t1/2 (minutes ± S.D.) and Emax (% ± S.D.), which are reported in Table 5.
TABLE 5.
Effect of cocaine on [3H]DA accumulation
[3H]DA uptake was assessed at various time points in the absence and presence of the indicated concentrations of cocaine. The combined data from all three experiments (Fig. 7A) (180 data points) were fit to the equation described in Table 4. The time course data for each drug condition were separately applied to the above equation, each of which differed only in the names of the corresponding parameters: Emax1, Emax2, Emax3, Emax4, and Emax5 and t1/2-1, t1/2-2, t1/2-3, t1/2-4, and t1/2-5. Thus, the unconstrained fit led to the reported parameter values. The data were then fit with the constraint that the Emax values were all equal. This resulted in a significant increase in the sum-of-squares (F = 6.34, P < 0.001). Fitting the data with the constraint that the t1/2 values were equal led to a highly significantly increased sum-of-squares (F = 89, P = 0). Constraining the Emax values to equal 100% also led to a modestly significant increase in sum-of-squares (F = 4.19, P = 0.003).
| [Cocaine] | t1/2 | Emax | Fold Increase in t1/2 |
|---|---|---|---|
| nM | min | % | |
| 0 | 40 ± 2 | 100 ± 2 | 0 |
| 100 | 63 ± 5* | 112 ± 4* | 1.57 |
| 300 | 121 ± 15* | 121 ± 15 | 3.0 |
| 600 | 266 ± 24* | 132 ± 9* | 6.65 |
| 1200 | 440 ± 165* | 140 ± 43 | 11.0 |
P < 0.05 vs. control (unpaired Student's t test).
Discussion
Our previously published papers identified three quinazolinamine DAT allosteric modulators: SRI-20040, SRI-20041, and SRI-9804 (see Fig. 1, A and B; and Supplemental Fig. 1 for structures). While two of the quinazolinamine modulators (SRI-9804 and SRI-20040) partially inhibit both uptake of [3H]DA (forward transport) and DAT-mediated release of preloaded [3H]DA (reverse transport), the third compound (SRI-20041) inhibits substrate uptake but has no appreciable effect on efflux (Rothman et al., 2009). This latter compound appeared to be the first DAT ligand to differentially affect substrate uptake versus release, suggesting that the two functional modes of substrate translocation are unique, and that it may be possible to design compounds selectively affecting a single part of the NSS translocation cycle. The experiments reported here significantly extend these findings.
The first-generation DAT allosteric modulators partially inhibited DAT uptake and DAT binding (measured using [125I]RTI-55) with micromolar potency. A major advance made in this study is the development of second-generation compounds with nanomolar potency for partial inhibition of DAT uptake. Unlike the first-generation compounds, the second-generation compounds were generally 100- to 1000-fold less potent inhibitors of DAT binding when compared with DAT uptake. Some agents, such as SRI-29574 and SRI-30522 [2-([1,2,4]triazolo[1,5-a]pyridin-6-yl)-N-(2,2-diphenylethyl)quinazolin-4-amine], were inactive as inhibitors of DAT binding. To our knowledge, these compounds are the first compounds that discriminate inhibition of DAT uptake from inhibition of DAT binding, providing strong support for the hypothesis that these second-generation DAT allosteric modulators bind to a site on the DAT distinct from the cocaine-binding site. Interestingly, the second-generation DAT allosteric modulators also partially inhibited SERT and NET uptake. This observation suggests that further research should be aimed at developing allosteric modulators selective for DAT, SERT, and NET.
The data reported here clearly demonstrate that the second-generation DAT allosteric modulators and standard DAT inhibitors, such as cocaine and GBR12935, interact differently with the DAT. A defining difference is that the second-generation DAT allosteric modulators partially inhibit DAT uptake with nanomolar potency. Moreover, as noted above, the second-generation DAT allosteric modulators are much less potent in inhibiting DAT binding, unlike standard DAT uptake inhibitors (Table 2). Perhaps most interesting, with the exception of one agent (SRI-29213), the second-generation DAT allosteric modulators fail to significantly alter d-amphetamine–induced, DAT-mediated release of [3H]MPP+ or [3H]DA (Table 3) when tested using concentrations of “blockers” 20–25 times higher than the corresponding IC50 value for inhibiting DAT uptake (Table 3). SRI-29213 reduced the Emax value for d-amphetamine–induced release, similar to what had been observed with the first-generation agents SRI-9804 and SRI-20040. These results suggest that the second-generation SRI compounds affect forward transport (i.e., uptake) but not reverse transport (i.e., release), indicating that these two processes are separable and independently regulated, as has previously been suggested (Cao and Reith, 2002). This possibility is supported by the finding that the phosphorylation of Thr53 of DAT can partially reduce forward transport by DAT but completely eliminate reverse transport produced by amphetamine (Foster et al., 2012). Similar findings were reported for site-directed mutagenesis of Thr62 (Fraser et al., 2014), and by Khoshbouei et al. (2003), who reported that N-terminal phosphorylation shifts DAT from a "reluctant" state to a "willing" state for d-amphetamine–induced DA efflux, without affecting inward transport. Moreover, a recent study showed that impairing the interaction of phosphatidylinositol 4,5-bisphosphate with DAT impairs amphetamine-induced DA efflux without affecting DA uptake (Hamilton et al., 2014).
In summary, this paper reports a new generation of potent DAT allosteric modulators that partially inhibit DAT uptake without altering DAT-mediated reverse transport and with minimal inhibition of DAT binding. The molecular mechanism by which these second-generation allosteric modulators alter DAT function remains to be determined. As reviewed elsewhere (Schmitt et al., 2013), the current understanding of this complex mechanism is still evolving. It is possible that some of the compounds reported here will help elucidate these mechanisms. Some possibilities for future research include radiolabeling the most potent ligands, such as SRI-31040 [2-(7-methylimidazo[1,2-a]pyridin-6-yl)-N-(2-phenyl-2-(1H-pyrrol-2-yl)ethyl)quinazolin-4-amine], to allow the direct study of the hypothesized allosteric binding site, which would aid future structure-activity studies and in vivo investigations with selected compounds to explore their behavioral/therapeutic effects. Finally, in silico molecular modeling experiments could be used to identify the putative allosteric binding site on DAT, as was recently accomplished for SERT (Kortagere et al., 2013).
Supplementary Material
Abbreviations
- BAT
biogenic amine transporter
- BSA
bovine serum albumin
- DA
dopamine
- DAT
dopamine transporter
- GBR12935
1-(2-diphenylmethoxyethyl)-4-(3-phenylpropyl)piperazine
- 5-HT
serotonin
- MPP+
1-methyl-4-phenylpyridinium
- NE
norepinephrine
- NET
norepinephrine transporter
- NSS
neurotransmitter/sodium symporter
- RTI-55
3β-(4′-iodophenyl)tropan-2β-carboxylic acid methyl ester
- SERT
serotonin transporter
- SRI-20040
N-(2,2-diphenylethyl)-2-phenyl-4-quinazolinamine
- SRI-20041
N-(3,3-diphenylpropyl)-2-phenyl-4-quinazolinamine
- SRI-29213
N-(3,3-diphenylpropyl)-2-(1H-indol-5-yl)quinazolin-4-amine
- SRI-29574
N-(2,2-diphenylethyl)-2-(imidazo[1,2-a]pyridin-6-yl)quinazolin-4-amine
- SRI-29786
2-(4-(dimethylamino)phenyl-N-(2,2-diphenylethyl)quinazolin-4-amine
- SRI-29986
N-(2,2-diphenylethyl)-2-(indolin-5-yl)quinazolin-4-amine
- SRI-30522
2-([1,2,4]triazolo[1,5-a]pyridin-6-yl)-N-(2,2-diphenylethyl)quinazolin-4-amine
- SRI-30835
2-(4-(dimethylamino)phenyl)-N-(2-morpholino-2-phenylethyl)quinazolin-4-amine
- SRI-31040
2-(7-methylimidazo[1,2-a]pyridin-6-yl)-N-(2-phenyl-2-(1H-pyrrol-2-yl)ethyl)quinazolin-4-amine
- SRI-31335
2-(4-(dimethylamino)phenyl)-N-phenethylquinazolin-4-amine
- SRI-9804
N-(diphenylmethyl)-2-phenyl-4-quinazolinamine
- WIN35428
2β-carbomethoxy-3β-(4-fluorophenyl)tropane
Authorship Contributions
Participated in research design: Rothman, Ananthan, Partilla, Baumann.
Conducted experiments: Partilla.
Contributed new reagents or analytic tools: Ananthan, Saini, Moukha-Chafiq, Pathak.
Performed data analysis: Rothman, Ananthan, Partilla.
Wrote or contributed to the writing of the manuscript: Rothman, Ananthan, Partilla, Baumann, Saini, Moukha-Chafiq, Pathak.
Footnotes
This work was supported by the Intramural Research Program of the National Institutes of Health [National Institute on Drug Abuse]; and the National Institutes of Health National Institute on Drug Abuse [Grant R33-DA029962 (to S.A.)].
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Cao BJ, Reith ME. (2002) Nitric oxide inhibits uptake of dopamine and N-methyl-4-phenylpyridinium (MPP+) but not release of MPP+ in rat C6 glioma cells expressing human dopamine transporter. Br J Pharmacol 137:1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felten A, Montag C, Markett S, Walter NT, Reuter M. (2011) Genetically determined dopamine availability predicts disposition for depression. Brain Behav 1:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest LR, Krämer R, Ziegler C. (2011) The structural basis of secondary active transport mechanisms. Biochim Biophys Acta 1807:167–188. [DOI] [PubMed] [Google Scholar]
- Foster JD, Yang JW, Moritz AE, Challasivakanaka S, Smith MA, Holy M, Wilebski K, Sitte HH, Vaughan RA. (2012) Dopamine transporter phosphorylation site threonine 53 regulates substrate reuptake and amphetamine-stimulated efflux. J Biol Chem 287:29702–29712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser R, Chen Y, Guptaroy B, Luderman KD, Stokes SL, Beg A, DeFelice LJ, Gnegy ME. (2014) An N-terminal threonine mutation produces an efflux-favorable, sodium-primed conformation of the human dopamine transporter. Mol Pharmacol 86:76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Caron MG. (2003) Monoamine transporters: from genes to behavior. Annu Rev Pharmacol Toxicol 43:261–284. [DOI] [PubMed] [Google Scholar]
- Gether U, Andersen PH, Larsson OM, Schousboe A. (2006) Neurotransmitter transporters: molecular function of important drug targets. Trends Pharmacol Sci 27:375–383. [DOI] [PubMed] [Google Scholar]
- Greengard P. (2001) The neurobiology of slow synaptic transmission. Science 294:1024–1030. [DOI] [PubMed] [Google Scholar]
- Hamilton PJ, Belovich AN, Khelashvili G, Saunders C, Erreger K, Javitch JA, Sitte HH, Weinstein H, Matthies HJ, Galli A. (2014) PIP2 regulates psychostimulant behaviors through its interaction with a membrane protein. Nat Chem Biol 10:582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshbouei H, Wang H, Lechleiter JD, Javitch JA, Galli A. (2003) Amphetamine-induced dopamine efflux. A voltage-sensitive and intracellular Na+-dependent mechanism. J Biol Chem 278:12070–12077. [DOI] [PubMed] [Google Scholar]
- Kortagere S, Fontana AC, Rose DR, Mortensen OV. (2013) Identification of an allosteric modulator of the serotonin transporter with novel mechanism of action. Neuropharmacology 72:282–290. [DOI] [PubMed] [Google Scholar]
- Kurian MA, Li Y, Zhen J, Meyer E, Hai N, Christen HJ, Hoffmann GF, Jardine P, von Moers A, Mordekar SR. (2011) Clinical and molecular characterisation of hereditary dopamine transporter deficiency syndrome: an observational cohort and experimental study. Lancet Neurol 10:54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montague PR, Berns GS. (2002) Neural economics and the biological substrates of valuation. Neuron 36:265–284. [DOI] [PubMed] [Google Scholar]
- Nightingale B, Dersch CM, Boos TL, Greiner E, Calhoun WJ, Jacobson AE, Rice KC, Rothman RB. (2005) Studies of the biogenic amine transporters. XI. Identification of a 1-[2-[bis(4-fluorophenyl)methoxy]ethyl]-4-(3-phenylpropyl)piperazine (GBR12909) analog that allosterically modulates the serotonin transporter. J Pharmacol Exp Ther 314:906–915. [DOI] [PubMed] [Google Scholar]
- Pariser JJ, Partilla JS, Dersch CM, Ananthan S, Rothman RB. (2008) Studies of the biogenic amine transporters. 12. Identification of novel partial inhibitors of amphetamine-induced dopamine release. J Pharmacol Exp Ther 326:286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith ME, Blough BE, Hong WC, Jones KT, Schmitt KC, Baumann MH, Partilla JS, Rothman RB, Katz JL. (2015) Behavioral, biological, and chemical perspectives on atypical agents targeting the dopamine transporter. Drug Alcohol Depend 147:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS. (2001) Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse 39:32–41. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Dersch CM, Ananthan S, Partilla JS. (2009) Studies of the biogenic amine transporters. 13. Identification of “agonist” and “antagonist” allosteric modulators of amphetamine-induced dopamine release. J Pharmacol Exp Ther 329:718–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Katsnelson M, Vu N, Partilla JS, Dersch CM, Blough BE, Baumann MH. (2002) Interaction of the anorectic medication, phendimetrazine, and its metabolites with monoamine transporters in rat brain. Eur J Pharmacol 447:51–57. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Vu N, Partilla JS, Roth BL, Hufeisen SJ, Compton-Toth BA, Birkes J, Young R, Glennon RA. (2003) In vitro characterization of ephedrine-related stereoisomers at biogenic amine transporters and the receptorome reveals selective actions as norepinephrine transporter substrates. J Pharmacol Exp Ther 307:138–145. [DOI] [PubMed] [Google Scholar]
- Salamone JD, Correa M, Farrar AM, Nunes EJ, Pardo M. (2009) Dopamine, behavioral economics, and effort. Front Behav Neurosci 3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt KC, Reith ME. (2010) Regulation of the dopamine transporter: aspects relevant to psychostimulant drugs of abuse. Ann N Y Acad Sci 1187:316–340. [DOI] [PubMed] [Google Scholar]
- Schmitt KC, Rothman RB, Reith ME. (2013) Nonclassical pharmacology of the dopamine transporter: atypical inhibitors, allosteric modulators, and partial substrates. J Pharmacol Exp Ther 346:2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitte HH, Freissmuth M. (2015) Amphetamines, new psychoactive drugs and the monoamine transporter cycle. Trends Pharmacol Sci 36:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitte HH, Huck S, Reither H, Boehm S, Singer EA, Pifl C. (1998) Carrier-mediated release, transport rates, and charge transfer induced by amphetamine, tyramine, and dopamine in mammalian cells transfected with the human dopamine transporter. J Neurochem 71:1289–1297. [DOI] [PubMed] [Google Scholar]
- Tanda G, Newman AH, Katz JL. (2009) Discovery of drugs to treat cocaine dependence: behavioral and neurochemical effects of atypical dopamine transport inhibitors. Adv Pharmacol 57:253–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.