

Abstract
Tumor microenvironments include a number of fibrin clots due to the microbleeding caused by cancer cell invasion into blood vessels, which suggests the potential utility of a platelet vector for systemic cancer treatment. We previously reported that inactivated Sendai virus (hemagglutinating virus of Japan; HVJ) envelope (HVJ-E) activates anti-tumor immunity and induces cancer cell-selective apoptosis. The hemagglutination activity that blocks the systemic administration of HVJ-E was dramatically attenuated by incorporation into platelets. Platelets incorporating HVJ-E (PH complex) were then injected into the tail veins of B16F10 melanoma-bearing mice. The PH complex primarily accumulated in tumor tissues and caused the significant accumulation of various immune cells in the tumor bed. Injections of the PH complex to the melanoma-bearing mouse significantly reduced the tumor size, and the tumor growth was ultimately arrested. Secretion of the chemokine regulated upon activation normal T-expressed and presumably secreted (RANTES) was upregulated following PH stimulation. The RANTES-depletion in melanoma-bearing mice significantly attenuated the cytotoxic T lymphocyte activity and led to a dramatic abrogation of the mouse melanoma suppression induced by the PH complex. Thus, a platelet vector incorporating viral particles, a Trojan horse for cancer treatment, will provide a new approach for cancer therapy using oncolytic viruses.
Introduction
Although there have been significant advances in the field of cancer treatments in the past decades, there are still some limitations with regard to the selectivity of anticancer reagent delivery to cancer cells and the tumor microenvironment. Methods for the cancer-selective delivery of therapeutic molecules have been desired since the concept of targeted drug delivery was proposed by Paul Ehrlich as a “magic bullet” in 1906.1 One approach, passive targeting, is based on the enhanced permeability and retention (EPR) effect. Because the structure of the tumor vasculature is immature and leaky,2 small (less than 100 nm in diameter) drug delivery vectors are believed to reach tumor cells by passing through the spaces between endothelial cells. This concept has been generally applied to non-viral vectors. Recently, mesenchymal stem cells3 have been used as tumor-targeting vehicles based on the finding that these cells are recruited to tumor tissues, most likely through the action of chemokines produced by the tumor tissue. A non-pathogenic obligate anaerobic bacterium, Bifidobacterium longum, has been shown to specifically accumulate in tumor tissues after systemic delivery.4 The specific accumulation of this bacterium occurs due to the anaerobic conditions of tumor tissues. These results highlight the importance of the particular characteristics of the tumor microenvironment for achieving specific delivery of therapeutic molecules to tumors. When examining the characteristics of the tumor microenvironment, Matsumura et al. proposed the utility of cancer stromal-targeting therapy using an anti-fibrin antibody5 because these authors found many areas of blood coagulation in tumor tissues due to microhemorrhaging caused by tumor invasion into tumor blood vessels.
Based on the observation of extensive blood coagulation in the tumor microenvironment, we hypothesized that a platelet vector could specifically target tumor tissues. Platelets have a physiologically important role in the removal of foreign materials from blood.6 Based on this characteristic, platelets have been reported to take up a variety of particles in vivo, such as liposomes and viral particles.6,7 Therefore, we attempted to incorporate an inactivated Sendai virus (HVJ; hemagglutinating virus of Japan) particle termed HVJ-envelope (HVJ-E) into a platelet vector.8 The intratumor injection of HVJ-E alone strongly induced an anti-tumor immune response; however, a limitation of HVJ-E is that it cannot be administered systemically due to the agglutination of erythrocytes by HN (hemagglutinin) glycoproteins on the envelope.9
Here, we developed a novel cancer-targeting vector based on platelets, and we demonstrated the utility of an HVJ-E-incorporating platelet vector in cancer immunotherapy.
Results
Platelet as a vector; infusion and release of HVJ-E and fluorescent particles
First, we examined whether HVJ-E particles could be infused into platelets. For this purpose, we determined the conditions necessary to infuse particles into platelets using FITC-beads. Murine platelets isolated from fresh blood were incubated with the beads at 37 °C for 2 hours, and then washed with PBS. Subsequently, numerous FITC-beads were observed in the platelets (Figure 1a). Next, using the same conditions, HVJ-E was also incubated with the platelets. Using electron microscopy, HVJ-E particles were detected in the platelets (Figure 1b). Immunofluorescence analysis confirmed that HVJ-E particles were infused inside the mouse platelets (with Triton X-100 treatment) (Supplementary Figure S1a). Additionally, human platelets successfully incorporated HVJ-E particles (Supplementary Figure S1b).
Figure 1.

Infusion and release of HVJ-E or fluorescent particles from platelets. (a) Infusion of FITC-beads (green) into platelets (red). Platelet membrane was stained with DiI (di-alkyl indocarbocyanine). (b) Transmission electron microscopic analysis of HVJ-E-infused platelets (8,000×). (c) Fluorescence intensity measurements of SPHERO fluorescent particles released from thrombin-stimulated SPHERO/platelet complexes. The data are shown as the mean ± SEM of four independent experiments. **P < 0.01 (versus 0U/ml). (d) Western blot analysis of HVJ-E M (matrix protein of HVJ) protein after the thrombin stimulation of PH complexes. HVJ-E samples from 1 to 100 HAU (haemagglutinating units) were loaded as standard samples. The PH supernatant contained the released HVJ-E particles, and the PH pellet contained the HVJ-E particles remaining inside the platelets after thrombin stimulation. In sample P (platelets only), no thrombin was added.
Next, we tested whether the infused HVJ-E particles were released from the platelets by stimulation with thrombin. First, we determined the thrombin concentration required to release SPHERO fluorescent particles that were previously infused into platelets. We found that a thrombin concentration greater than 1 U/ml was necessary for the release (Figure 1c). For platelets infused with HVJ-E, the viral particles were released from the platelets at a thrombin concentration of 100 U/ml (Figure 1d). Additionally, thrombin activity was high enough to specifically activate the PH complex in B16F10 tumor tissue whereas the thrombin activity was hardly detected in plasma. (Supplementary Figure S1c). These data indicate that HVJ-E particles could be infused into platelets to form PH complexes and could be released by platelet activation via thrombin stimulation.
Targeting tumor tissue in tumor-bearing mice via the systemic administration of the PH complex
The disadvantage of HVJ-E is that its systemic delivery is limited due to the hemagglutination caused by the HN protein. The injection of HVJ-E particles into the blood-stream directly can cause the agglutination of red blood cells. To avoid the coagulation caused by systemic administration, HVJ-E particles were infused in a platelet vector. The free HVJ-E particles or HVJ-E particle attached to the surface of platelet vectors were measured by a hemagglutinating assay (HA). The hemagglutinating activity of HVJ-E disturbs the systemic administration HVJ-E. When HVJ-E was mixed with chicken erythrocytes, hemagglutination was induced (H100 and H1000) (Figure 2a). However, after the PH complexes were washed twice with PBS, the hemagglutination activity of the PH complexes was dramatically decreased and largely undetectable (Figure 2a). HVJ-E particles localized to the blood vessels (PH1), whereas some HVJ-E particles were dispersed into the tumors (PH2) (Figure 2b).
Figure 2.

HA assay of PH complexes. (a) The hemagglutinating activity of the PH complexes (containing 100 or 1,000 HAU of HVJ-E) was reduced by washing with buffer (washing once or twice). (b) Tumor tissue sections were stained with FITC-albumin (green) and anti-F protein to visualize the tumor blood vessels and HVJ-E, respectively, after the administration of PH complexes (PH), HVJ-E, platelets (Plt), or NaCl solution (NaCl).
Next, PH complexes were injected into the tail veins of B16F10 melanoma tumor-bearing mice. The colocalization of HVJ-E particles with activated platelets was observed only in the PH-treated tumors (Figure 3a). No PH complexes were found in other normal organs such as the liver, spleen and kidney (data not shown). Fibrin clots were consistently observed in B16F10 tumor groups (Figure 3b), and large numbers of activated platelets (CD62P) that colocalized with the fibrin clots in the PH-treated tumors were also observed. Additionally, tumor blood vessels were stained with FITC-albumin. Collectively, these observations indicate that the PH complexes injected into the mouse tail veins successfully reached the tumor vessels and were trapped by fibrin clots and that some complexes appeared to be associated with tumor cells.
Figure 3.

The administration of PH complexes (PH), HVJ-E, platelets (Plt), or NaCl solution (NaCl) to B16F10 melanoma-bearing mice and the accumulation of delivered HVJ-E particles in tumor tissues. (a) Tumor sections were stained with anti-CD62P (red) and anti-F protein (green) to visualize the activated platelets and HVJ-E localization, respectively. (b) Tumor sections were stained with anti-CD62P (red) and anti-fibrin (green) to visualize the activated platelets and fibrin localization, respectively.
In vivo F10 tumor suppression by PH treatment
PH complexes were injected five times into the tail veins of B16F10 melanoma tumor-bearing mice. The tumor size in the PH complex-treated group was significantly reduced (Figure 4a). Mice in the platelet-, HVJ-E- and NaCl-treated groups began dying after approximately 18 days, whereas all PH-treated mice were alive through 40 days (Supplementary Figure S2). The accumulation of immune cells, such as CD4+ T cells, CD8+ T cells (Figure 4b,c), NK cells (CD49b), and DCs (CD11c) (Supplementary Figure S3a,b), was observed in the PH complex-treated B16F10 tumors at 48 hours after the fifth injection.
Figure 4.

Suppression of tumor growth by PH complex treatment in B16F10 melanoma-bearing mice and the accumulation of immune cells in the tumor. (a) Tumor growth curve of B16F10 melanoma-bearing mice treated with PH complexes (PH), HVJ-E, platelets (Plt), or NaCl solution (NaCl). The data are shown as the mean ± SEM of four animals per group. †The date when the number of mouse in the group became less than 2. (b,c) B16F10 tumor tissue sections from tumor-bearing mice treated with PH complexes (PH), HVJ-E, platelets (Plt) or NaCl solution were stained with anti-CD4 (red) and anti-CD8 (red) antibodies to visualize the localization of CD4+ and CD8+ T cells, respectively, in the tumor tissues.
Cytotoxicity of the PH complex or HVJ-E against cancer cells and tumor endothelial cells
The cytotoxicity of the PH complex and HVJ-E against cancer cells and tumor endothelial cells (TECs) from mouse F10 melanoma tissues was examined using an in vitro cell proliferation assay. PH complexes (different amounts of HVJ-E, from 100 to 2,000 HAU, were infused into the same number of platelets) and HVJ-E were added to B16F10 (mouse melanoma) cells and TECs (Figure 5a,b). PH complexes at higher concentrations suppressed the proliferation of the B16F10 cancer cells, but not the TECs, 48 hours after transfection. PH complexes and HVJ-E were also added to human pancreatic carcinoma cells (PC3), human umbilical vein endothelial cells (HUVECs) and human aortic endothelial cells (HAECs). Only the proliferation of PC3s was suppressed, and the proliferation of HUVECs and HAECs was not affected (Supplementary Figure S4a–c). B16F10 cells were treated with PH complexes after transfection with siRNA for RIG-I or MAVS. A proliferation assay of PH complex-treated B16F10 cells showed that cell death caused by the PH complexes was significantly suppressed by either RIG-I or MAVS siRNA, but not control siRNA (Supplementary Figure S5). This result indicates that similar to HVJ-E,10 PH complexes induced cell death through the RIG-I/MAVS pathway.
Figure 5.

Proliferation assay of B16F10 melanoma cells and isolated TECs from mouse F10 melanoma tumors. Different concentrations of HVJ-E (from 4 or 100 to 2,000 HAU) were infused into the same number of platelets (5.0 × 106 platelets) to construct various PH complexes. The PH complexes and different concentrations of HVJ-Es were added to (a) B16F10 or (b) TECs. The survival rates of the cells were measured. The data are shown as the mean ± SEM of four independent analyses.
The contribution of the HVJ-E-induced chemokine, RANTES/CCL5, to tumor suppression by activated anti-tumor immunity
Cytokine and chemokine arrays were used to identify the factors that contributed to the suppression of PH-treated tumors. TEC isolated from B16F10 tumors and B16F10 cells were stimulated with PH complexes, and RANTES was the most highly secreted chemokine in both cases (Figure 6a,b). PH complexes also stimulated HUVECs to secrete RANTES, IP-10 and IL-6 (Supplementary Figure S6a). RANTES secretions from TECs and F10 cells were also measured (Supplementary Figure S6a,b). IP-10 secretions from TECs and F10 cells was detected but there was no significant difference among any of the groups (NC, platelet, HVJ-E, and PH). IL-6 secretion from TEC and F10 cells was not detected. The HVJ-E and PH groups, but not the platelet group, showed the induction of RANTES secretion, which indicated that the HVJ-E particles were the sole inducer of RANTES.
Figure 6.
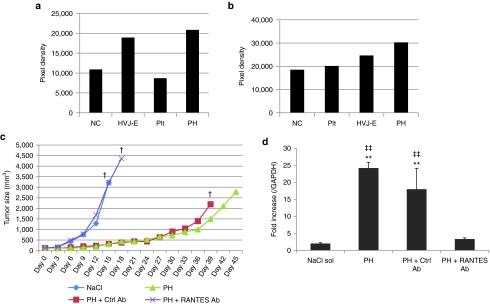
HVJ-E-induced RANTES production contributed to tumor suppression. (a,b) Chemokine and cytokine arrays of PH-treated TECs isolated from (a) tumors and (b) B16F10 cells. (c) Tumor growth curves of B16F10 melanoma-bearing mice treated with PH complexes (PH), PH complexes + RANTES-neutralizing antibody (PH + RANTES Ab), PH complexes + control IgG (PH + Ctrl Ab) or NaCl solution (NaCl). †The date when the number of mouse in the group became less than 2. The data are shown as the mean ± SEM of four animals per group. (d) RT-PCR of RANTES mRNA level in B16F10 tumor tissues. The F10 tissue samples were collected 48 hours after three injections. The data are shown as the mean ± SEM of four independent analyses. ‡‡P < 0.01 (versus PH + RANTES-neut ab); **P < 0.01 (versus NaCl).
A RANTES-neutralizing antibody was administered with the PH complexes to mice bearing B16F10 melanomas to verify the tumor suppression effect. The suppression effect of the PH complexes was abrogated by the RANTES-neutralizing antibody (Figure 6c). The expression of RANTES in melanoma tissue was enhanced by the PH complexes and was abolished in the RANTES-neutralizing antibody group (Figure 6d). Mice in the RANTES-depletion group started dying after approximately 18 days, similar to the NaCl-injected group, whereas all PH-treated mice remained alive through 40 days (Supplementary Figure S7).
Robust production of interferon (IFN)-γ from mouse splenocytes was achieved in response to melanoma stimulation, suggesting the activation of CTLs specific to the melanoma cells by the systemic administration of the PH complexes to B16F10-bearing mice (Figure 7a). An ELISPOT (IFN-γ) assay was performed using mouse splenocytes stimulated with B16F10 or MSC-1 (mesenchymal stromal cell from C57BL/6 mouse) cells (Supplementary Figure S8a). The splenocytes isolated from PH-treated F10-bearing mice specifically responded to the B16F10 cells, but not the MSC-1 cells. Anti-CD4 (clone GK1.5) or anti-CD8 (clone 53–7.62) antibodies were injected into the melanoma-bearing mice to deplete CD4+ or CD8+ T cells, respectively. In both the CD4- and CD8-depleted groups, the tumor suppression by the PH complex was significantly abrogated compared with the control IgG-injected group (Figure 7b,c). NK cells were also depleted by the injection of an anti-asialo GM1 antibody into B16F10-bearing mice. The NK cell-depleted group had significant attenuation of tumor suppression by the PH complex compared with the control IgG-injected group (Figure 7d). These results strongly suggest that both CD4+ and CD8+ T cells are necessary to activate CTLs to respond to B16F10 cells and that NK cells also contribute to tumor killing following PH complex treatment.
Figure 7.
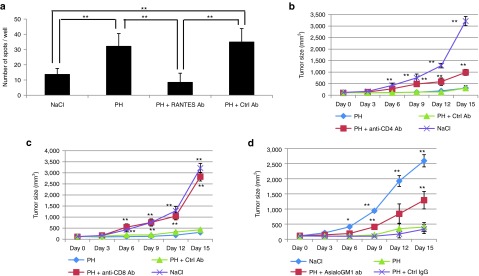
Induction of the anti-tumor immune response by T cells and NK cells in PH-treated tumor-bearing mice. (a) ELISPOT assay. Induction of IFN-γ from splenocytes isolated from PH-treated F10 tumor-bearing mice after 48 hours of incubation with or without B16F10 cells. The data are shown as the mean ± SEM of six independent analyses. **P < 0.01. (b–d) Tumor sizes of (b) CD4+ T cell-, (c) CD8+ T cell-, or (d) NK cell-depleted B16F10-bearing mice. The data are shown as the mean ± SEM of four animals per group. *P < 0.05 (versus PH), **P < 0.01 (versus PH).
A CTL assay (51Cr-release assay) using mouse splenocytes isolated from B16F10-bearing mice was performed (Supplementary Figure S8b). The splenocytes from the PH-treated mice demonstrated specific cytotoxicity against B16F10 cells, but not against MSC-1 cells. Collectively, these observations suggest that the PH complex induces anti-tumor immunity via the activation of both CTLs and NK cells and that RANTES plays a major role in the promotion of anti-tumor immunity.
In summary, as illustrated in Figure 8, systemically administered PH complexes targets the fibrin clots that dominantly present in tumor blood vessels and presumably release HVJ-E after activation in the fibrin clots. In not only B16F10 but also B16BL6 (mouse melanoma) and LL/2 (mouse Lewis lung carcinoma) tumor tissue sections, many thrombi (CD41-positive blood clots) were observed in the tumor vasculatures (stained with CD31 antibody) (Supplementary Figure S9a). The three different tumor tissue samples were collected at three different time points/tumor sizes (7 days/5 mm, 14 days/10 mm and 21 days/15 mm). Staining of the tumor sections with an anti-fibrin antibody revealed fibrin clots in all sample sections (Supplementary Figure S9b–d). Therefore, the systemic injection of PH complexes can induce similar anti-tumor effects in other murine tumor models. The HVJ-E particles, which were released from activated platelet vectors, induced the secretion of RANTES from TECs and B16F10 cells. Collectively, the combination of HVJ-E and RANTES accelerates tumor regression by enhancing anti-tumor immunity.
Figure 8.

Model depicting the mechanism by which the PH complex targets and induces chemokine production in F10 tumor tissues. The PH complex is inactive in normal blood vessels; however, when it interacts with fibrin clots in tumor vessels, the complex begins to release the HVJ-E particles (1), which induces TECs or cancer cells to secrete chemokines, mainly RANTES (2). The cytokines recruits immune cells to the tumor tissue (3), which enhance anti-tumor immunity.
Discussion
Here, we demonstrated that the systemic administration of a platelet vector containing HVJ-E dominantly accumulates in tumor tissues and results in significant tumor regression in melanoma-bearing mice. Platelets were the only factor that was not detected by CD62P immunostaining in the tumor tissues; however, the PH detected by CD62P was well merged with the fibrin clots (Figure 2b). This finding suggests that PH was likely activated within the tumors to release HVJ-E from the platelets into the tumor microenvironment. Notably, some, but not all, of the PH overlapped with FITC-albumin. This result suggests that some HVJ-E may escape from tumor vessels to directly associate with the tumor cells. As shown in Supplementary Figure S4a, PH reduced the survival of cultured cancer cells. We previously reported that HVJ-E directly induced apoptosis in cultured cancer cells by viral RNA fragment-mediated RIG-I activation. The PH complex appears to enhance direct tumor cell killing via the same signaling pathway utilized by HVJ-E. In tumor-bearing mice, the direct killing of melanoma cells may also contribute to PH-mediated tumor suppression and the enhancement of anti-tumor immunity. However, when melanoma-bearing mice are treated with PH, the contribution of the direct cancer cell-killing activity of PH to the suppression of melanoma growth may be much less than that of its anti-tumor immune activity as the number of HVJ-E particles (189 HAU/mouse) introduced into the tumor-bearing mice was much smaller than the number of HVJ-E particles needed for the direct killing of cancer cells.
PH complexes successfully delivered HVJ-E particles and induced RANTES secretion, which enhanced the inhibition of tumor growth. In the in vitro assay, HVJ-E was released from the platelets via thrombin treatment. Although we had no clear evidence that HVJ-E was also released from platelets in the tumor tissues, activated platelets were detected with CD62P in the tumor tissues only when they contained HVJ-E. However, the reason for the activation of platelets containing HVJ-E in vivo remains to be elucidated.
Our results also demonstrated that platelets possess the ability to suppress the hemagglutination activity of HVJ-E, which is a limitation of HVJ-E. The intratumor injection of HVJ-E is being clinically tested in Japan for the treatment of patients with melanoma and prostate cancer. The systemic administration of HVJ-E using platelets will expand the applications for HVJ-E in cancer therapy.
After five injections of PH complexes or NaCl into B16F10-bearing mice, the serum was isolated and an antibody to HVJ-E was measured by mouse anti-Sendai virus (HVJ) IgG ELISA. The PH treatment group showed evidence of the production of anti-Sendai virus IgG (data not shown). However, we already reported that even in the presence of anti-HVJ antibodies, gene transfer using HVJ-E is not suppressed.11,12
In this study, we revealed that the RANTES chemokine, one of the major chemokines, induces a strong anti-tumor immune response. RANTES (CCL5), which is a member of the CC chemokine family, has strong chemotactic activity toward multiple immune cells, such as eosinophils, basophils, mast cells, monocytes, CTLs, naïve CD4+ T cells and memory CD45RO+ T cells.13,14,15,16 Several studies have investigated the efficacy of expressing RANTES for the promotion of anti-tumor immunity. These studies showed that the ectopic, intratumoral expression of RANTES attracted and activated tumor-specific and tumor-non-specific immune cells, such as DCs, CD4+ T cells, CD8+ T cells, and NK cells.17,18,19 PH complexes reached the fibrin-rich area of the tumor and released HVJ-E particles. Subsequently, the released HVJ-E particles stimulated the secretion of RANTES from the endothelial and/or tumor cells, and consequently, the secreted RANTES contributed to attracting anti-cancer immune cells.
However, when only recombinant RANTES was injected into the melanoma mass, tumor suppression was not significantly achieved (data not shown). Several studies have reported that RANTES exhibits pleiotropic activities in tumor development: it not only attracts immune cells to the tumor tissue, but also promotes tumor proliferation and invasion. Other studies have reported the expression of RANTES in primary human tumors, such as renal, prostate, breast and ovarian cancers, and melanoma.20,21,22,23,24,25,26 In the RANTES-expressing MCF-7 human breast cancer cell line, RANTES promoted cell proliferation by an mTOR-dependent process.27 Thus, the roles of RANTES in tumor biology remain somewhat controversial and are not fully understood. Therefore, our study of melanoma treatment using the PH complex shows that the presence of HVJ-E in melanoma tissue is likely to be essential for enhancing anti-tumor immunity in vivo, in addition to inducing the secretion of RANTES.
The incorporation of anti-cancer compounds and other viral particles into platelet vectors appears to be feasible. Although solving the problems related to the mass production and long-term storage of platelets for clinical use is important, we have already succeeded in incorporating HVJ-E into freeze-dried platelets. As shown in Supplementary Figure S1b, HVJ-E particles were incorporated into human platelets, which indicate the clinical utility of the PH complex.
Thus, platelet vectors are promising tools for cancer treatment with minimum toxicity.
Materials and Methods
Virus. HVJ (VR-105 parainfluenza1 Sendai/52, Z strain) was purchased from the American Type Culture Collection (ATCC; Manassas, VA) and amplified in the chorioallantoic fluid of 10- to 14-day-old chick eggs and purified by centrifugation, as previously described.28,29
Mice. Female C57BL/6N mice and Fox Chase SCID C.B-17/lcr-scid/scidJcl mice were purchased from Clea Japan (Tokyo, Japan) and were maintained in a temperature-controlled, pathogen-free room. All the animals were handled according to the approved protocols and guidelines of the Animal Committee of Osaka University (Suita, Japan).
Platelet purification and infusion of virus and fluorescent particles. Anticoagulated mouse blood was centrifuged at 200g for 3 minutes, and the upper, platelet-rich plasma (PRP) was collected. The PRP was centrifuged at 200g for 3 minutes, and the platelets were resuspended in modified Tyrode's buffer (134 mmol/l NaCl, 2.9 mmol/l KCl, 0.34 mmol/l Na2HPO4, 1 mmol/l MgCl2, 10 mmol/l HEPES, and 5 mmol/l D-glucose, pH 7.4). Prostaglandin I2 sodium salt was added to the PRP (1 pg/ml final concentration) and incubated for 2 minutes. The platelets were washed by the addition of 1 ml of Tyrode's buffer. Human platelets were isolated using the same procedure as mouse platelets from human blood that was donated by a human volunteer.
HVJ-E, Fluoresbrite plain YG (0.1 µm) microparticles (Polysciences, Warrington, PA) or SPHERO fluorescent particles (0.04–0.06 µm; Spherotech, Lake Forest, IL) were incubated with the platelets at 37 °C for 2 hours and were washed twice with modified Tyrode's buffer.
Transmission electron microscopy. Platelets and PH complexes were fixed with 2.5% glutaraldehyde in 0.1 mol/l phosphate buffer (pH 7.4) at 4 °C and were post-fixed in 1% OsO4 solution at 4 °C for 1 hour. The samples were dehydrated in a graded ethanol series and embedded in Quetol 812 epoxy resin (Nissin EM, Tokyo, Japan). Ultrathin sections (80 nm) were cut with a Reichert ultramicrotome (Ultracut E, Leica Microsystems K.K., Tokyo, Japan), stained with uranyl acetate and lead citrate and examined under a Hitachi H-7650 electron microscope (Hitachi, Tokyo, Japan).
Thrombin stimulation of HVJ-E or fluorescent/platelet complex. HVJ-E/platelet complexes were stimulated with thrombin (GE Healthcare UK Limited) for 4 hours at 37 °C and centrifuged at 200g to collect the platelets. The supernatant, which contained the released HVJ-E particles, was centrifuged at 20,400g. The platelets and released HVJ-E particle samples were analyzed by Western blot.
Fluorescent particles (SPHERO/platelet complexes) were stimulated with thrombin for 14 hours at RT and centrifuged at 200g. The fluorescence intensity of the supernatant was measured using a Mithras LB 940 (Berthold Japan K.K., Tokyo, Japan).
Western blot analysis. The samples were subjected to SDS-PAGE on 12% gels, and the proteins were transferred to Immobilon-P transfer membranes (Millipore, Billerica, MA). To detect the proteins, anti-M (Hokkaido System Science) or anti-β-actin (Sigma-Aldrich Japan, Tokyo, Japan) immunoglobulin G (IgG) was used as the primary antibody. ECL horseradish peroxidase-conjugated donkey anti-rabbit IgG (GE Healthcare UK) was used as a secondary antibody for the detection of M (matrix protein of HVJ), and ECL horseradish peroxidase-conjugated sheep anti-mouse IgG (GE Healthcare UK) was used as a secondary antibody for the detection of β-actin. The ECL Western Blotting Detection Reagent (GE Healthcare UK) was used to detect the signal for each protein.
Hemagglutination (HA) assay. PH complexes were serially diluted in PBS in a 96-well U-bottom plate. A 50-μl aliquot of a suspension of chicken red blood cells (0.5% RBCs) (Nacalai Tesque, Kyoto Japan) was then added to each well, and the samples were incubated at 4 °C for 16 hours. The agglutination titer was determined as the last dilution at which agglutination was detected.
Cell culture and proliferation assay. B16F10 (mouse melanoma), B16BL6 (mouse melanoma), LL/2 (mouse Lewis lung carcinoma) and MSC-1 (mouse mesenchymal stromal cells) cells were maintained in Dulbecco's modified Eagle's medium (Nacalai Tesque) Murine splenocytes were maintained in RPMI-1640 medium (Nacalai Tesque). All media were supplemented with 10% FBS (BioWest, Nuaillé, France), 100 U/ml penicillin and 0.1 mg/ml streptomycin (Penicillin-Streptomycin Mixed Solution, Nacalai Tesque). β-Mercaptoethanol (4 ng/ml) was added to the medium for the splenocyte culture. B16F10 cells (1.0 × 103 cells/well) were seeded into 96-well plates, and HVJ-E or PH complexes were added to the wells. After 24 or 48 hours of incubation, 20 µl of Cell Titer 96 AQueous One Solution (Promega, Madison, WI) was added to each well to measure the viability of the cancer cells. The plates were incubated at 37 °C for 2 hours, and the absorbance was measured at a wavelength of 490 nm according to the manufacturer's instructions.
In vivo tumor volume measurement and RANTES depletion. Viable B16F10 melanoma cells (1.0 × 106 cells) were resuspended in 50 µl of PBS and were intradermally injected into the backs of female C57BL/6N mice. When each tumor had grown to 3–5 mm in diameter, the mice were treated with an intravenous tail injection of platelets (2.5 × 105), HVJ-E (1.89 × 109 particles), PH complexes (1.89 × 109 particles of HVJ-E infused into 2.5 × 107 platelets, for a total volume of 50 µl), or PBS on days 0, 3, 6, 9 and 12. The tumor volume was measured in a blinded manner using slide calipers and was calculated using the following formula: tumor volume (mm3) = length × (width)2/2. To deplete RANTES, an anti-RANTES antibody (ab9915; Abcam, Cambridge, MA) was administered intravenously at the time of PH complex administration. Rabbit IgG (R&D Systems, Minneapolis, MN) was used as a control for the anti-RANTES antibody.
Immunostaining of immune cells in F10 tumor tissues. The PH complex-treated and other F10 melanoma tumor sections were fixed with 4% paraformaldehyde solution and blocked with 3% BSA. The sections were stained with anti-CD4 and anti-CD8 primary antibodies (Sigma-Aldrich Japan). The secondary antibodies included an Alexa Fluor 488-conjugated rabbit anti-mouse IgG (Life Technologies Corporation, Carlsbad, CA). The sections were mounted in Vectrashield mounting medium (Vector Laboratories, Burlingame, CA) and imaged with a confocal laser microscope (Radiance 2100; Bio-Rad Laboratories, Drive Hercules, CA) equipped with the Laser Sharp 2000 software program. Anti-CD62P (550289; BD Bioscieces Pharmingen, San Diego, CA) and anti-fibrin (D01P; Abnova, Taipei City, Taiwan) primary antibodies were used to detect activated platelets and fibrin, respectively, in the F10 tumor tissues.
Isolation of vascular endothelial cells from B16F10 tumors and cytokine and chemokine arrays. Tumor vascular endothelial cells were isolated from B16F10-bearing mice. The B16F10 tumor tissue was minced and enzymatically digested in PBS supplemented with 3% collagenase (Sigma-Aldrich Japan) using an orbital shaker for 30 minutes at 37 °C. The digested tissue was filtered through a 40-mm nylon mesh cell strainer (BD Biosciences, Bedford, MA). The sample was placed on top of a Percoll solution (Sigma-Aldrich Japan) and centrifuged at 400g for 30 minutes. The second layer, which contained endothelial cells, was transferred to a 100-mm culture dish and incubated at 37 °C in 5% CO2 for 1.5 hours. This procedure allowed the fibroblasts and preadipocytes contained in the mixture to attach to the culture dish. The medium, which contained suspended endothelial cells, was carefully transferred to another tube. The endothelial cells were further purified using an autoMACS Pro Separator with an anti-CD105 antibody (Miltenyi Biotech, Bergisch Gladbach, Germany). The isolated ECs or B16F10 cells were seeded into six-well plates (1.5 × 106 cells/well), and platelets (2.5 × 105), HVJ-E (1.89 × 109 particles), or PH complexes (1.89 × 109 particles) were added to the medium. After 24 hours, the media were collected and used as samples for cytokine and the chemokine arrays (Proteome Profiler Panel A Array Kit; RD Systems, Minneapolis, MN). The pixel densities of the spots on the array membranes were quantified using Image Quant TL software (GE Healthcare Japan, Tokyo, Japan).
Preparation of splenocytes and ELISPOT (mouse IFN-γ) assay. Spleens were removed aseptically from PH complex-treated mice after the fifth injection. Antigen-specific IFN-γ-producing cells were identified using an enzyme-linked immunosorbence spot (ELISPOT) assay. Briefly, ELISPOT plates (IP Sterile Clear Plate, MAIPS4510; Millipore, Bedford, MA) were coated with an anti-mouse IFN-γ capture Ab (Mouse IFN-γ ELISpot Development Module; RD Systems, Minneapolis, MN). Spleen cells (2.0 × 107 cells) and mitomycin C (Nacalai Tesque)-treated B16F10 melanoma cells (2.0 × 106 cells) were co-incubated for 12 hours. The pre-stimulated spleen cells (5.0 × 105 cells) were placed onto an antibody-coated plate with B16F10 cells (2.0 × 103 cells). After 48 hours of incubation at 37 °C, the cells were removed by washing the plates, and the sites of cytokine secretion were detected using biotinylated anti-mouse IFN-γ detection Ab and streptavidin-alkaline phosphatase conjugate (ELISpot Blue Color Module). The enzymatic reaction was developed with BCIP-NBT substrate.
Depletion of CD4, CD8, and NK cells in B16F10-bearing mice. Anti-CD4 (clone GK1.5) and anti-CD8 (clone 53–7.62) antibodies were kindly provided by Murakami (Osaka University, Suita, Japan), and the anti-asialo GM1 antibody was purchased from Wako Pure Chemical Industries. To deplete the CD4 T cells, CD8 T cells, or NK cells, each antibody [anti-CD4 (200 mg), anti-CD8 (500 mg), or anti-asialo GM1 (20 mg), respectively] was administered intraperitoneally on days -1, 0, 1, 2, 4, 6, 8, 11, and 14. Rat IgG (Sigma) was used as a control for the anti-CD4 and anti-CD8 antibodies, and rabbit IgG (R&D Systems) was used as a control for the anti-asialo GM1 antibody.
Statistical analysis. The statistical analyses were conducted using the Tukey-Kramer test or Student's unpaired t-test, and P values less than 0.05 were considered statistically significant.
SUPPLEMENTARY MATERIAL Figure S1. Immunostaining of infused HVJ-E F-protein in mouse and human platelets. Figure S2. Survival rate of PH complex-treated B16F10-bearing mice. Figure S3. Tumor sections were stained with anti-CD49b and CD11c to visualize the localization of NK cells and DCs. Figure S4. Proliferation assay of HVJ-E and PH complex-treated PC3s and human endothelial cells (HUVECs and HAECs). Figure S5. Proliferation assay of PH-treated RIG-I and MAVS siRNA-transfected B16F10 cells. Figure S6. Human cytokine and chemokine array of PH complex-stimulated HUVEC, TEC, and B16F10 cells. Figure S7. Survival rate of PH complex and RANTES antibody-treated B16F10-bearing mice. Figure S8. Specific antitumor effects induced by PH complex injection into B16F10-bearing mice, analyzed by ELISPOT and 51Cr-release assay. Figure S9. Thrombus and fibrin clot localization in various tumors (B16F10, B16BL6, and LL/2). Supplementary Materials and Methods
Acknowledgments
This work was financially supported by Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science. The authors thank Masaaki Murakami (Hokkaido University, Sapporo, Japan) for providing anti-CD4 and anti-CD8 antibodies. The work was conducted in Suita, Osaka, Japan. The authors declare no conflict of interest.
Supplementary Material
References
- Ehrlich P. Collected Studies on Immunity. J. Wiley & sons; New York; 1906. [Google Scholar]
- Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- Altaner C, Altanerova V, Cihova M, Ondicova K, Rychly B, Baciak L, et al. Complete regression of glioblastoma by mesenchymal stem cells mediated prodrug gene therapy simulating clinical therapeutic scenario. Int J Cancer. 2014;134:1458–1465. doi: 10.1002/ijc.28455. [DOI] [PubMed] [Google Scholar]
- Taniguchi S, Fujimori M, Sasaki T, Tsutsui H, Shimatani Y, Seki K, et al. Targeting solid tumors with non-pathogenic obligate anaerobic bacteria. Cancer Sci. 2010;101:1925–1932. doi: 10.1111/j.1349-7006.2010.01628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y. Cancer stromal targeting (CAST) therapy. Adv Drug Deliv Rev. 2012;64:710–719. doi: 10.1016/j.addr.2011.12.010. [DOI] [PubMed] [Google Scholar]
- Male R, Vannier WE, Baldeschwieler JD. Phagocytosis of liposomes by human platelets. Proc Natl Acad Sci U S A. 1992;89:9191–9195. doi: 10.1073/pnas.89.19.9191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaujac C, Boukour S, Cramer-Bordé E. Platelets and viruses: an ambivalent relationship. Cell Mol Life Sci. 2010;67:545–556. doi: 10.1007/s00018-009-0209-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda Y, Nakajima T, Nishikawa T, Yamamoto S, Ikegami H, Suzuki N, et al. Hemagglutinating virus of Japan (HVJ) envelope vector as a versatile gene delivery system. Mol Ther. 2002;6:219–226. doi: 10.1006/mthe.2002.0647. [DOI] [PubMed] [Google Scholar]
- Kaneda Y. Virosome: a novel vector to enable multi-modal strategies for cancer therapy. Adv Drug Deliv Rev. 2012;64:730–738. doi: 10.1016/j.addr.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Matsushima-Miyagi T, Hatano K, Nomura M, Li-Wen L, Nishikawa T, Saga K, et al. TRAIL and Noxa are selectively upregulated in prostate cancer cells downstream of the RIG-I/MAVS signaling pathway by nonreplicating Sendai virus particles. Clin Cancer Res. 2012;18:6271–6283. doi: 10.1158/1078-0432.CCR-12-1595. [DOI] [PubMed] [Google Scholar]
- Kaneda Y, Yamamoto S., Nakajima T. Development of HVJ Envelope Vector and its Application to Gene Therapy. Elsevier Academic Press; Amsterdam, The Netherlands; 2005. [Google Scholar]
- Mima H, Tomoshige R, Kanamori T, Tabata Y, Yamamoto S, Ito S, et al. Biocompatible polymer enhances the in vitro and in vivo transfection efficiency of HVJ envelope vector. J Gene Med. 2005;7:888–897. doi: 10.1002/jgm.735. [DOI] [PubMed] [Google Scholar]
- Alam R, Stafford S, Forsythe P, Harrison R, Faubion D, Lett-Brown MA, et al. RANTES is a chemotactic and activating factor for human eosinophils. J Immunol. 1993;150 8 Pt 1:3442–3448. [PubMed] [Google Scholar]
- Bischoff SC, Krieger M, Brunner T, Rot A, von Tscharner V, Baggiolini M, et al. RANTES and related chemokines activate human basophil granulocytes through different G protein-coupled receptors. Eur J Immunol. 1993;23:761–767. doi: 10.1002/eji.1830230329. [DOI] [PubMed] [Google Scholar]
- Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature. 1990;347:669–671. doi: 10.1038/347669a0. [DOI] [PubMed] [Google Scholar]
- Pietrzak A, Misiak-Tłoczek A, Brzezińska-Błaszczyk E. Interleukin (IL)-10 inhibits RANTES-, tumour necrosis factor (TNF)- and nerve growth factor (NGF)-induced mast cell migratory response but is not a mast cell chemoattractant. Immunol Lett. 2009;123:46–51. doi: 10.1016/j.imlet.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Fuentes-Beltrán A, Montes-Vizuet R, Valencia-Maqueda E, Negrete-García MC, García-Cruz Mde L, Teran LM. Chemokine CC-ligand 5 production and eosinophil activation into the upper airways of aspirin-sensitive patients. Clin Exp Allergy. 2009;39:491–499. doi: 10.1111/j.1365-2222.2008.03190.x. [DOI] [PubMed] [Google Scholar]
- Sallusto F, Lenig D, Mackay CR, Lanzavecchia A. Flexible programs of chemokine receptor expression on human polarized T helper 1 and 2 lymphocytes. J Exp Med. 1998;187:875–883. doi: 10.1084/jem.187.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JJ, Qin S, Unutmaz D, Soler D, Murphy KE, Hodge MR, et al. Unique subpopulations of CD56+ NK and NK-T peripheral blood lymphocytes identified by chemokine receptor expression repertoire. J Immunol. 2001;166:6477–6482. doi: 10.4049/jimmunol.166.11.6477. [DOI] [PubMed] [Google Scholar]
- Mrowietz U, Schwenk U, Maune S, Bartels J, Küpper M, Fichtner I, et al. The chemokine RANTES is secreted by human melanoma cells and is associated with enhanced tumour formation in nude mice. Br J Cancer. 1999;79:1025–1031. doi: 10.1038/sj.bjc.6690164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke F, Relf M, Negus R, Balkwill F. A cytokine profile of normal and malignant ovary. Cytokine. 1996;8:578–585. doi: 10.1006/cyto.1996.0077. [DOI] [PubMed] [Google Scholar]
- Niwa Y, Akamatsu H, Niwa H, Sumi H, Ozaki Y, Abe A. Correlation of tissue and plasma RANTES levels with disease course in patients with breast or cervical cancer. Clin Cancer Res. 2001;7:285–289. [PubMed] [Google Scholar]
- Luboshits G, Shina S, Kaplan O, Engelberg S, Nass D, Lifshitz-Mercer B, et al. Elevated expression of the CC chemokine regulated on activation, normal T cell expressed and secreted (RANTES) in advanced breast carcinoma. Cancer Res. 1999;59:4681–4687. [PubMed] [Google Scholar]
- Vaday GG, Peehl DM, Kadam PA, Lawrence DM. Expression of CCL5 (RANTES) and CCR5 in prostate cancer. Prostate. 2006;66:124–134. doi: 10.1002/pros.20306. [DOI] [PubMed] [Google Scholar]
- Wilcox RA, Wada DA, Ziesmer SC, Elsawa SF, Comfere NI, Dietz AB, et al. Monocytes promote tumor cell survival in T-cell lymphoproliferative disorders and are impaired in their ability to differentiate into mature dendritic cells. Blood. 2009;114:2936–2944. doi: 10.1182/blood-2009-05-220111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König JE, Senge T, Allhoff EP, König W. Analysis of the inflammatory network in benign prostate hyperplasia and prostate cancer. Prostate. 2004;58:121–129. doi: 10.1002/pros.10317. [DOI] [PubMed] [Google Scholar]
- Murooka TT, Rahbar R, Fish EN. CCL5 promotes proliferation of MCF-7 cells through mTOR-dependent mRNA translation. Biochem Biophys Res Commun. 2009;387:381–386. doi: 10.1016/j.bbrc.2009.07.035. [DOI] [PubMed] [Google Scholar]
- Kurooka M, Kaneda Y. Inactivated Sendai virus particles eradicate tumors by inducing immune responses through blocking regulatory T cells. Cancer Res. 2007;67:227–236. doi: 10.1158/0008-5472.CAN-06-1615. [DOI] [PubMed] [Google Scholar]
- Fujihara A, Kurooka M, Miki T, Kaneda Y. Intratumoral injection of inactivated Sendai virus particles elicits strong antitumor activity by enhancing local CXCL10 expression and systemic NK cell activation. Cancer Immunol Immunother. 2008;57:73–84. doi: 10.1007/s00262-007-0351-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.