

Abstract
We have previously shown that for the majority of antigens, adenoviral vaccines expressing the target antigen fused to the MHC associated invariant chain (Ii) induce an accelerated, augmented, and prolonged transgene-specific CD8+ T-cell response. Here we describe a new adenoviral vaccine vector approach where the target antigen fused to Ii is expressed from the adenoviral E1 region and IL-2 is expressed from the E3 region. Immunization of mice with this new vector construct resulted in an augmented primary effector CD8+ T-cell response. Furthermore, in a melanoma model we observed significantly prolonged tumor control in vaccinated wild type (WT) mice. The improved tumor control required antigen-specific cells, since no tumor control was observed, unless the melanoma cells expressed the vaccine targeted antigen. We also tested our new vaccine in immunodeficient (CD80/86 deficient) mice. Following vaccination with the IL-2 expressing construct, these mice were able to raise a delayed but substantial CD8+ T-cell response, and to control melanoma growth nearly as efficaciously as similarly vaccinated WT mice. Taken together, these results demonstrate that current vaccine vectors can be improved and even tailored to meet specific demands: in the context of therapeutic vaccination, the capacity to promote an augmented effector T-cell response.
Introduction
Over time, many strategies to induce potent tumor-specific T-cell responses have been devised. However, most potential cancer vaccines fail to provide convincing results in human patients with advanced disease, thus leaving plenty of room for improvement. Compared to vaccination against pathogens, tumor therapy is extremely demanding as one often has to cope with a pre-established state of immune dysfunction induced by the tumor and its environment.1 CD8+ T cells are generally believed to play a key role in tumor control, and, consequently, the induction of a potent tumor-specific CD8+ T-cell response represents a major goal of any tumor vaccine. Replication deficient adenoviruses, particularly of the group C subtype, have been found to be very efficient inducers of potent CD8+ T-cell responses as demonstrated in both mice and primates.2 Interestingly, in recent studies we showed that a vaccine expressing an exogenous tumor antigen (the glycoprotein of lymphocytic choriomeningitis virus (LCMV GP)) fused to the MHC class II associated Invariant chain (Ii) (Ad5-IiGP) elicits an augmented CD8+ T-cell response.3 Furthermore, when this vaccine was tested in a tumor model against hard-to-treat GP-expressing melanomas, significantly prolonged tumor control was observed.4 Nevertheless, although tumor growth could be dramatically delayed using improved adenoviral vaccines, non-eradicated melanoma cells still tended to rebound, and long-term survival of tumor challenged mice remained unachievable unless additional therapy was provided.5 The mechanism underlying this consistent tumor re-growth is unknown, but it could theoretically be caused by both immune escape and/or the gradual establishment of an immunosuppressive tumor microenvironment.6 Accordingly, we hypothesized that adenoviral vaccine vectors co-expressing a tumor Ag alongside a cytokine, which promotes effector T-cell differentiation, might increase the antitumor efficacy. Accordingly, we redesigned our Ad5 vector expressing LCMV GP fused to Ii to co-express interleukin-2 (Ad5-IiGP-IL-2).
Based on in vitro studies, IL-2 was initially considered a unique T-cell growth factor and a “master cytokine” in relation to T-cell activation.7,8 The role of IL-2 signaling in vivo, on the other hand, has until recently been quite difficult to define.9 One reason for this is that besides serving as an immune-enhancer, IL-2 may also promote activation-induced cell death10 and exert a positive feed-back on regulatory T cells (Tregs).11,12 However, a growing body of experimental evidence now unequivocally point to a non-redundant role of IL-2 in promoting the differentiation and accumulation of cytolytic effector CD8+ T cells in vivo.13,14,15,16,17,18,19,20,21 Since IL-2 has also been found to overcome the proliferation block of anergic cells22 as well as inhibition through the PD1:PD-L pathway,23 a clear rationale for administration of IL-2 in cancer immunotherapy can be found in the literature. Indeed, systemic IL-2 therapy represents a contemporary treatment option and is associated with complete responses against a variety of cancers.24 Notably, unremitting complete responses, i.e., cure from disease, are frequently reported,24 but unfortunately, systemic delivery of IL-2 is hampered by dose-limiting toxicities.25 As a consequence, low dose IL-2 has been suggested as an alternative treatment modality. However, this results in poor therapeutic scores with decreased protection, duration and quality of the responses.26 Furthermore, the short half-life of IL-2 (minutes),27 necessitates frequent and recurrent administrations, unless complexed to an antibody.28
An alternative delivery method, co-encoding the cytokine of interest with the vaccine antigen is not a completely new concept, and IL-2 is one among many cytokines that have been successfully explored as DNA vaccine adjuvants.29 However, viral vectors come with an intrinsic adjuvanticity, and only a few reports have claimed improved efficacy of viral vectored vaccines through cytokine co-administration. Our results demonstrate that current adenoviral vaccine vectors can be improved and even tailored to meet specific demands, such as an augmented generation of antigen-specific effector CD8+ T cells.
Results
Co-expression of IL-2 augments the Ag-specific CD8+ T-cell response in WT mice
With the aim to study the impact of IL-2 expression from antigen-expressing adenoviral vectors on the immunogenicity of these vaccines, three new vaccine constructs were generated targeting either the GP or, for control, the nucleoprotein (NP) of LCMV (Ad5-IiGP-IL2, Ad5-GP-IL2, Ad5-NP-IL2). Initially we performed in vitro analyses to ascertain the functionality of our newly designed vaccine constructs in terms of IL-2 secretion and the biological activity of the secreted product. To this end, we infected immortalized APC-like JAWII cells30 at high multiplicity. As expected, cells infected with any of our IL-2 encoding constructs, acquired the capacity to secrete equal and biological functional amounts of IL-2 as demonstrated not only by standard ELISA titration (Supplementary Figure S1a), but also in a bioasssay of T cell proliferation (Supplementary Figure S1b) using in vivo generated virus-primed T-cell blasts as indicators of IL-2 activity.31 Regarding the importance of MOI on IL-2 secretion, cf. Supplementary Figure S1c.
Next, we studied whether vector induced expression of IL-2 would augment the Ag-specific T-cell response elicited in WT mice. WT mice were vaccinated using either the conventional Ad5-IiGP construct or a similar construct co-expressing IL-2, and on days 7, 11, 14, 21, 48, and 300 p.i., numbers of Ag-specific CD8 T cells were determined. As can be seen in Figure 1a, the IL-2 encoding construct significantly augmented the primary CD8+ T-cell response compared to that elicited in Ad5-IiGP vaccinated mice, resulting in a significant increase in numbers of IFNγ producing Ag-specific T cells in the spleen 11–21 days post vaccination (p.v.) (for a set of representative dot plots, cf. Supplementary Figure S2).
Figure 1.

Co-expression of IL-2 augments the Ag-specific T cell response in WT mice. WT mice were vaccinated in the hind footpad with 2 × 107 IFU Ad5-IiGP ± IL-2 in 30 μl PBS. (a) At the indicated time points subsequent to vaccination, spleens were harvested, and numbers of GP33-specific CD8+ T cells was quantified by intracellular IFNγ staining. (b) As in a, but the fraction of antigen-specific memory precursor effector cells (MPEC), double positive (DP) cells, double negative (DN) cells and short-lived effector cells (SLEC) are depicted. MPECs are characterized as CD127highKLRG-1low. DPs are CD127highKLRG-1high. DNs are CD127lowKLRG-1low, and SLECs are CD127lowKLRG-1high.
Biologically relevant levels of vector produced IL-2 are restricted to the local environment surrounding infected APCs
An important question connected with the finding of an increased effector T cell response is whether the frequency of circulating Tregs is similarly increased as this might potentially annul the beneficial effect of co-expressing IL-2 from APCs. However, when vaccinated mice were analyzed 11, 14, or 21 days after vaccination (Figure 2a), numbers of Tregs in the spleen were not significantly different whether the mice had been vaccinated using the IL-2 encoding construct or the conventional construct. Consistent with this absence of evidence for a systemic effect of IL-2 in mice vaccinated with the IL-2 expressing vector, we could not detect IL-2 in the circulation of these mice when serum was analyzed 3 days after local vaccination (<20 pg/ml serum), a time point at which protein expression is expected to be near its peak.
Figure 2.

Analyzing the action range of vector produced IL-2. (a) WT mice were vaccinated in the hind footpad with 2 × 107 IFU Ad5-IiGP ± IL-2 in 30 μl PBS. After 11, 14, and 21 days, spleens were harvested, and numbers of Tregs (FoxP3+CD25+ CD4+) were quantified by flow cytometry. (b,c) groups of WT mice were co-infected in the same footpad with two different antigen-expressing constructs (NP or GP), only one of which co-expressed IL-2 at a time; controls were mice vaccinated with both antigens in the absence of vector encoded IL-2. Eleven days later numbers of GP- and NP-specific CD8 T cells were determined by intracellular IFNγ staining. Data are pooled from three independent experiments.
To further study the action range of vector produced IL-2, we performed additional experiments in which groups of mice were co-infected in the same footpad with two different antigen-expressing constructs (NP or GP), only one of which co-expressed IL-2 at a time; as controls we used mice vaccinated with both antigens in the absence of vector encoded IL-2. As can be seen in Figure 2b,c, a statistically significant increase in numbers of antigen-specific effector CD8+ T cells was only observed when antigen and IL-2 were co-expressed from the same vector, indicating that the level of IL-2 produced from infected APCs in vivo was only sufficient to significantly impact T-cell activation in the immediate vicinity of these cells.
Prolonged CD25 expression on antigen-specific CD8+ T cells in the lymph node draining the vaccination site
The determinants for generation of an increased number of effector CD8+ T cells could be a number of factors including increased cell proliferation and/or decreased susceptibility to apoptosis. However, when we analyzed antigen-specific CD8+ T cells harvested from the spleen on day 9 p.v., at which time the difference between mice vaccinated with or without IL-2 encoded in the vector is still increasing (cf. Figure 1a), we did not observe a difference in the expression of neither the survival marker Bcl-2, nor the apoptotic marker Annexin-V (Figure 3a), though expression of both factors are known to be influenced by IL-2 stimulation.32,33
Figure 3.

Prolonged CD25 expression on antigen-specific CD8 T cells in draining lymph nodes. (a) WT mice were vaccinated in the hind footpad with 2 × 107 IFU Ad5-IiGP ± IL-2 in 30 μl PBS. Nine days later spleens and popliteal lymph nodes were harvested and cells were stained with CD8 and CD44 specific Abs in addition to GP33- and GP34-specific tetramers (to define antigen-specific CD8 T cells) followed by staining for the apoptotic marker Annexin V, the anti-apoptotic marker BCL-2 or CD25; cellular distribution with regard to the indicated markers is depicted. Filled graph: Ad5-IiGP-IL-2; full line: Ad5-IiGP. (b) 107 splenocytes from CD8+ or CD4+ TCR transgenic donors (TCR318 and SMARTA, respectively) were adoptively transferred into naive recipients subsequent to CFSE labeling. One day after cell transfer, the mice were vaccinated with 2 × 107 IFU Ad5-IiGP ± IL-2. Seventy hours later the popliteal lymph nodes were harvested, and donor cell distribution with regard to CFSE dilution was analyzed. Full line: Ad5-IiGP-IL-2; filled graph: Ad5-IiGP. Results are representative data from two to four independent experiments with four to six animals per group per experiment.
To address if increased IL-2 production from APCs accelerated early T-cell proliferation, we made use of a transfer model where CSFE labeled TCR transgenic CD8+ or CD4+ T cells from either CD45.1 or CD45.2 donors are transferred into recipients expressing CD45.2 or CD45.1, respectively. One day later recipients were vaccinated using either construct, and 70 hours later CSFE dilution patterns in the draining lymph nodes were compared. As evident from Figure 3b, we could not detect altered proliferation patterns following IL-2 expression from the vector; neither in CD8+ nor CD4+ T cells. The latter has therapeutic relevance, since the timing of co-administered IL-2 is essential for proper CD4+ T-cell activation17 and incorrect timing may impact the CD4 T-cell response negatively.
Lastly, extended IL-2 secretion from the APCs may facilitate prolonged expression of the IL-2 receptor-alpha (CD25), which is recycled upon stimulation,34 hence causing a sustained activation phase. A likely net result from such a feed-back would be an increased number of circulating Ag-specific T cells at the peak of the adaptive immune response as we observed in our kinetics analysis (see Figure 1a), even if increased proliferation is not clearly discernible in our proliferation assay which, notably, is performed using high avidity TCR transgenic cells and at a higher concentration of antigen-specific cells than normally found at least during the early phase of antigen stimulation. In accordance with this hypothesis, we witnessed an up-regulation of CD25 expression on antigen-specific CD8+ T cells in the vaccine draining lymph nodes of IL-2 vaccinated mice on day 9 p.v.(Figure 3a). Of therapeutic importance, increased CD25 expression was not observed in the spleen, making this a local phenomenon restricted to the antigen presenting microenvironment, consistent with our findings regarding the short action range of the locally augmented IL-2 activity (Figure 2b,c).
Notably, extended up-regulation of CD25 tends to favor terminal-effector differentiation,13,18 which could cause an augmented effector response at the expense of reduced T-cell memory. It is therefore important to note that expression of IL-2 from the vaccine construct did not significantly influence the phenotypes of the generated T cells nor did it have a negative impact on the number of antigen-specific CD8+ T cell found late after vaccination (Figure 1a,b, respectively). Similarly, regarding cytokine profiles these were identical with most cells producing IFNγ in combination with TNF-α as normally expected for primary effector CD8+ T cells induced by adenoviral immunization (data not shown).
IL-2 encoding Ad5 constructs are less dependent on CD4 T-cell help
Normally, vaccination with adenoviral vectors requires CD4+ T-cell help for induction of a good CD8+ T-cell response.35 The above results seem to suggest that in Ad5 vaccinated WT mice the availability of IL-2 represents a limiting factor concerning the magnitude of the induced response. Since a key function of CD4+ Thelper cells during help-dependent primary CD8+ T-cell responses is to deliver IL-2,36,37,38 we next sought to address if employing an IL-2 encoding vaccine construct would diminish the necessity for CD4+ T-cell help during vaccination with adenoviral vectors. Importantly, in the construct used as reference so far, fusion of the expressed Ag to Ii, has previously been shown to minimize the requirement for CD4+ T-cell help in vivo.35 Accordingly, we vaccinated MHC-II deficient knockout mice employing a weaker vaccine construct encoding only GP, but not Ii, and again, either with or without co-expression of IL-2. As can be seen in Figure 4a, the inclusion of IL-2 in the vaccine construct reduced the requirement for CD4+ T-cell help, and the antigen-specific CD8+ T-cell response elicited with the IL-2 encoding construct in CD4 T-cell deficient mice roughly matched that induced in WT mice in the absence of IL-2. Thus, together our results with the IL-2 expressing construct suggest that availability of IL-2 is a key factor in regulating the magnitude of adenovector induced CD8+ T-cell responses and that a central role of CD4+ T cells is to produce sufficient amounts of endogenous IL-2.
Figure 4.

Co-expression of IL-2 reduces the requirement for CD4 T-cell help and increases the expansion of CD25 expressing cells. (a) WT or MHC-II-/- mice were vaccinated with Ad5-GP, either with or without IL-2. Fourteen days after vaccination GP33-specific splenic T cells were quantified by intracellular cytokine staining. (b) Lethally irradiated B6.SJL (CD45.1) mice were reconstituted with a mixture of bone-marrow cells from CD25-/- (CD45.2) and B6.SJL (CD25+/+) mice. Ten weeks after irradiation and cell transfer, the mice were vaccinated with the indicated construct, and 11 days later, GP33-specific splenic CD8+ T cells were quantified by intracellular cytokine staining. The data represent one out of two independent experiments.
To further support a direct effect of IL-2 on the CD8+ T cells, we reconstituted lethally irradiated B6.SJL (CD45.1) mice with a mixture of bone marrow from CD25-/- (CD45.2) and CD25+/+ B6.SJL (CD45.1) mice. Ten weeks after reconstitution, the mice were vaccinated with Ad5-IiGP either with or without IL-2, and 11 days later antigen-specific CD8+ T cells of each genotype were enumerated (Figure 4b). While similar low numbers of effector cells were generated from CD25-/- donor cells, irrespectively of the vaccine construct used for immunization, expansion of WT cells tended to be greater following vaccination with the IL-2 expressing construct.
IL-2 increases the Ag-mediated survival of WT mice challenged with B16.F10-GP melanoma cells
Previously, we have shown that the Ad5-IiGP induced CD8+ T-cell mediated control of subcutanous (s.c.) GP33-expressing melanomas could parallel that associated with stimulation by live LCMV virus, which in many respects stands as a gold standard for CD8+ T-cell activation.4 However, long-term survivors are still a rare phenomenon. Consequently, to see if we could improve the vaccine induced tumor control, we tested our IL-2 expressing construct under similar conditions. Thus we challenged WT mice with 106 B16.F10-GP melanoma cells s.c. followed by therapeutic vaccination with Ad5-IiGP or Ad5-IiGP-IL-2 5 days later. As can be seen in Figure 5a, the GP-specific, IL-2 encoding construct tended to prolong the survival.
Figure 5.
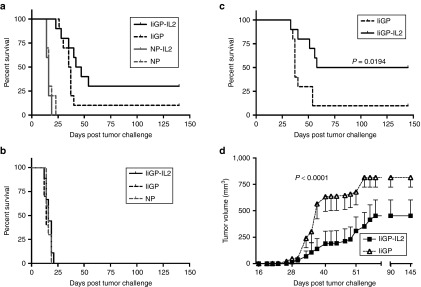
Inclusion of IL-2 in the vaccine construct improves both tumor control and overall survival. (a) WT mice were challenged with 106 B16.F10-GP melanoma cells at day 0. Five days later, all mice were vaccinated with vectors expressing either a relevant (GP) or an irrelevant (NP) Ag together with or without IL-2. (b) WT mice were challenged with 106 B16.F10 melanoma cells at day 0. Five days later mice were vaccinated with either Ad5-IiGP or Ad5-NP, both irrelevant antigens, or with Ad5-IiGP-IL-2. (c) WT mice were challenged with 105 B16.F10-GP melanoma cells at day 0. Five days later, all mice were vaccinated the Ad5-IiGP construct, either with or without inclusion of IL-2. (a–c) Mortality of tumor bearing mice as a function of time. (d) As in c, except that tumor volume as a function of time is depicted; data are presented as mean ± SEM. Representative data of two to three independent experiments; all with 10 mice per group. Mantel-Cox Log-Rank test were used for statistical evaluation of survival curves; two-way analysis of variance is used for comparison of tumor volume.
This effect is mediated by antigen-specific CD8+ T cells and not through a general, non-specific immune activation for two reasons. First, a similar construct encoding IL-2, but combined with an irrelevant antigen, LCMV NP, did not cause prolonged survival (Figure 5a). Similarly, if we challenged mice given the Ad-IiGP vaccine (±IL-2) with B16.F10 melanoma cells not expressing GP, there was no therapeutic effect of either vaccine (Figure 5b).
While the difference between Ad5-IiGP and Ad5-IiGP-IL-2 vaccinated mice regarding survival following challenge with 106 tumor cells only reached the level of statistical significance if data from several experiments were pooled, we could obtain significant results in individual experiments when the tumor challenge dose was lowered 10-fold (Figure 5c,d); thus, by expressing IL-2 from the vaccine vector, we managed not only to prolong the survival time, but also to increase the proportion of long-term survivors to 50% as opposed to 10% without IL-2 expression from the vector.
IL-2 by-passes the lack of co-stimulation in CD80/86-/- mice
The above results encouraged us to test the potency of the IL-2 encoding construct in a model mimicking the immunodeficiency seen in the context of many human tumors. For this purpose, mice deficient in co-stimulation, CD80/86-/- mice, seemed appropriate: both APCs in the tumor environment and the tumor cells themselves usually show low expression of CD80 and CD86,1,6 and mice lacking these molecules are known to have a blunted immune response.39,40 However, because CD80/86 engagement of CD28 is a direct mediator of IL-2 transcription,22 we hypothesized that the IL-2 encoding construct could reverse the immunodeficiency in these mice. Indeed, when CD80/86-/- mice were vaccinated with the IL-2 encoding construct and antigen-specific CD8+ T cells in the spleen were enumerated 14 days later, these mice were found to harbor nearly as many primary effector CD8+ T cells as did matched WT mice, and at least as many as WT mice given the conventional vaccine (Figure 6b). However, at 11 days p.v.(Figure 6a) the response in Ad5-IiGP-IL-2 vaccinated CD80/86-/- mice was rather low compared to that in matched WT mice, pointing to a delayed, but still functional, CD8+ T-cell response in the former mice when vaccinated with the Ad-IiGP-IL-2 vector.
Figure 6.
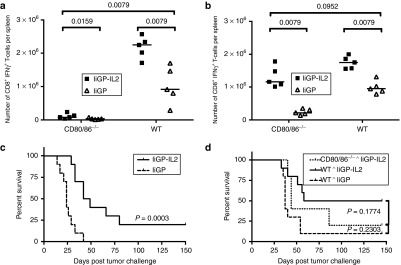
IL-2 expression from the vaccine construct dramatically increases immune competence in CD80/86-/- mice. (a,b) WT and CD80/86-/- mice were vaccinated with Ad5-IiGP, either with or without IL-2. (a) Eleven days or (b) 14 days after vaccination, GP33-specific splenic CD8+ T cells were quantified by intracellular staining. (c) CD80/86-/- mice were challenged with 105 B16.F10-GP melanoma cells at day 0. Five days later, all mice were vaccinated as indicated, tumor-induced mortality is depicted. (d) Comparison of the IL-2 encoding vaccine's potential in WT vs. CD80/86-/- mice. The WT mice are from the same experiment as depicted in Figure 2b. Representative data of two to three independent experiments, 5–10 mice per group. Statistic evaluation: (a,b) Mann–Whitney U-test; (c,d) Mantel-Cox Log-Rank Test.
To test the therapeutic potential of the vaccine under these conditions, we challenged CD80/86-/- mice with 105 B16.F10-GP melanoma cells followed by therapeutic vaccination 5 days after tumor challenge. As can be seen in Figure 6c, the IL-2 encoding vector dramatically increased the survival time of tumor challenged CD80/86-/-mice. Indeed, the survival of IL-2 vaccinated CD80/86-/- mice clearly matched that of WT mice vaccinated with the conventional construct, although it might not completely reach the level found in WT mice vaccinated with the IL-2 encoding construct (Figure 6d). Interestingly, by comparing the results obtained in Ad5-IiGP vaccinated CD80/86-/- mice to those observed in similarly vaccinated WT mice, it may be noted that this vaccine, which previously have been shown to operate almost independently of CD4+ Thelper cells3,35 still requires co-stimulation to be efficient (Figure 6a–d).
Numbers of Ag-specific CD8+ T cells is increased in tumor bearing and tumor challenged mice, respectively, subsequent to IL-2 vaccination
In agreement with previous studies, the prolonged survival of tumor challenged mice vaccinated using the IL-2-encoding construct appeared to require Ag-specific CD8+ T cells.4,41 In order to formally test if tumor challenged mice vaccinated with the IL-2 encoding construct had augmented CD8+ T-cell responses, we challenged WT mice with 106 B16.F10-GP melanoma cells and, 5 days later, vaccinated half the mice with Ad5-IiGP and the other half with Ad5-IiGP-IL-2. Ten days after vaccination, the CD8+ T-cell response in the spleen was studied. Both vaccines caused a significant increase in numbers of antigen-specific CD8+ T-cell in the spleen compared to unvaccinated controls. Importantly, and as expected, numbers of functional antigen-specific CD8+ T cells were higher in IL-2 vaccinated mice (Figure 7a), whereas numbers of FoxP3+CD25+CD4+ T cells remained unchanged (cf. Supplementary Figure S3); thus, notably, the ratio of Treg vs. GP-specific Tconv cells was markedly reduced in IL-2 vaccinated mice (Figure 7b).
Figure 7.

IL-2 inclusion in the vaccine construct increases anti-tumor function in tumor bearing mice. (a) WT mice were challenged with 106 B16.F10-GP melanoma cells in the right flank at day 0. Five days later, the mice were vaccinated as indicated, and after further 10 days, the spleens were harvested, and numbers of tumor-specific, GP33-specific T cells were determined by intracellular IFNγ staining. (b) As in A, except that the ratio between Tregs and GP33-specific Tconv cells are depicted. (c) Mice were vaccinated as indicated in the figure, followed by intravenous injection of 5 × 105 B16.F10-GP melanoma cells 11 days after vaccination. Forty-six hours later, the lungs were isolated, and numbers of tumor-specific, GP33-specific CD8+ T cells recovered were determined by intracellular IFNγ staining.
Additionally, a clinical important factor in tumor treatment is the number of Ag-specific T cells that manage to enter the tumor site. To analyze this, WT mice were challenged intravenously (i.v.) with 105 B16.F10-GP melanoma cells 11 days after vaccination with either vaccine construct (Ad5-IiGP or Ad5-IiGP-IL-2). Following i.v. injection of melanoma cells, a significant fraction of these cells will lodge in the lung vasculature, hence providing an opportunity to investigate the capacity of Ag-specific T cells to migrate to a site of newly implanted tumor cells. Accordingly, 46 hours after challenge we harvested the lungs, and numbers of IFNγ producing GP33-specific T cells found in this organ was determined. As evident from Figure 7c, both vaccines caused significantly more Ag-specific CD8+ T cells to be recovered from the lungs as compared to unvaccinated controls. Furthermore, significantly more cells were recovered from the lungs of Ad5-IiGP-IL-2 vaccinated tumor bearing mice compared to Ad5-IiGP immunized mice, indicating that the increased CD8+ T-cell response so far only demonstrated in the spleen of mice vaccinated with the IL-2 encoding construct is reflected locally as an increased invasion of the tumor site.
IL-2 inclusion in the vaccine does not alter the proliferative capacity of memory cells upon vaccinia challenge
As mentioned earlier, IL-2 is expected to promote a more terminal effector T-cell differentiation.13,18 Hence, the improved tumor control and augmented primary CD8+ T cell response could potentially be at the expense of an impaired capacity for immunological recall. However, as depicted in Figure 1b, phenotypes of recently activated Ag-specific T cells, in addition to the numbers of Ag-specific CD8+ T cells remaining following contraction, do not appear to be appreciably affected by IL-2 production from the vector.
To formally address the ability of vaccine-induced long-term memory CD8+ T-cells to expand and protect following rechallenge, WT mice previously vaccinated with a vector with or without IL-2 were challenged with vaccinia virus (VV) expressing the glycoprotein of LCMV (VV-GP) 60 days later. As shown in Figure 8a, prior vector-encoded IL-2 stimulation was not found to impair the vaccinia-driven secondary expansion of GP33-specific T cells. Even 275 days after vaccination, VV-GP induced CD8+ T-cell expansion in vaccinated mice was similar regardless of whether the vaccine had co-expressed IL-2 or not. Similarly, when it came to controlling vaccinia infection in the ovaries, no difference was observed (data not shown).
Figure 8.
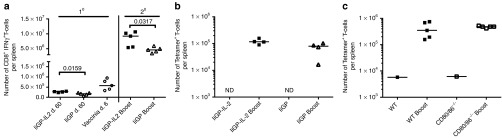
Vaccine encoded IL-2 does not impair Ag-specific CD8+ T-cell memor. (a) WT mice were vaccinated with Ad5-IiGP either with or without IL-2, and 60 days later some of the vaccinated mice and naive controls were challenged with 2 × 106 VV-GP i.p. Six days post challenge, numbers of GP33-specific T cells in the spleen were determined by intracellular IFNγ staining. (b) WT mice were vaccinated with Ad5-IiGP either with or without IL-2, and 120 days later 104 GP-specific CD8+ splenocytes were adoptively transferred into naive B6.SJL recipients. Prior to transfer the splenocytes from 5 donor mice per group were pooled and enriched for CD8+ T cells. One day after adoptive transfer the mice were challenged with 2 × 106 VV-GP. Six days after challenge, the spleens were isolated and the number of GP-specific T cells was quantified by tetramer staining. (c) 104 GP-specific splenocytes from day 60 Ad5-IiGP-IL-2 vaccinated mice were adoptively transferred into naive B6.SJL recipients. Subsequent procedures mirrors b, except that in this case the donor mice were either WT or CD80/86-/- as indicated in the figure.
In order to stringently eliminate any impact variations in numbers of vaccine primed GP-specific T cells present prior to challenge might have on the outcome of the memory response, WT mice were vaccinated and 120 days later, we adoptively transferred equal numbers of GP-specific T cells from Ad5-IIGP-IL2 and Ad5-IiGP vaccinated donors (CD45.2) into matched groups of naive recipients (CD45.1). Subsequent to cell transfer, the recipients were challenged with VV-GP and the expansion of antigen-specific CD45.2+CD8+ T cells was evaluated. Again, the recall response was of similar magnitude irrespectively of prior vaccination regimen (Figure 8b), suggesting similar recall capacity of the Ag-specific cells on a per cell basis, regardless of excess IL-2 stimulation during priming.
Following contraction, numbers of vaccine-induced memory CD8+ T cells in Ad5-IiGP-IL-2 vaccinated CD80/86-/- mice settled at a level close to that in similarly vaccinated WT mice (data not shown). To directly compare the proliferative potential of Ad5-IiGP-IL-2 induced memory cells in CD80/86-/- mice to those found in WT mice given the same vaccine, we again applied the adoptive transfer approach for an appropriate comparison. Equal numbers of antigen-specific CD8+ T cells from WT and CD80/86-/- mice were transferred into groups of naive recipients, and 1 day later the mice were challenged with VV-GP as previously described. Importantly, when T-cell expansion was analyzed following transfer into naive WT recipients, GP33-specific memory T cells generated with co-expression of IL-2 in the immunodeficient milieu of CD80/86-/- mice expanded as efficiently as GP33-specific T cells generated under similar condition in WT mice (Figure 8c), documenting the generation of a functional memory CD8+ T cell population (i.e., a T-cell subset capable of undergoing normal secondary expansion) in these mice, when vaccinated using the IL-2 expressing vaccine.
Discussion
The magnitude of a primary CD8+ T-cell response is decided by a number of factors, including the degree of co-stimulation and availability of CD4 T-cell help. Indeed, CD4 T cells seem to promote CD8+ T-cell responses via two interconnected synergistic pathways:36,38 First, via CD40L to license APCs to become potent inducers of CD8+ T-cell activation, which is associated with increased expression of the IL-2Rα chain. Second, by providing IL-2, which improves CD8+ T-cell survival and promotes effector cell differentiation. It is well known that many live infections are capable of inducing strong CD8+ T-cell responses in the absence of CD4 T-cell help. However, other infections and in particular immunization with non-live antigens typically requires CD4 help.
Regarding immunization with non-replicating adenoviral vectors it has previously been demonstrated that an efficient CD8+ T-cell response requires some form of CD4 T-cell help. An earlier study from our group demonstrated that this requirement may be alleviated if the antigen of interest is fused to Ii.35 The reason for this is not clear, but may be related to the fact that antigen presentation on the surface of the involved APCs is increased,35 thereby causing CD8+ T cells to require less co-stimulation and/or produce their own IL-2 in sufficient quantities for cell survival and differentiation.42 Interestingly, this type of construct also directly increased CD8+ T-cell responses, and significantly improved tumor control in mice therapeutically vaccinated with a vector encoding the tumor antigen fused to Ii.3,4
In this study, we extend our modifications of adenoviral vectors to include the expression of IL-2 from the E3 region of Ad5. When infected with this vector, APCs secrete increased amounts of IL-2 and, importantly, the functionality of the secreted product was validated in a T-cell proliferation assay using virus-induced CD8+ T cells. As expected when mice were vaccinated with this vector, we observed a transient, but significant improvement of the primary transgene-specific CD8+ T-cell response, and, notably, the augmentation of the primary response comes without any associated impairment of long-term memory, as might have been feared based on recent studies indicating that CD25 ligation causes a more terminal effector differentiation of the involved CD8+ T cells.13,18 The implications of these observations are two-fold. First, the augmented response suggests that IL-2 is a rate limiting factor during normal adenoviral vaccination of WT animals. Second, that perhaps a prolonged, but relatively small increase in availability of IL-2 may not be sufficient to negatively impact the generation of CD8+ T-cell memory, while still augmenting the effector response. Importantly, the improved effector T-cell response came without a concomitant increase in circulating Tregs. This probably reflects the very limited and localized increase in release of IL-2, which appears to be confined to the immediate vicinity of infected APCs. Regarding the use of the IL-2 encoding vector as an improved cancer vaccine, we found a moderate improvement in the survival of WT mice best demonstrated in mice with a low tumor load. However, tumor bearing individuals are typically not normal in their immune capacity, and impaired or blocked co-stimulation may represent an important problem.1 For this reason we decided also to test our new vaccine concept in CD80/86-/- deficient mice. Since it well-known that ligation of CD28, the receptor for CD80 and 86, represent a key trigger for IL-2 production,22 it is perhaps not surprising to find that using a vector, which allows IL-2 production from the same APCs that present the antigen, very substantially restores the immune function of these mice. The CD8+ T-cell response appears to be slightly delayed compared to that in WT mice, but still of substantial magnitude to significantly delay the growth of hard-to-treat melanomas. Whether other mechanism than an augmented CD8+ T-cell response, also contribute in this respect can be debated. However, we saw absolutely no anti-tumor effect when using an IL-2 expressing vector without the relevant target antigen encoded, nor when we tested the vaccine against melanoma cells not expressing the target antigen. Both of these findings strongly indicate that if additional immune mechanisms such as NK cell activation play any relevant role in mediating the improved tumor control, this is only in conjunction with antigen-specific CD8+ T cells.
Current attempts to vaccinate against cancer have met with little success. One reason for this is the presence of an immunodeficient state in patients with advanced disease. Various inhibitory mechanisms tend to abort the anti-tumor response and prevent the effector T cells nevertheless generated from efficiently controlling the tumor growth.1 This makes it unlikely that any single type of intervention will ever suffice in the clinical setting. Thus, active tumor therapy probably has to be assisted by immunomodulatory regimens directed at blocking inhibitory signals and/or augmenting stimulating pathways.1 We have previously shown that by combining active Ad5-IiGP vaccination with antagonistic anti-CTLA-4 and agonistic anti-CD40, the frequency of long-term non-progressors can be significantly improved, although more than half of the treated mice still succumb to a challenge dose of 106 melanoma cells s.c.5 In this study, we present an improved vaccine construct expressing IL-2 in addition to the tumor antigen of interest. Importantly, this vaccine also seemed to work quite efficiently in immunodeficient mice (and even induce relevant levels of CD8+ T-cell memory), suggesting that a similar strategy might be relevant in human patients with advanced disease. However, we do not envision a vaccine of this kind to be used on its own. Rather we foresee the vaccine to be applied in patients in which the primary tumor is removed by surgery. If used immediately prior to surgery it might be possible to completely control tumor metastasis and prevent death. Alternatively, the vaccine could be applied in combination with other modalities of tumor therapy such as immunomodulating antibodies or chemotherapy.
Materials And Methods
Ethics statement. Experiments were conducted in accordance with national guidelines regarding animal experiments as approved by the national ethics committee on experimental animal welfare.
Mice. All mice used in this study were maintained on a C57BL/6 (B6) background. Female WT B6 (CD45.2) and MHC II-/- mice were obtained from Taconic M&B (Ry, Denmark). WT B6.SJL (CD45.1) and CD80/86-/- mice were bred locally from breeding pairs originally obtained from the Jackson Laboratory (Bar Harbor, ME), while CD25-/- mice came directly from the Jackson laboratory. Transgenic mice (TCR318 and SMARTA) expressing a TCR for LCMV gp33-41 or gp61-80, respectively, were the progeny of breeding pairs provided by H Pircher, A Oxenius, and RM Zinkernagel (University of Zürich, Zürich, Switzerland). All mice used in this study were 7–12 weeks old at initiation of the experiments; they were housed in individually ventilated cages in a specific pathogen-free facility.
Adenoviral vectors and vaccination. The plasmid, pBHG10, utilized in this study for generation of our replication deficient Ad5 vaccine vector, has previously been described.43 Gene of interest (LCMV glycoprotein (GP) or LCMV nucleoprotein (NP) with or without linkage to Invariant chain (Ii)) is located in the E1 reading frame adjacent to a CMV-promoter. The IL-2 gene is inserted in the E3 reading frame bordering a PGK-promoter. All adenoviral vaccine vectors were produced as previously described.3 Prior to vaccination, all constructs were tested in vitro for the capacity to induce the secretion of biologically active IL-2 (see Figure 1). For vaccination, mice were briefly anesthetized using isoflurane, and 2 × 107 HEK293 infectious units (IFU) of Ad5 vaccine, diluted in 30 μl PBS, was administered in the right hind footpad.
Validating IL-2 production. Aliquots of 106 JAWII cells30 were infected with the relevant vector constructs at the indicated multiplicities of infection (MOI), and the cells were subsequently grown in 25 cm2 cell culture flasks with 5 ml of culture medium. Three days later cell-free culture supernatants were harvested, and vector-induced production of IL-2 was verified using a commercially available standard IL-2 ELISA (eBioscience, cat. no. 88–7024) and a bioassay based on the proliferation of virus-induced T-cell blasts as previously described.31 Briefly, mice were infected with LCMV virus and 6 days later splenocytes were harvested, and single-cell suspensions were incubated with tissue culture supernatant for 48 hours. Cultures were marked by adding [3H]-thymidin for the last 6 hours. After incubation, cells were harvested with a FilterMate 196 harvester (PerkinElmer), and [3H]-thymidin uptake was assayed by liquid scintillation counting on a TopCount (PerkinElmer) counter.
Tumor cell lines and tumor inoculation. B16.F10 and B16.F10-GP (expressing the minimal epitope of the LCMV glycoprotein, GP33-41) melanoma cells were cultured in DMEM 1965 supplemented with 10% FCS, glutamine, streptomycin, and penicillin. Additionally, in the case of B16.F10-GP cells G418 (0.8 mg/ml) was added to the culture medium. Both cell lines were kind gifts from Hanspeter Pircher (University of Freiburg, Germany).44
To study the capacity to resist solid tumors, mice were injected subcutaneously (s.c.) with 105 or 106 melanoma cells (diluted in 200 μl PBS) in the right flank at day 0. Tumor growth was measured every 2–3 days, and tumor volumes were calculated as length × width2 × 0.5236. When the tumors reached the size of ≥12 mm, the mice were euthanized for ethical reasons. For i.v. injections 300–500 μl PBS containing relevant tumor cells were injected via a tail vein
Vaccinia virus infection. Vaccinia virus expressing LCMV GP (VV-GP), grown on 13 CV-1 cells at low multiplicity of infection, was originally obtained from DHL Bishop (Oxford University, Oxford, UK) via A. Oxenius (ETH, Zürich, Switzerland). For infection of mice, 2 × 106 plaque-forming units (PFU) of VV-GP were administered intraperitoneal (i.p) in 300 μl PBS.
Cell preparation and enumeration. Single-cell suspensions of spleen, lung, or popliteal lymph node derived lymphocytes were obtained by pressing the organs through a fine steel mesh (mesh size, 70 μm), followed by centrifugation and two washes in Hanks buffered salt solution before resuspension in RPMI 1640 cell culture medium containing 10% fetal calf serum supplemented with NaHCO3, 2-mercaptoethanol, L-glutamine, and penicillin-streptomycin. All samples were subsequently counted on a Countess cell counter, Invitrogen.
Bone marrow chimeric mice. B6.SJL mice were lethally irradiated (9 Gray), and 1 day later reconstituted using single cell suspensions of bone marrow cells prepared from the femurs of CD25-/-and B6.SJL (CD25+/+) donor mice. A total of 107 cells were adoptively transferred (5 × 106 of each genotype, mixed and resuspended) into each recipient. Ten weeks after irradiation and cell transfer, the mice were used for vaccination.
Flow cytometry. Intracellular cytokine staining, or surface staining using GP-specific tetramers, was used to determine the frequencies of epitope specific CD8+ T cells. For enumeration of GP-specific T cells with tetramers, cells were stained with Abs for CD8+ and CD44 in combination with GP33-and GP34-specific PE/APC-conjugated tetramers, kindly provided by Søren Buus (University of Copenhagen, Denmark).45,46
Intracellular cytokine staining was performed after 5 hours of incubation with relevant peptides (0.1 µg/ml GP33) at 37 °C and 5% CO2. After incubation, the cells were stained according to our standard protocols46,47 using the following Abs (Nordic Biosite): CD4: PerCP-Cy5.5/APC-Cy7; CD8: PerCP-Cy5.5/PE-Cy7; CD44: APC-Cy7; KLRG-1: PE; CD127: PE-Cy7; Annexin V: PE; BCL-2: PE; CD25 PE/Bv421; FoxP3: FITC; B220: PacBlue; CD45.1:PE; CD45.2: PerCP-Cy5.5; and IFN-γ: APC. Samples were run on a LSRII flow cytometer (BD biosciences) and analyzed using FlowJo software (Tree Star).
CFSE labeling, transfer of TCR-transgenic splenocytes, and CFSE dilution analysis. Splenocytes from TCR-transgenic CD8+ (TCR318) or CD4+ (SMARTA) mice (CD45.1 and CD45.2, respectively) were labeled with CFSE at a concentration of 1 mol/l followed by 10 minutes of incubation in a 37 °C water bath. The reaction was terminated by adding FCS to reach a final concentration of 10%. Subsequently, the cells were washed three times in PBS before the cell numbers were determined using a cell counter (Countess, Invitrogen). A total of 107 CFSE-labeled cells were adoptively transferred i.v. into WT recipients (recipients were of the opposite CD45.1/2 background compared to the relevant donors). The following day, recipients were vaccinated with 2 × 107 IFU of the indicated Ad5-GP constructs in the right hind footpad. Seventy hours p.v., the draining popliteal lymph nodes were harvested and donor derived cells were identified and analyzed as described.
Evaluation of memory cell expansion in vivo. Spleen cells from vaccinated C57BL/6 (CD45.2) mice were negatively selected for CD4+ and Ig-positive cells (CD8+ T-cell enrichment) using relevant antibodies and magnetic beads. Subsequent to CD8+ enrichment, the cells were transferred to naive B6.SJL (CD45.1) recipients. Each recipient received 104 GP-specific CD8+ T cells, as enumerated by tetramer staining. One day after adoptive cell transfer, recipient mice were challenged i.p. with 2 × 106 PFU VV-GP. Six days later, the spleens were harvested, and single suspensions of splenocytes were stained with GP-specific tetramers in combination with anti-CD8, anti-CD44, anti CD45.1 and anti-CD45.2 to determine donor cell numbers.
Statistical analysis. Comparisons among groups in survival experiments were analyzed by the Log-Rank test (Mantel-Cox). Tumor volume are depicted as mean ± SE of mean and analyzed by two-way analysis of variance. A nonparametric Mann–Whitney U-test was used to compare quantitative data. GraphPad Prism (version 6) software was used for statistical analysis (GraphPad Software).
SUPPLEMENTARY MATERIAL Figure S1. All IL-2 expressing constructs secret equal amounts of biologically active IL-2. Figure S2. Co-expression of IL-2 augments the Ag-specific T-cell response in WT mice. Figure S3. Numbers of Tregs in the spleen of tumor-bearing mice are independent of the vaccination regimen.
Acknowledgments
The authors thank Søren Buus, University of Copenhagen, for providing relevant tetramers. This work was funded in part by the Danish Medical Research Council and The Novo Nordisk Foundation. Together with University of Copenhagen, the authors J.P.C., P.J.H., and A.R.T. are holding a patent on the invariant chain fusion strategy.
Supplementary Material
All IL-2 expressing constructs secret equal amounts of biologically active IL-2.
Co-expression of IL-2 augments the Ag-specific T-cell response in WT mice.
Numbers of Tregs in the spleen of tumor-bearing mice are independent of the vaccination regimen.
References
- Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. 2013;25:268–276. doi: 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- Colloca S, Barnes E, Folgori A, Ammendola V, Capone S, Cirillo A, et al. Vaccine vectors derived from a large collection of simian adenoviruses induce potent cellular immunity across multiple species. Sci Transl Med. 2012;4:115ra2. doi: 10.1126/scitranslmed.3002925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst PJ, Sorensen MR, Mandrup Jensen CM, Orskov C, Thomsen AR, Christensen JP. MHC class II-associated invariant chain linkage of antigen dramatically improves cell-mediated immunity induced by adenovirus vaccines. J Immunol. 2008;180:3339–3346. doi: 10.4049/jimmunol.180.5.3339. [DOI] [PubMed] [Google Scholar]
- Sorensen MR, Holst PJ, Pircher H, Christensen JP, Thomsen AR. Vaccination with an adenoviral vector encoding the tumor antigen directly linked to invariant chain induces potent CD4(+) T-cell-independent CD8(+) T-cell-mediated tumor control. Eur J Immunol. 2009;39:2725–2736. doi: 10.1002/eji.200939543. [DOI] [PubMed] [Google Scholar]
- Sorensen MR, Holst PJ, Steffensen MA, Christensen JP, Thomsen AR. Adenoviral vaccination combined with CD40 stimulation and CTLA-4 blockage can lead to complete tumor regression in a murine melanoma model. Vaccine. 2010;28:6757–6764. doi: 10.1016/j.vaccine.2010.07.066. [DOI] [PubMed] [Google Scholar]
- Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis S, Smith KA. Long term culture of tumour-specific cytotoxic T cells. Nature. 1977;268:154–156. doi: 10.1038/268154a0. [DOI] [PubMed] [Google Scholar]
- Smith KA. Interleukin-2: inception, impact, and implications. Science. 1988;240:1169–1176. doi: 10.1126/science.3131876. [DOI] [PubMed] [Google Scholar]
- Bachmann MF, Oxenius A. Interleukin 2: from immunostimulation to immunoregulation and back again. EMBO Rep. 2007;8:1142–1148. doi: 10.1038/sj.embor.7401099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenardo MJ. Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature. 1991;353:858–861. doi: 10.1038/353858a0. [DOI] [PubMed] [Google Scholar]
- Klebb G, Autenrieth IB, Haber H, Gillert E, Sadlack B, Smith KA, et al. Interleukin-2 is indispensable for development of immunological self-tolerance. Clin Immunol Immunopathol. 1996;81:282–286. doi: 10.1006/clin.1996.0190. [DOI] [PubMed] [Google Scholar]
- Papiernik M, de Moraes ML, Pontoux C, Vasseur F, Pénit C. Regulatory CD4 T cells: expression of IL-2R alpha chain, resistance to clonal deletion and IL-2 dependency. Int Immunol. 1998;10:371–378. doi: 10.1093/intimm/10.4.371. [DOI] [PubMed] [Google Scholar]
- Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32:79–90. doi: 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DM, Ravkov EV, Williams MA. Distinct roles for IL-2 and IL-15 in the differentiation and survival of CD8+ effector and memory T cells. J Immunol. 2010;184:6719–6730. doi: 10.4049/jimmunol.0904089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MA, Tyznik AJ, Bevan MJ. Interleukin-2 signals during priming are required for secondary expansion of CD8+ memory T cells. Nature. 2006;441:890–893. doi: 10.1038/nature04790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann MF, Wolint P, Walton S, Schwarz K, Oxenius A. Differential role of IL-2R signaling for CD8+ T cell responses in acute and chronic viral infections. Eur J Immunol. 2007;37:1502–1512. doi: 10.1002/eji.200637023. [DOI] [PubMed] [Google Scholar]
- Blattman JN, Grayson JM, Wherry EJ, Kaech SM, Smith KA, Ahmed R. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat Med. 2003;9:540–547. doi: 10.1038/nm866. [DOI] [PubMed] [Google Scholar]
- Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin-2Ralpha expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity. 2010;32:91–103. doi: 10.1016/j.immuni.2009.11.010. [DOI] [PubMed] [Google Scholar]
- D'Souza WN, Schluns KS, Masopust D, Lefrançois L. Essential role for IL-2 in the regulation of antiviral extralymphoid CD8 T cell responses. J Immunol. 2002;168:5566–5572. doi: 10.4049/jimmunol.168.11.5566. [DOI] [PubMed] [Google Scholar]
- D'Souza WN, Lefrançois L. IL-2 is not required for the initiation of CD8 T cell cycling but sustains expansion. J Immunol. 2003;171:5727–5735. doi: 10.4049/jimmunol.171.11.5727. [DOI] [PubMed] [Google Scholar]
- Cheng LE, Greenberg PD. Selective delivery of augmented IL-2 receptor signals to responding CD8+ T cells increases the size of the acute antiviral response and of the resulting memory T cell pool. J Immunol. 2002;169:4990–4997. doi: 10.4049/jimmunol.169.9.4990. [DOI] [PubMed] [Google Scholar]
- Powell JD, Ragheb JA, Kitagawa-Sakakida S, Schwartz RH. Molecular regulation of interleukin-2 expression by CD28 co-stimulation and anergy. Immunol Rev. 1998;165:287–300. doi: 10.1111/j.1600-065x.1998.tb01246.x. [DOI] [PubMed] [Google Scholar]
- Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, et al. PD-1:PD-L inhibitory pathway affects both CD4(+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol. 2002;32:634–643. doi: 10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Coventry BJ, Ashdown ML. The 20th anniversary of interleukin-2 therapy: bimodal role explaining longstanding random induction of complete clinical responses. Cancer Manag Res. 2012;4:215–221. doi: 10.2147/CMAR.S33979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg C, Létourneau S, Pantaleo G, Boyman O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proc Natl Acad Sci USA. 2010;107:11906–11911. doi: 10.1073/pnas.1002569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrescu DT, Ichim TE, Riordan NH, Marincola FM, Di Nardo A, Kabigting FD, et al. Immunotherapy for melanoma: current status and perspectives. J Immunother. 2010;33:570–590. doi: 10.1097/CJI.0b013e3181e032e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohue JH, Rosenberg SA. The fate of interleukin-2 after in vivo administration. J Immunol. 1983;130:2203–2208. [PubMed] [Google Scholar]
- Tomala J, Chmelova H, Mrkvan T, Rihova B, Kovar M. In vivo expansion of activated naive CD8+ T cells and NK cells driven by complexes of IL-2 and anti-IL-2 monoclonal antibody as novel approach of cancer immunotherapy. J Immunol. 2009;183:4904–4912. doi: 10.4049/jimmunol.0900284. [DOI] [PubMed] [Google Scholar]
- Senovilla L, Vacchelli E, Garcia P, Eggermont A, Fridman WH, Galon J, et al. Trial watch: DNA vaccines for cancer therapy. Oncoimmunology. 2013;2:e23803. doi: 10.4161/onci.23803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen TN, Haase C, Michelsen BK. Treatment of an immortalized APC cell line with both cytokines and LPS ensures effective T-cell activation in vitro. Scand J Immunol. 2002;56:492–503. doi: 10.1046/j.1365-3083.2002.01166.x. [DOI] [PubMed] [Google Scholar]
- Andersson EC, Christensen JP, Scheynius A, Marker O, Thomsen AR. Lymphocytic choriomeningitis virus infection is associated with long-standing perturbation of LFA-1 expression on CD8+ T cells. Scand J Immunol. 1995;42:110–118. doi: 10.1111/j.1365-3083.1995.tb03633.x. [DOI] [PubMed] [Google Scholar]
- Akbar AN, Borthwick NJ, Wickremasinghe RG, Panayoitidis P, Pilling D, Bofill M, et al. Interleukin-2 receptor common gamma-chain signaling cytokines regulate activated T cell apoptosis in response to growth factor withdrawal: selective induction of anti-apoptotic (bcl-2, bcl-xL) but not pro-apoptotic (bax, bcl-xS) gene expression. Eur J Immunol. 1996;26:294–299. doi: 10.1002/eji.1830260204. [DOI] [PubMed] [Google Scholar]
- Kuroda K, Yagi J, Imanishi K, Yan XJ, Li XY, Fujimaki W, et al. Implantation of IL-2-containing osmotic pump prolongs the survival of superantigen-reactive T cells expanded in mice injected with bacterial superantigen. J Immunol. 1996;157:1422–1431. [PubMed] [Google Scholar]
- Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26:453–479. doi: 10.1146/annurev.immunol.26.021607.090357. [DOI] [PubMed] [Google Scholar]
- Holst PJ, Christensen JP, Thomsen AR. Vaccination against lymphocytic choriomeningitis virus infection in MHC class II-deficient mice. J Immunol. 2011;186:3997–4007. doi: 10.4049/jimmunol.1001251. [DOI] [PubMed] [Google Scholar]
- Wiesel M, Joller N, Ehlert AK, Crouse J, Spörri R, Bachmann MF, et al. Th cells act via two synergistic pathways to promote antiviral CD8+ T cell responses. J Immunol. 2010;185:5188–5197. doi: 10.4049/jimmunol.1001990. [DOI] [PubMed] [Google Scholar]
- Wilson EB, Livingstone AM. Cutting edge: CD4+ T cell-derived IL-2 is essential for help-dependent primary CD8+ T cell responses. J Immunol. 2008;181:7445–7448. doi: 10.4049/jimmunol.181.11.7445. [DOI] [PubMed] [Google Scholar]
- Umeshappa CS, Xie Y, Xu S, Nanjundappa RH, Freywald A, Deng Y, et al. Th cells promote CTL survival and memory via acquired pMHC-I and endogenous IL-2 and CD40L signaling and by modulating apoptosis-controlling pathways. PLoS ONE. 2013;8:e64787. doi: 10.1371/journal.pone.0064787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borriello F, Sethna MP, Boyd SD, Schweitzer AN, Tivol EA, Jacoby D, et al. B7-1 and B7-2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity. 1997;6:303–313. doi: 10.1016/s1074-7613(00)80333-7. [DOI] [PubMed] [Google Scholar]
- Carreno BM, Collins M. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu Rev Immunol. 2002;20:29–53. doi: 10.1146/annurev.immunol.20.091101.091806. [DOI] [PubMed] [Google Scholar]
- Jensen BA, Pedersen SR, Christensen JP, Thomsen AR. The availability of a functional tumor targeting T-cell repertoire determines the anti-tumor efficiency of combination therapy with anti-CTLA-4 and anti-4-1BB antibodies. PLoS ONE. 2013;8:e66081. doi: 10.1371/journal.pone.0066081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kündig TM, Shahinian A, Kawai K, Mittrücker HW, Sebzda E, Bachmann MF, et al. Duration of TCR stimulation determines costimulatory requirement of T cells. Immunity. 1996;5:41–52. doi: 10.1016/s1074-7613(00)80308-8. [DOI] [PubMed] [Google Scholar]
- Bett AJ, Haddara W, Prevec L, Graham FL. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc Natl Acad Sci USA. 1994;91:8802–8806. doi: 10.1073/pnas.91.19.8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prévost-Blondel A, Zimmermann C, Stemmer C, Kulmburg P, Rosenthal FM, Pircher H. Tumor-infiltrating lymphocytes exhibiting high ex vivo cytolytic activity fail to prevent murine melanoma tumor growth in vivo. J Immunol. 1998;161:2187–2194. [PubMed] [Google Scholar]
- Leisner C, Loeth N, Lamberth K, Justesen S, Sylvester-Hvid C, Schmidt EG, et al. One-pot, mix-and-read peptide-MHC tetramers. PLoS ONE. 2008;3:e1678. doi: 10.1371/journal.pone.0001678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensen MA, Holst PJ, Steengaard SS, Jensen BA, Bartholdy C, Stryhn A, et al. Qualitative and quantitative analysis of adenovirus type 5 vector-induced memory CD8 T cells: not as bad as their reputation. J Virol. 2013;87:6283–6295. doi: 10.1128/JVI.00465-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grujic M, Bartholdy C, Remy M, Pinschewer DD, Christensen JP, Thomsen AR. The role of CD80/CD86 in generation and maintenance of functional virus-specific CD8+ T cells in mice infected with lymphocytic choriomeningitis virus. J Immunol. 2010;185:1730–1743. doi: 10.4049/jimmunol.0903894. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
All IL-2 expressing constructs secret equal amounts of biologically active IL-2.
Co-expression of IL-2 augments the Ag-specific T-cell response in WT mice.
Numbers of Tregs in the spleen of tumor-bearing mice are independent of the vaccination regimen.