

Abstract
Nucleic acid-based vaccines such as viral vectors, plasmid DNA, and mRNA are being developed as a means to address a number of unmet medical needs that current vaccine technologies have been unable to address. Here, we describe a cationic nanoemulsion (CNE) delivery system developed to deliver a self-amplifying mRNA vaccine. This nonviral delivery system is based on Novartis's proprietary adjuvant MF59, which has an established clinical safety profile and is well tolerated in children, adults, and the elderly. We show that nonviral delivery of a 9 kb self-amplifying mRNA elicits potent immune responses in mice, rats, rabbits, and nonhuman primates comparable to a viral delivery technology, and demonstrate that, relatively low doses (75 µg) induce antibody and T-cell responses in primates. We also show the CNE-delivered self-amplifying mRNA enhances the local immune environment through recruitment of immune cells similar to an MF59 adjuvanted subunit vaccine. Lastly, we show that the site of protein expression within the muscle and magnitude of protein expression is similar to a viral vector. Given the demonstration that self-amplifying mRNA delivered using a CNE is well tolerated and immunogenic in a variety of animal models, we are optimistic about the prospects for this technology.
Introduction
Prophylactic vaccination has revolutionized the practice of medicine. However, despite advances, there remain many medical needs that current technologies have not been able to overcome.1 Nucleic acid–based vaccines have long held the promise of vaccines that can be produced quickly in response to public health emergencies, are safe, and elicit protective immune responses, particularly in the case of cell-mediated responses. Yet, despite decades of research and development, there is still no licensed nucleic acid–based vaccine for human use. Recombinant viral vector technologies efficiently deliver nucleic acids, but their utility is often hampered by antivector immunity, production limitations, and safety concerns. Although plasmid DNA (pDNA) vaccines have proven safe and broadly effective in small animal models, they often require multiple, high doses in larger species and generally are less potent in humans than conventional vaccines based on live-attenuated organisms and recombinant proteins. Significant advancements in pDNA vaccines have been made through optimization of pDNA constructs, coexpression of immune-stimulatory molecules, and improved delivery technologies. Ongoing clinical trials will ultimately determine if these improvements in pDNA vaccines are sufficient to generate practical human vaccines.2,3
Recently, mRNA has emerged as an alternative to pDNA with a number of high profile reports using mRNA for vaccine and gene therapy applications.4,5,6 As a vaccine, mRNA has some clear advantages over pDNA. First, mRNA need only be delivered into the host cell cytoplasm to be translated, whereas pDNA must be transported across the nuclear membrane and transcribed, a process known to be inefficient.7 Second, safety concerns about pDNA integration into the host genome post-transfection, albeit low probability, are obviated by RNA as integration of RNA is not possible. Several nonviral means of delivering mRNAs have been explored, including injecting naked mRNA (formulated in buffer), device-mediated delivery such as the gene gun or electroporation; or formulating with synthetic delivery vehicles, such as liposomes, lipoplexes, and cationic polymers.4
Self-amplifying mRNA, i.e., RNAs that encode not only an antigen of interest but also a viral RNA-dependent RNA polymerase to amplify the RNA in the cytoplasm of transfected cells, lead to significantly greater immune responses than conventional RNAs.5 There has been extensive work on delivery of self-amplifying mRNA using viral replicon particles (VRPs), where RNA is packaged in a viral particle (e.g., alphavirus). As VRPs do not encode the structural proteins needed to spread from cell to cell, they are single-cycle infectious particles. Such VRPs have safely elicited potent immune responses in multiple animal models and in humans.8,9,10,11,12,13,14,15
We previously described the SAM vaccine platform, in which delivery of a self-amplifying mRNA was facilitated by a nonviral lipid nanoparticle.5 Potency, as measured by antibody and T-cell responses, was shown to be as good as, or better than that of VRP or pDNA delivered by electroporation. Here, we describe an alternative nonviral delivery system based on a cationic nanoemulsion (CNE), which binds to the SAM RNA, enhances its delivery, and thereby substantially increases the potency of the vaccine. CNE is composed of the cationic lipid DOTAP (1,2-dioleoyl-sn-glycero-3-phosphocholine) chosen for its previous clinical use and fixed positive charge emulsified with the constituents of the emulsion adjuvant MF59, which has been tested in more than 100 clinical trials, is licensed in 30 countries, has a well-established safety profile, and is well tolerated in children and adults, including the elderly.16 We report here the performance of CNE-delivered SAM RNA in three animal species (mouse, rabbit, nonhuman primate), with antigens from three pathogens that represent unmet medical needs, namely the fusion (F) glycoprotein of respiratory syncytial virus (RSV), the envelope glycoprotein B (gB) and a fusion protein (pp65 –IE1) of phosphoprotein 65 (pp65) and immediate early protein 1 (IE-1) from human cytomegalovirus (hCMV) and gp140 envelope glycoprotein (env) of human immunodeficiency virus (HIV).
Results
Characterization of CNE delivery system before and after the addition of RNA
CNE was prepared by mixing an aqueous phase containing buffer and Tween 80 with an oil phase containing Span 85, DOTAP, and squalene. The resulting mixture was then homogenized into a coarse emulsion using an overhead mixer and processed through a microfluidizer. Figure 1a shows a pictorial representation of the emulsion bound to RNA. Electron cryomicroscopy (cryo-EM) images of the resulting emulsion showed spherical droplets between 25 and 150 nm in diameter, with most particles in the 80–100 nm size range (Figure 1b). Mean particle size and poly-dispersity for CNE was determined by dynamic light scattering (Figure 1c). The number-weighted mean diameter was 69 nm and Z-average diameter was 101 nm, with a poly-dispersity index of 0.098. The size distribution is characterized by a single peak with a low poly-dispersity index, indicating a relatively mono-disperse size distribution, and are consistent with the size determinations from the cryo-EM images. After the addition of RNA to CNE, changes in particle size were measured by dynamic light scattering. The number-weighted mean diameter increased to 86 nm and Z-average diameter to 129 nm with a poly-dispersity index of 0.117. Particle size data and zeta potential for mRNA and pDNA complexed to CNE are summarized in Supplementary Table S1.
Figure 1.

Physical characterization of cationic nanoemulsion (CNE)-formulated SAM. (a) Schematic illustration and role of components of CNE in complex with SAM RNA. (b) Cryo-EM of CNE prior to RNA addition. (c) Particle size measured by dynamic light scattering of CNE before (red line) and after (green line) SAM RNA addition. Data are reported as the Z-average (Z-ave) with the poly-dispersity index (pdi). (d) Denaturing RNA agarose gel electrophoresis showing protection of SAM RNA from RNAse: molecular weight ladder (lane 1), SAM RNA (lane 2), SAM RNA after incubation with RNase (lane 3), SAM RNA after extraction from CNE (lane 4), CNE with SAM RNA after RNase exposure, inactivation of RNase, and RNA extraction from CNE (lane 5).
RNase-mediated degradation of an RNA vaccine in tissues after administration may be a limiting factor in delivering an intact transcript to the cell cytoplasm. To evaluate the protective effect of CNE on RNA stability, self-amplifying mRNA expressing RSV-F was incubated with RNase A in the presence or absence of CNE. Agarose gel electrophoresis demonstrated that unformulated SAM RNA treated with RNase was fully degraded (Figure 1d), whereas SAM RNA bound to CNE was protected, despite being adsorbed to the surface of the delivery particle where it may be accessible to the RNase. When pDNA was evaluated for DNAse stability upon binding to CNE, similar protection was observed (data not shown). As gel electrophoresis did not test the biological activity of the RNA, SAM RNA encoding RSV F was complexed to CNE (± RNAse exposure) and transfected into baby hamster kidney (BHK) cells to quantify the number of positive cells. Sixteen hours post-transfection, cells were trypsinized and immunostained for RSV F protein expression and analyzed by flow cytometry. The percentage of RSV F antigen-positive BHK cells after transfection with the untreated CNE-RNA complex was 44.9 ± 2.4% and with an RNAse-treated complex was 44.0 ± 1.6% (Supplementary Figure S1), indicating that CNE fully protected the functionality of the RNA vaccine. No changes were observed regarding the stability (particle size and in vivo immunogenicity) of the CNE/RNA complex when stored on ice for 24 hours (data not shown).
Immunogenicity of an RSV SAM vaccine in mice
The RSV F glycoprotein is a conserved target of neutralizing antibodies and a promising RSV vaccine antigen.17 As a proof of concept for the CNE delivery system, a SAM RNA expressing the RSV F antigen was used to immunize BALB/c mice intramuscularly (i.m.). F-specific serum IgG (Figure 2a) and RSV-neutralizing antibody titers (Figure 2b) were measured as markers of immunogenicity. As benchmarks, an RSV F vaccine based on MF59-adjuvanted recombinant subunit protein, VRP, CNE-formulated mRNA, and CNE-formulated pDNA were used. After two immunizations, CNE-formulated SAM RNA was immunogenic at all the doses tested (0.015–15 µg), as measured by F-specific IgG titers (Figure 2a). The lowest dose tested (0.015 µg of SAM RNA) elicited a geometric mean titer (GMT) of 4.9 × 103, comparable to a 1,000-fold higher dose (15 µg) of unformulated SAM RNA (GMT 7.4 × 103) or pDNA (GMT 3.5 × 103). Unformulated mRNA did not elicit a measurable F-specific IgG titer. The RSV SAM vaccine at the 15 µg dose was significantly more immunogenic (GMT 1.8 × 105) than CNE-formulated mRNA (GMT below detection limit) and pDNA (GMT 1.3 × 103) (P = 0.001, ANOVA, Tukey's post-test). The RSV SAM vaccine (15 µg) elicited titers equivalent to those induced by 1 × 106 infectious units (IU) of VRP (GMT 9.2 × 104) and 3 µg of an MF59-adjuvanted RSV-F subunit vaccine (GMT 4.5 × 105), and consistently higher F-specific IgG titers than the alternative nucleic acid technologies at all doses tested.
Figure 2.

Immunogenicity of RSV SAM vaccine and comparators in mice. Groups of eight BALB/c mice were immunized i.m. on days 0 and 21 with 3 µg RSV-F subunit antigen with MF59, 1 × 106 IU RSV F-expressing VRP, 15 µg SAM RNA expressing RSV F in phosphate-buffered saline (PBS), 15 µg mRNA expressing RSV F in PBS, 15 µg pDNA expressing RSV F in PBS, 0.015 µg SAM RNA expressing RSV F formulated with cationic nanoemulsion (CNE), 0.15 µg SAM RNA expressing RSV F formulated with CNE, 1.5 µg SAM RNA expressing RSV F formulated with CNE, 15 µg SAM RNA expressing RSV F formulated with CNE, 1.5 µg mRNA expressing RSV F formulated with CNE, 15 µg mRNA expressing RSV F formulated with CNE, 1.5 µg pDNA expressing RSV F formulated with CNE, and 15 µg pDNA expressing RSV F formulated with CNE. Control mice were not injected with any formulation. Sera were collected 2 weeks after the second vaccination. (a) F-specific IgG titers measured by enzyme-linked immunosorbent assay (ELISA). Data are from individual mice (depicted as dots), and the geometric mean titers (GMTs) are solid lines. (b) RSV 60% neutralization titers 2 weeks after the second vaccination. Data are from pooled sera (two pools per group, depicted as dots), and the GMTs as solid lines. The dotted lines indicate assay limits of detection (titers of 25 for ELISA, 20 for neutralizing antibodies). For calculation of GMTs, titers below the limits of detection were assigned a value of 5 for ELISA and 10 for neutralization. NS, not statistically different from 15 µg SAM RNA expressing RSV F formulated with CNE (analysis of variance (ANOVA), Tukey's post-test), * statistically different from 15 µg SAM RNA expressing RSV F formulated with CNE (P = 0.001 ANOVA, Tukey's post-test).
Neutralizing antibody titers were detected in animals immunized with 3 µg subunit adjuvanted with MF59, with 1 × 106 IU of VRP, or with 0.15 µg or more of CNE-formulated SAM RNA (Figure 2b). No detectable neutralizing titers were elicited by mRNA or pDNA formulated with CNE or with mRNA, pDNA, or SAM RNA in phosphate-buffered saline (PBS). In general, the relative neutralizing titers elicited by each preparation paralleled the relative F-specific IgG enzyme-linked immunosorbent assay (ELISA) titers. However, the 15 µg dose of CNE SAM vaccine elicited higher neutralizing antibody titers than either the MF59 adjuvanted subunit vaccine (P > 0.05, analysis of variance (ANOVA), Tukey's post-test), or VRP (P < 0.05, ANOVA, Tukey's post-test).
Immunogenicity of an HIV SAM vaccine in rabbits
For pDNA-based vaccines, immune responses elicited by vaccination of larger species have generally been lower than those elicited in small rodent species, and much larger doses of pDNA have been required in large animals.18 We chose the New Zealand white rabbit as our first large nonrodent species for proof of concept for the CNE delivery system as it is an accepted model to differentiate HIV vaccine candidates and an established model for vaccine tolerability.19 We generated a SAM RNA encoding HIV gp140 env from the TV1 viral isolate to evaluate immunogenicity in rabbits. Immunogenicity was measured by gp140-specific serum IgG (Figure 3a) and HIV neutralizing antibody titers after i.m. administration (Figure 3b). Corresponding HIV gp140 vaccines based on MF59-adjuvanted subunit protein, VRP, CNE-formulated mRNA, and CNE-formulated pDNA vaccines were used as benchmarks. After two immunizations, CNE SAM was immunogenic at both the 5 and 25 µg doses as measured by gp140-specific IgG titers (Figure 3a). At the lowest dose of CNE SAM RNA tested (5 µg), the GMT (1.4 × 105) was > 80-fold higher (P = 0.001, ANOVA, Tukey's post-test) than a fivefold higher dose (25 µg) of unformulated SAM RNA (GMT 1.7 × 103). At the highest dose of CNE SAM RNA tested, antibody titers were within twofold of those elicited by the adjuvanted subunit (GMT 3.0 × 105 versus 5.0 × 105) and within fourfold of those elicited by VRP (GMT 3.0 × 105 versus 1.1 × 106) and were not statistically different (P > 0.05, ANOVA, Tukey's post-test). CNE-formulated mRNA and pDNA responses were below the limit of detection for all but one animal after two immunizations.
Figure 3.

Immunogenicity of HIV SAM vaccine and comparators in rabbits. Groups of five New Zealand white rabbits were immunized i.m. on days 0 and 21 with PBS, 1 × 108 IU HIV gp140 expressing VRP, 25 µg HIV gp140 env with MF59, 25 µg SAM RNA expressing HIV gp140 in PBS, 5 µg SAM RNA expressing HIV gp140 formulated with cationic nanoemulsion (CNE), 25 µg SAM RNA expressing HIV gp140 formulated with CNE, 25 µg mRNA expressing HIV gp140 formulated with CNE, or 25 µg pDNA expressing HIV gp140 formulated with CNE. Sera were collected 4 weeks post first (4wp1) immunization (open circles) and 2 weeks post second (2wp2) immunization (closed circles). (a) gp140-specific IgG titers measured by enzyme-linked immunosorbent assay (ELISA). Data are from individual rabbits (depicted as dots), and the geometric mean titers (GMTs) are solid lines. (b) HIV MW965.26 serum neutralizing antibody titers elicited by 1 × 108 IU HIV gp140 expressing VRP, 25 µg SAM RNA expressing HIV gp140 in PBS, 25 µg SAM RNA expressing HIV gp140 formulated with CNE, 25 µg mRNA expressing HIV gp140 formulated with CNE, and 25 µg pDNA expressing HIV gp140 formulated with CNE. Data are from the individual sera (depicted as dots) tested 2 weeks after the second vaccination, and the GMT is a solid line. The dotted lines indicate assay limits of detection (400 for ELISA, 20 for neutralization). For calculation of GMTs, titers below the limit of detections were assigned a value of 200 for ELISA and 10 for neutralization. NS, not statistically different from 25 µg SAM RNA expressing HIV gp140 formulated with CNE (analysis of variance (ANOVA), Tukey's post-test), * statistically different from 15 µg SAM RNA expressing RSV F formulated with CNE (P = 0.001 ANOVA, Tukey's post-test).
Sera from a subset of the groups, 1 × 108 IU VRP, 25 µg HIV SAM RNA in PBS, 25 µg SAM RNA expressing gp140 formulated with CNE, and 25 µg gp140-expressing mRNA or pDNA formulated with CNE, were tested for neutralizing titers against the neutralization sensitive MW965.26 tier 1 strain of HIV (Figure 3b). Serum from all animals in the CNE SAM group neutralized the virus (GMT 2.5 × 102), whereas in the VRP and unformulated SAM RNA groups, only four out of five sera from animals neutralized the virus (GMTs of 1.2 × 102 and 7.0 × 101, respectively). As observed for the ELISA titers, neutralizing titers were not detected in serum from animals receiving mRNA or pDNA.
Immunogenicity of an hCMV SAM vaccine in nonhuman primates
Rhesus macaques are used in CMV research because infection with rhesus CMV mimics human infection with CMV, and they are relatively easy to handle. To test the immunogenicity of the SAM vaccine in rhesus macaques, constructs encoding the hCMV envelope glycoprotein B (gB) and a fusion protein (pp65 –IE1) of phosphoprotein 65 (pp65) and immediate early protein 1 (IE-1) were used. The two SAM RNAs were formulated separately with CNE and administered as i.m. injections into separate thighs of animals at a 75 µg dose of RNA for each antigen. Immunogenicity was evaluated by measuring gB-specific serum IgG (Figure 4a) and hCMV-neutralizing antibody titers (Figure 4b). Antigen-specific immune responses, both total anti-gB IgG and neutralizing titers (GMT 5.9 × 103 and 1.0 × 103, respectively), were detected in all animals after a single immunization and were boosted threefold after a second immunization.
Figure 4.

Immunogenicity in rhesus macaques. A group of six rhesus macaques was immunized intramuscularly on days 0 and 28 with 75 µg SAM RNA expressing hCMV gB formulated with cationic nanoemulsion (CNE) and 75 µg of SAM RNA expressing hCMV pp65-IE1 formulated with CNE. (a) gB-specific IgG titers measured by enzyme-linked immunosorbent assay (ELISA). Data are presented as geometric mean titers (GMTs) ± standard error of the mean (SEM) 2 weeks post first immunization (2wp1), 2 weeks post second immunization (2wp2), and 8 weeks post second immunization (8wp2). (b) CMV clinical isolate 8819 neutralization titers on ARPE-19 cells; data are presented as GMTs ± SEM, 2wp1 immunization, 2wp2 immunization, and 8wp2 immunization. The dotted lines indicate preimmunization titers (2,381 for gB ELISA, 422 for CMV neutralization).
After two immunizations, all animals (n = 5) had measureable CD4+ (0.30% ± 0.08 SD) and CD8+ (0.66% ± 0.18 SD) T-cell responses, with CD4+ responses to individual antigens more consistent than CD8+ responses (Figure 5). All animals generated CD4+ T-cell responses against all antigens administered with gB generating the strongest response in all animals. While CD8+ T-cell responses were also measured in all animals, they were less uniform with two animals having stronger responses against gB, two animals having stronger responses to IE-1, and one animal having a stronger response to pp65.
Figure 5.

Circulating antigen-specific T cells in immunized rhesus macaques. Blood was drawn and peripheral blood monocytes (PBMCs) were cryopreserved from six rhesus macaques that were immunized i.m. on days 0 and 28 with 75 µg SAM RNA expressing hCMV gB formulated with cationic nanoemulsion (CNE) and 75 µg of SAM RNA expressing hCMV pp65-IE1 formulated with CNE (same animals as Figure 4). PBMCs were thawed, stimulated, and analyzed as described in Materials and Methods. PBMCs were available for five of the six rhesus. Percentages of cytokine + antigen-specific CD4+ and CD8− T cells in individual rhesus after stimulation in vitro with a hCMV gB peptide mix (black checkered), hCMV pp65 peptide mix (black), or hCMV IE-1 peptide mix (gray). (a) 21 days prior to the first immunization and (b) 14 days after the second immunization. Percentages of antigen-specific CD8+ CD4− T cells in individual rhesus after stimulation in vitro with a CMV gB peptide mix (black checkered), CMV pp65 peptide mix (black), or CMV IE-1 peptide mix (gray) (c) 21 days prior to the first immunization and (d) 14 days after the second immunization.
Expression profile of CNE-formulated SAM RNA in mice and nonhuman primates
To better understand the differences between the expression profiles of three antigen-expressing nucleic acids (SAM RNA, mRNA, and pDNA), the reporter gene firefly luciferase was cloned into each vector and bioluminescence was measured over 10 weeks after a single bilateral i.m. injection of BALB/c mice with 1.5 or 15 µg. Major differences in the kinetics of expression were observed (Figure 6a). Animals that received SAM RNA formulated with CNE had the strongest bioluminescence intensity, which peaked at day 7 and persisted for 56 days at both doses tested. Expression from the mRNA peaked within 6 hours of administration and fell to baseline within 3 days of administration (Figure 6b). Expression from VRPs peaked within 24 hours of administration and returned to baseline by day 42. Luciferase expression from pDNA was low but consistent throughout the 10-week experiment at the 15 µg dose and did not return to baseline prior to the end of the experiment.
Figure 6.

Luciferase activity in mice. Groups of five BALB/c mice were inoculated i.m. on day 0 with either phosphate-buffered saline (PBS) (closed grey diamond), 5 × 106 IU luc expressing VRP (open black diamond), 1.5 µg SAM RNA expressing luciferase formulated with CNE at a 1.5 dose (open dark gray circle) or 15 µg dose (closed dark gray circle), mRNA expressing luciferase formulated with CNE at a 1.5 µg dose (open gray square) or 15 µg dose (closed gray square), or pDNA expressing luciferase formulated with CNE at a 1.5 µg dose (open light gray triangle) or 15 µg dose (closed light gray triangle). Average total flux (photons/second) of bioluminescence emanating from the injection site was measured repeatedly over the course of 10 weeks. Total flux throughout the whole 10-week experiment (a), total flux throughout the first 5 days of the experiment (b), peak expression as measured by total flux, time of peak expression is noted in parenthesis (c).
To evaluate expression of CNE-formulated SAM in nonhuman primates, we compared 75 µg of SAM RNA formulated with CNE to the previously tested human dose of 1 × 108 IU of VRP for expression of a secreted alkaline phosphatase (SEAP) reporter gene in rhesus macaques after i.m. administration (Supplementary Figure S2). SEAP enzyme activity in the sera collected on days 0, 3, 7, and 14 was determined by using a chemiluminescence assay. The CNE-delivered SAM RNA and VRP displayed similar expression profiles, with a peak at 3 days postadministration and measurable but lower levels of enzyme activity extending to 14 days. SEAP activity was undetectable in sera from animals that received a SAM-vector encoding an irrelevant (or control) antigen.
CNE recruits immune cells to transfected muscle cells
The mechanism of action of MF59 has been extensively studied and is marked by a strong cellular infiltrate in the muscle shortly after immunization.20,21,22,23 To assess if CNE had similar effects, a reporter gene encoding green fluorescent protein (GFP) was cloned into the SAM RNA and was formulated with CNE. Mice were injected i.m. with PBS, 15 µg of GFP-expressing SAM RNA in PBS, 1 × 106 IU of GFP-expressing VRP, MF59 −/+ 15 µg GFP-expressing SAM RNA, or CNE −/+ 15 µg formulated GFP-expressing SAM RNA. Leukocyte infiltration 72 hours after immunization was assessed by staining collagenase-dissociated muscle with a variety of antibodies to lineage and activation markers followed by flow cytometry. Mice immunized with MF59 −/+ SAM RNA and CNE −/+ SAM RNA showed a strong cellular infiltrate of CD11b+ cells compared to groups immunized with PBS, PBS-SAM RNA, or VRP (Figure 7a). Despite similar levels of infiltrating cells in the MF59-formulated and CNE-formulated groups, the cellular compositions differed (Figure 7b–h). Animals that received CNE showed a lower percentage of infiltrating eosinophils when compared with animals that received MF59. In contrast, percentages of monocytes and neutrophils present in the infiltrates were higher in animals that received CNE compared with animals that received MF59. Interestingly, the proportions of myeloid dendritic cells (mDCs)—a key antigen-presenting cell—were lower in all animals that received GFP-expressing SAM RNA as compared to their respective controls (PBS ± SAM, MF59 ± SAM, and CNE ± SAM). This may indicate mDC egression from the vaccination site to transport antigens toward draining lymph nodes. This effect was especially evident in animals that received CNE-formulated GFP-expressing SAM RNA (5%) compared to those that received CNE alone (18%). No GFP-positive cells were detected by flow cytometry suggesting that antigen expression most likely does not take place in infiltrating (or resident) immune cells but in cell types present within the injection site at time of vaccination.
Figure 7.
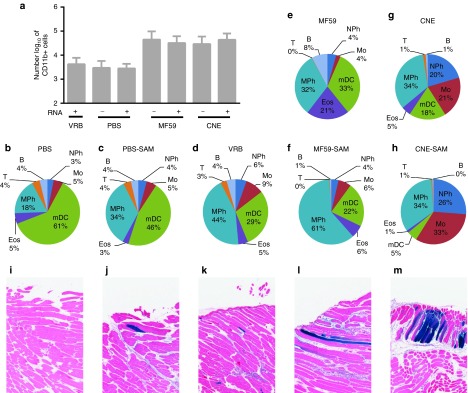
Antigen expression, localization, and cell infiltration. BALB/c mice were inoculated i.m. with phosphate-buffered saline (PBS) (n = 12), 15 µg SAM RNA expressing green fluorescent protein (GFP) in PBS (n = 4), 5 × 106 IU VRP expressing GFP (n = 4), MF59 (n = 8), 15 µg SAM RNA expressing GFP formulated with MF59 (n = 8), cationic nanoemulsion (CNE) (n = 8), or 15 µg SAM RNA expressing GFP formulated with CNE (n = 8). (a) CD11b+ cells found in collagenase dissociated muscle cell suspensions 3 days after immunization. (b–h) Leukocyte infiltrate composition 3 days after administration of (b,c) PBS (−/+ 15 µg SAM RNA expressing GFP), (d) 5 × 106 IU VRP expressing GFP, (e,f) MF59 (−/+ 15 µg SAM RNA expressing GFP), (g,h) CNE (−/+ 15 µg SAM RNA expressing GFP); B, B-cell; Eos, eosinophil; Mo, monocyte; mDC, dendritic cell; MPh, macrophage; NPh, neutrophil; T, T-cell. (i–m) Representative histological tissue sections stained for β-gal 3 days postinnoculation of BALB/c mice after administration with (i) PBS, (j) 15 µg SAM RNA expressing β-gal formulated with PBS, (k) 1 × 106 IU VRP expressing β-gal, (l) 15 µg SAM RNA expressing β-gal formulated with MF59, (m) 15 µg SAM RNA expressing β-gal formulated with CNE.
To identify the productively transfected cells, β-galactosidase (â-gal) was cloned into SAM RNA, and mice were immunized with SAM RNA expressing â-gal in PBS, delivered by VRP, formulated with CNE, or formulated with MF59. At 72 hours after the immunization, mice were sacrificed, and excised quadriceps muscles were stained for β-gal expression using X-gal followed by hematoxylin and eosin (H&E) staining (Figure 7i–m). Myocytes appeared to be the only Xgal-stained cells after i.m. administration of the vaccines (Figure 7 j–m). The strongest staining in this study was observed in the CNE-SAM group. Mononuclear infiltrates were observed in all groups and were most prevalent in the MF59-SAM and CNE-SAM groups, consistent with the cell infiltration results.
To better understand the necessity of a cationic lipid in generating immune responses, groups of 8 BALB/c mice were immunized i.m. with SAM RNA expressing RSV F in PBS, formulated with MF59 or formulated with CNE. Sera was drawn 2 weeks after two immunizations and F-specific serum IgG was measured (Figure 8). The CNE formulated SAM vaccine elicited responses that were significantly more immunogenic (GMT 1.1 × 104) than SAM RNA in PBS (GMT 3.6 × 102) and MF59 formulated SAM RNA (GMT 1.6 × 102) (P = 0.001, ANOVA, Tukey's post-test).
Figure 8.

Comparison of immunogenicity of SAM RNA delivered with MF59 or cationic nanoemulsion (CNE). Groups of 8 BALB/c mice were immunized i.m. on days 0 and 21 with 1 µg SAM RNA expressing RSV F in phosphate-buffered saline (PBS), 1 µg SAM RNA expressing RSV F formulated with MF59, or 1 µg SAM RNA expressing RSV F formulated with CNE. Sera were collected two weeks after the second vaccination. F-specific serum IgG titers measured by enzyme-linked immunosorbent assay (ELISA). Data are from individual mice (depicted as dots), and the geometric mean titers (GMTs) are solid lines. Error bars depict the standard error of the mean. * statistically different from RSV F SAM RNA in PBS and SAM RNA expressing RSV F formulated with MF59 (P = 0.001 analysis of variance, Tukey's post-test).
Discussion
Nanoemulsions have been used extensively throughout the pharmaceutical industry for delivery of poorly water soluble drugs, and the vaccine industry has optimized them for use as vaccine adjuvants.16,24 The use of CNEs have been described previously for delivery of pDNA.25,26 The CNE delivery system contains the same components as MF59 with the addition of the cationic lipid DOTAP. DOTAP was chosen because of its previous clinical use, availability as GMP material, squalene solubility, and ability to hold a positive charge at a pharmaceutically relevant pH. There are a number of examples in which cationic lipids containing quaternary amines are being tested clinically as vaccine adjuvants and for delivery of DNA vaccines i.m. These lipids have been well tolerated in early clinical trials.27,28,29 In all our animal experiments, the site of injection was monitored, and no redness or soreness was observed (data not shown). The lack of significant changes in body weight and clinical blood chemistry indicated that CNE was well tolerated, although cationic lipids have been known to be poorly tolerated when used for systemic delivery of siRNA or pDNA.30
This report demonstrates that a CNE nonviral delivery system can also be effectively used to deliver a large single-stranded, self-amplifying mRNA. CNE was a very effective delivery system for SAM RNA, with a 1,000-fold SAM RNA dose-sparing effect relative to PBS-formulated SAM RNA. The enhancing effect of CNE was limited to SAM RNA, as there was no enhancement in the immune responses to pDNA when formulated with CNE. Possible reasons for this difference include inefficient delivery of pDNA across the nuclear membrane, a difference in binding affinity between RNA and pDNA to CNE, or the increased susceptibility to degradation of unformulated RNA compared to unformulated pDNA.31 These data, in addition to other published reports on the use of electroporation delivery of self-amplifying mRNA and pDNA, highlight that the optimal delivery technologies for pDNA and RNA vaccines are different.32 mRNA was unable to induce responses in our hands, perhaps due to the low dose being tested here. Previous reports with mRNA have shown that much higher doses (80 µg) of mRNA have been used to generate immune responses in mice.6
In rhesus macaques, CNE was an effective delivery system for the SAM vaccine. First, injection of a SEAP expressing SAM resulted in measureable SEAP enzyme activity in the sera of all six animals after a single administration, at concentrations equivalent to those elicited by VRP. The VRP dose (1 × 108) tested in this study was previously tested in humans and shown to be immunogenic as an experimental hCMV vaccine.11 Second, a single immunization in rhesus macaques with the hCMV SAM (gB + pp65-IE1) vaccine-induced gB-specific IgG and neutralizing antibodies that were boosted after a second immunization. In addition, after two immunizations, CD4+ and CD8+ T cells were detected in all animals. A limitation, thus far, for pDNA vaccines is that doses given in small animal species do not translate to larger species in which pDNA vaccines have not been able to match viral vectors in potency.2 In contrast, CNE SAM vaccines were potent for both antibody and T-cell responses in macaques at doses less than 100 µg. Both the humoral and cellular responses elicited by CNE SAM vaccine were comparable to other experimental CMV vaccines including a pDNA prime/modified vaccinia virus boost in nonhuman primates (neutralizing titers of ~1,000), a VRP in phase 1 clinical trials (neutralizing antibody titers of ~600, 0.05% CD4+ T cells, and 0.2% CD8+ T cells), and an MF59 adjuvanted subunit vaccine in phase 2 trials (ELISA titers of ~100,000).11,33,34 Despite the similarities noted, here, sera would need to be analyzed by using the same assays to make definitive conclusions of the magnitude of responses between these studies.
The CNE-delivered SAM vaccine had the protein expression profile of VRP-delivered SAM RNA, but elicited a cellular infiltrate at the site of injection comparable in magnitude to that elicited by the emulsion adjuvant, MF59. Luciferase reporter gene expression by SAM RNA delivered with CNE showed prolonged high levels of expression in a dose-dependent manner for up to 6 weeks. In contrast, luciferase expression from pDNA was low, but prolonged, lasting throughout the 10-week experiment. Reporter gene expression from mRNA showed a peak within 6 hours of administration but returned to baseline levels within 3 days. These results are consistent with previously published data regarding the duration and magnitude of luciferase expression after delivery of pDNA and nonamplifying mRNA, and are similar to our previous reports of LNP delivery of self-amplifying mRNA.5,35,36 While trying to correlate the immunogenicity with luciferase expression, we noted the total magnitude of expression does not directly correlate to immunogenicity. Differences in the duration of expression or the total area under the curve may account at least partly for the differences in immunogenicity. It is important to note here that SAM RNA is known to interact with a number of intracellular pattern recognition receptors whose activation can adjuvant the adaptive immune response. Histologic data collected after 3 days postinoculation with SAM RNA encoding β-gal showed that VRP and CNE delivered RNAs predominantly expressed antigen from myocytes, consistent with previous reports for pDNA and mRNA.36
The mechanism of action of MF59 has been studied extensively.20,21,22,23 MF59 elicits a cellular infiltrate that is important to its adjuvant activity.22 In this report, we demonstrate that the total number of infiltrating leukocytes elicited by CNE and MF59 injection were comparable, despite the presence of a cationic lipid in CNE. When SAM RNA was formulated with MF59 immunogenicity was reduced, generating antibody titers below those elicited by unformulated SAM RNA. This result demonstrates that a strong cellular infiltrate alone does not improve immune responses and that adsorption of the RNA to the surface of the particle by interaction with a cationic lipid is required for efficient cellular transfection and subsequent immune responses. In contrast to delivery by CNE, inoculation with VRPs leads to very low levels of cellular infiltration similar to those induced by the unformulated SAM RNA. The difference in cellular infiltration indicates that, although the VRPs and CNE-delivered SAM vaccines elicit comparable immune responses, the mechanism of action of the vaccines may be different. Pollard et al.37 have reported that delivery of conventional mRNA with DOTAP:DOPE liposomes induced potent T-cell responses in mice. However, the resulting adjuvant response impacted the T-cell response, and the authors suggested that one must balance the immune stimulation as type I interferon could reduce expression from RNA vaccines, ultimately impacting the immune response. Others have shown that condensed mRNA has adjuvant properties, and particle formation is necessary for induction of a potent immune response.38 Taken together with these reports and the data presented here indicate that the immunostimulatory effects of the RNA vaccine may be important for optimal non-viral delivery of RNA vaccines.39,40,41 Further studies are currently underway to better understand the balance between the adjuvant effects and immunogenicity of SAM vaccines.
In this report, CNE was demonstrated to be an effective nonviral delivery system for a large single-stranded, self-amplifying mRNA vaccine. Induction of immune responses was shown in multiple animal species, including rhesus macaques, at comparable levels to responses elicited by an adjuvanted subunit vaccine or VRP delivery of the same RNA, and at doses much lower than those required for pDNA vaccines. The prospects for this novel nucleic acid vaccine technology are encouraging and could enable a new generation of potent, versatile, and easily produced SAM vaccines to address health challenges of the 21st century.
Materials and Methods
RNA synthesis. DNA plasmids encoding self-amplifying mRNAs were constructed using standard molecular techniques. Plasmids were grown in Escherichia coli and purified using Qiagen Plasmid Maxi Kits (Qiagen, Valencia, CA). DNA was linearized immediately following the 3′ end of the self-amplifying mRNA sequence by restriction endonuclease digestion. Linearized DNA templates were transcribed into RNA using the MEGAscript T7 kit (Life Technologies, Carlsbad, CA) and purified by LiCl precipitation. RNA was then capped using the Vaccinia Capping System (New England BioLabs, Ipswich, MA) and purified by LiCl precipitation before formulation. cDNAs encoding the antigens (RSV F, HIV gp140, hCMV gB, or hCMV PP65-IE1) or reporter sequences (SEAP, eGFP, or luciferase) were inserted after the nonstructural protein sequence of the vector as previously described.5 Conventional mRNA was prepared the same way using a previously published sequence.42 Briefly, cDNAs encoding the antigens (RSV F or HIV gp140) or the reporter sequence (luciferase) were inserted into a plasmid, each gene of interest was flanked by the untranslated 5′ and 3′ sequences from Xenopus Laevis β-globin gene. mRNA was prepared following the same procedure as described above for SAM RNA.
Preparation of CNE. CNE was prepared similar to charged MF59 as previously described except the DOTAP was directly solubilized in the oil phase.26 Briefly, squalene, DOTAP, and sorbitan trioleate were combined and heated to 37 °C. The resulting oil phase was then combined with an aqueous phase consisting of polysorbate 80 in 10 mmol/l citrate buffer at pH 6.5. The final weight by weight percentages of squalene, DOTAP, sorbitan trioleate, and polysorbate 80 were 4.3, 0.4, 0.5, and 0.5% respectively. This mixture was homogenized for 2 minutes using a T25 homogenizer (IKA, Wilmington, NC) at 24K RPM to produce a primary emulsion. This primary emulsion was then passed through a M-110P Microfluidizer (Microfluidics, Newton, MA) with an ice bath cooling coil at a homogenization pressure of 20K PSI approximately eight times. The formulation was stored at 4 °C prior to use.
Nucleic acid complexation. Nucleic acids (self-amplifying mRNA, mRNA, and pDNA) were complexed to CNE at a 7:1 nitrogen/phosphate (N/P) ratio. N/P ratio was determined by calculating the number of moles of protonatable nitrogens per milliliter of CNE and calculating the number of moles of phosphate present in the molecule, assuming a constant of 3 nmols of phosphate per microgram of nucleic acid. Nucleic acid was prepared at 300 µg/ml and was added to an equal volume of CNE, mixed, and allowed to complex on ice for 30 minutes to 2 hours. Prior to administration, formulations were diluted to dosing concentrations.
RNase protection assay. To assess the ability of CNE to protect RNA from RNase degradation, the RNA/CNE complex was exposed to 6.4 mAU of RNase A (Ambion, Austin, TX) per microgram of RNA for 30 minutes at room temperature. RNase was inactivated by proteinase K (Novagen, Darmstadt, Germany) and incubating the sample at 55 °C for 10 minutes. To extract the remaining RNA from CNE, 25:24:1 v/v/v, phenol:chloroform:isoamyl alcohol (Fluka, St Louis, MO) was added to the solution at equal volume, mixed, and centrifuged at 12K RPM for 15 minutes. The aqueous phase containing RNA was removed and used to analyze RNA integrity by denaturing gel electrophoresis. A 1% denaturing agarose gel was prepared according to the manufactures instructions (Bio-Rad, Hercules, CA). Four hundred and sixty nanograms of RNA were loaded per lane. Millennium markers (Ambion) were used to approximate the molecular weight of the RNA. The gel was run at 130 V and then stained using 0.1% SYBR gold according to the manufacturer's guidelines (Invitrogen, Carlsbad, CA) in water by rocking at room temperature for 1 hour. Gel images were taken on a Bio-Rad Chemidoc XRS imaging system.
CNE particle size measurement. Dynamic light scattering was used to determine CNE particle size. CNE was diluted 1:100 in water and added to disposable low volume cuvettes (Malvern, Worcestershire, UK). Samples were measured on a Malvern NanoZs Zetasizer with a backward angle measurement using “water” as a dispersant (RI = 1.330) and analyzed with Mark-Houwink Parameters, D = kM^(−a), where D = diffusion coefficient (cgs), k = constant = 7.67e−5 (cm2/s), a = 0.428, M = molecular weight.
CNE transfection/RNase protection assay. RNA was complexed to CNE and then challenged with the RNase as described in the RNase protection assay method. Proteinase K was not used to inactivate RNase and RNA was not extracted from CNE to assess in vitro potency of the RNase challenged RNA/CNE complex. After 30 minutes of RNase exposure, 300 ng of RNA complexed to CNE complex was added to 750 µl of OptiMEM Reduced Serum Media (Invitrogen) and immediately added to 1 × 106 BHK cells in a six-well plate for 1 hour at 37 °C. The media was then removed and replaced with 2 ml of prewarmed 1% fetal bovine serum Dulbecco's modified Eagle's medium media for an overnight incubation. After a 16-hour incubation, the media was removed, cells were trypsinized, centrifuged, resuspended in fluorescence activated cell sorting (FACS) staining buffer (1× PBS, 0.25% bovine serum albumin, 0.2% NaN3), and stained with a fluorescently labeled anti-RSV F antibody (Maine Biotechnologies, Portland, ME, antibody tagged with Invitrogen Zenon Allophycocyanin Labeling kit). Cells were counted using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (Tree Star, Ashland, OR). A quadrant gate was drawn between the negative and positive populations yielding a percentage of positive cells. All samples were tested in duplicate.
Transmission electron cryomicroscopy. Each sample was preserved in vitrified ice supported by holey carbon films on 400-mesh copper grids. Each sample was prepared by applying a 3 μl drop of sample suspension to a cleaned grid, blotting away with filter paper, and immediately proceeding with vitrification in liquid ethane. Grids were stored under liquid nitrogen until transferred to the electron microscope for imaging. Electron microscopy was performed using an FEI Tecnai T12 electron microscope, operating at 120 keV equipped with an FEI Eagle 4k × 4k CCD camera. Vitreous ice grids were transferred into the electron microscope using a cryostage that maintains the grids at a temperature below −170 °C. Images of each grid were acquired at multiple scales to assess the overall distribution of the specimen.
Production of VRPs. To compare RNA vaccines to traditional RNA-vectored approaches for achieving in vivo expression of reporter genes or antigens, we utilized VRPs produced in BHK cells by the methods described by Perri et al. 8 In this system, the antigen (or reporter gene) replicons consisted of alphavirus chimeric replicons derived from the genome of Venezuelan equine encephalitis virus engineered to contain the 3′ terminal sequences (3′ UTR) of Sindbis virus and a Sindbis virus packaging signal (PS) (see Figure 2 of ref. 8). Self-amplifying RNA was packaged into VRPs by coelectroporating them into BHK cells along with defective helper RNAs encoding the Sindbis virus capsid and glycoprotein genes (see Figure 2 of ref. 8). The VRPs were then harvested and titrated by standard methods and inoculated into animals in culture fluid or other isotonic buffers.
In vivo evaluation. All animal studies were approved by the Novartis Institutional Animal Care and Use Committee in accordance with the requirements for the humane care and use of animals and all applicable local, state, and federal laws and regulations.
Mouse immunogenicity studies. Female BALB/c mice, aged 6–8 weeks and weighing ~20 g, were obtained from Charles River Laboratories (Wilmington, MA). Mice were immunized on days 0 and 21 with either 3 µg RSV-F subunit antigen with MF59, 1 × 106 IU RSV F-expressing VRP, 15 µg SAM RNA in PBS, 15 µg mRNA in PBS, 15 µg pDNA in PBS, 0.015 µg CNE-formulated SAM RNA, 0.15 µg CNE-formulated SAM RNA, 1.5 µg CNE-formulated SAM RNA, 15 µg CNE-formulated SAM RNA, 1.5 µg CNE formulated mRNA, 15 µg CNE formulated mRNA, 1.5 µg CNE formulated pDNA, or 15 µg CNE formulated pDNA. All nucleic acids expressed RSV F. Sera were collected 2 weeks after the second immunization. All vaccines were injected into the quadriceps muscles of the two hind legs (50 µl per site). Control mice were not injected with any formulation.
Rabbit immunogenicity studies. Immunization studies were conducted at Josman LLC (Napa, CA), a research facility that is licensed through the USDA (No. 93-R-0260) and has a Public Health Service Assurance from the NIH (No. A3404-01). Groups of five female New Zealand white rabbits were immunized i.m. on days 0 and 21 with PBS, VRP at a dose of 1 × 108 IU, 25 µg gp140 TV1.C Env adjuvanted with MF59, 25 µg SAM RNA in PBS, 5 µg CNE-formulated SAM RNA, 25 µg CNE-formulated SAM RNA, 25 µg CNE-formulated mRNA, or 25 µg CNE-formulated pDNA. All nucleic acids expressed gp140 TV1.C Env. Sera were collected prior to first immunization and 2 weeks following each immunization.
Rhesus macaque immunogenicity studies. Outbred female Indian rhesus macaques were housed at Advanced Biosciences Laboratories, Rockville, MD. The Institutional Animal Care and Use Committee at Advanced Biosciences Laboratories, which is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, approved all animal protocols in advance of any procedures. Six macaques were given two i.m. immunizations of 75 µg CNE-formulated SAM RNA expressing gB from hCMV into the right quadriceps muscle and 75 µg CNE-formulated SAM RNA expressing a fusion protein of phosphoprotein 65 and immediate early protein 1 (pp65 –IE1) into the left quadriceps at weeks 0 and 4. Animals were bled at 2 weeks following each immunization to collect serum and peripheral blood mononuclear cells (PBMC) for antibody and T-cell assays. All injection sites were examined for erythema, edema, and other dermal findings on days prior to injections and 24, 48, and 72 hours after injections.
Measurement of RSV F ELISA titers. F-specific serum IgG was assayed by ELISA as described previously.5 Serial dilutions of test serum were added to plates coated with of purified F protein. Bound antibody was detected with horseradish peroxidase-conjugated goat anti-mouse IgG (Southern Biotech, Birmingham, AL). Titers were defined as the reciprocal serum dilution at approximately OD450 nm = 0.5, normalized to a pool of high-titer control sera included on every plate.
Measurement of RSV F neutralizing antibodies. Neutralizing antibody responses against RSV F in mice were performed as described previously.17
Measurement of HIV gp140 ELISA titers. Antibodies specific for gp140 Env were determined in serum and mucosal samples using an ELISA technique, described previously.43 Plates were incubated with serum samples diluted in blocking buffer and further in serial dilutions on plates coated with purified gp140 Env. Antibody was detected with an alkaline phosphatase labeled goat anti-human IgG antibody (Sigma, St Louis, MO). Color intensity was measured on a BIORAD microplate reader at 405 nm (Biorad, model 680).
Measurement of HIV-neutralizing antibodies. Neutralizing antibody responses in rabbit immune sera were measured against HIV-1 subtype C MW965.26 pseudovirions employing a well-standardized assay with the TZM-bl cell line and a luciferase gene readout.44
Measurement of hCMV gB ELISA titers. hCMV ELISA was performed in 384-well microplates using purified hCMV gB. Plates were incubated with serum samples serially diluted in blocking buffer on plates coated with purified hCMV gB protein. After incubation, horseradish peroxidase-conjugated goat anti-monkey IgG (H+L), (Bethyl Laboratories, Montgomery, TX) was diluted and added to the plates for detection. The serum antibody titer is the inverse of the ED20% calculated by interpolation of dilution versus optical density on a four-parameter logistic regression.
Measurement of CMV hCMV neutralizing antibody. Neutralizing antibody responses in rhesus macaque were measured as previously described.45
Rhesus macaque T cell cytokine flow cytometry. Cryopreserved rhesus macaque PBMCs were used for all assays. Thawed PBMCs were seeded into 96-well U-bottom plates (1–2 × 106/well) and incubated overnight in culture medium (RPMI 1640 with L-glutamine, 10% heat inactivated fetal bovine serum, and penicillin/streptomycin). The following morning, the plates were centrifuged, supernatants were aspirated, and cell pellets were resuspended in culture medium containing 1 µg/ml anti-CD28 (clone CD28.2, BioLegend, San Diego, CA), 1 µg/ml anti-CD49d (clone 9F10, BioLegend), and 2 µg/ml antigen (either HCMV gB peptide mix, HCMV pp65 peptide mix, HCMV IE-1 peptide mix, or human actin peptide mix (all from JPT Peptide Technologies, Berlin, Germany). Duplicate cultures for each antigen were established. Cultures were then incubated for a total of 6 hours. Brefeldin A (GolgiPlug; BD Biosciences) was added after the initial 2 hours of culture. For multiparameter flow cytometry, cells were washed and stained with LIVE/DEAD Fixable Yellow Dead Stain (Invitrogen), followed by staining with a panel of antibodies against the surface markers CD4 (clone SK3, APC conjugate, eBioscience, San Diego, CA) and CD8 (SK1, APC-H7 conjugate, BD BioSciences). Following fixation and permeabilization using the BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit (BD Biosciences), cells were stained with a panel of antibodies against the intracellular markers CD3 (clone SP34, fluorescein isothiocyanate conjugate, BD Biosciences), IFN-γ (clone 4S.B3, PE-Cy7 conjugate, BD Biosciences), IL-2 (clone MQ1-17H12, Brilliant Violet 421 conjugate, BioLegend), IL-4 (clone MP4-25D2, PE conjugate, BD Biosciences), and TNF-α (clone MAb11, PerCP/Cy5.5 conjugate, BioLegend). Samples were acquired using an LSR II (BD Biosciences) and analyzed using FlowJo software (Tree Star).
RSV F subunit vaccine. The RSV F subunit vaccine candidate was previously described.17 The gene sequence for protein preparation was identical to the gene sequence for the nucleic acid vaccines except that the native transmembrane and cytoplasmic tail was included in the nucleic acid sequence. Antigen was expressed in Chinese hamster ovary cells and purified from the cell culture medium. MF59 was prepared as described previously.22 Immediately prior to immunization purified antigen was formulated with MF59.
HIV TV1.C subunit vaccine. Recombinant trimeric TV1 gp140 Env used in this study was expressed in a stable Chinese hamster ovary cell line and purified from the cell culture medium.46 The gene sequence for protein preparation was identical to the gene sequence for the nucleic acid vaccines. MF59 was prepared as described previously.22 Immediately prior to immunization purified antigen was formulated with MF59.
Luciferase activity in mice. Female BALB/c mice, aged 6–8 weeks and weighing about 20 g, were obtained from Charles River Laboratories. Prior to inoculation and 1 day prior to imaging mice were depilated at the injection site. Five mice per group were inoculated in the quadriceps in the two hind legs (50 µl per site) on day 0; bioluminescence was acquired using a Xenogen IVIS 200 imaging system (Caliper Life Sciences, Hopkinton, MA) imaging system at 6 hours, and 1, 2, 3, 7, 14, 21, 28, 35, 42, 49, 56, 63, and 70 days post injection. 5 mice per group were inoculated on day 0. Five minutes prior to imaging, mice were injected intraperitoneally with 8 mg/kg of luciferin solution (Caliper Life Sciences). Animals were then anesthetized (2% isoflurane in oxygen) and transferred to the IVIS 200. Image exposure times were kept constant; bioluminescence signal was collected with a cooled CCD camera. Data is reported as total flux (photons/second).
Detection of β-galacatosidase in mouse muscle tissue. Female BALB/c mice, aged 6–8 weeks and weighing ~20 g, were obtained from Charles River Laboratories. All inoculations were injected into the quadriceps muscles of the two hind legs (50 µl per site). Three days postinoculation, mice were euthanized by CO2 asphyxiation and quadriceps muscles were removed aseptically. Quadriceps muscles were placed in PBS containing 2 mmol/l MgCl2 (PBS/MgCl2) on ice. Muscle tissue was stained to reveal β-gal activity using the LacZ Tissue Staining kit following the manufacturer's protocol with modifications (InvivoGen, San Diego, CA). Briefly, quadriceps muscles were cut into, ~2 millimeter slices, fixed in 0.5% glutaraldehyde at room temperature for 1 hour, rinsed, and then stained in a phosphate-buffer solution containing 6 mmol/l ferricyanide, 6 mmol/l, ferrocyanide, and X-gal (5-bromo-4-chloro-3-indoyl-β-D-galactopyranoside) (1 mg/ml) for 2 hours at 37 °C. Following staining, the tissue slices were rinsed in PBS and were then postfixed in 4% paraformaldehyde at 4 °C for 24 hours. For light microscopic evaluation, the stained tissue slices were paraffin embedded, sliced into 5 µm sections, and stained with hematoxylin and eosin. Stained slides were scanned on an Aperio ScanScope, at 8× magnification (Aperio, Vista, CA).
Immune cell infiltrate. Female BALB/c mice, aged 6–8 weeks and weighing about 20 g, were obtained from Charles River Laboratories. Five mice per group were inoculated in the quadriceps muscles of the two hind legs (50 µl per site) on day 0. After 3 days, mice were euthanized by CO2 asphyxiation and the quadriceps muscles were removed aseptically and were placed in Hank's balanced salt solution on ice for further processing. Quadriceps muscles were then dissociated into single cell suspensions and analyzed by flow cytometry for immune cell infiltrate composition as outlined previously.22 Briefly, muscles were digested with 0.05% type II collagenase (Worthington Biochemicals, Lakewood Township, NJ) in Hank's balanced salt solution for 30 minutes at at 37 °C under constant agitation. The resulting cell suspensions were centrifuged, resuspended in Dulbecco's modified Eagle's medium (Gibco, Grand Island, NY) and filtered through 70 μm nylon mesh (Becton Dickinson, Franklin Lakes, NJ) before staining with the following fluorescently labeled antibodies: α-Ly6C-PerCpCy5.5, α-CD11b-PE-Cy7, α-Ly6G-PE, α-CD69-APC, α-CD4-PE, α-CD8-APC-Cy7, α-B220-APC (all from BD Pharmingen, Franklin Lakes, NJ) and α-I-A/I-E-AlexaFluor700, α-F4/80-eFluor450, α-CD11c-APC-AlexaFluor750, α-CD3-v450 (all from eBioscience). The stained cells were analyzed by using a FACS on an LSR II flow cytometer (Becton Dickinson) using BD DIVA software (BD Bioscience). Data were analyzed with FlowJo software (Tree Star).
SEAP expression in rhesus macaque. Outbred female Indian rhesus macaques were housed at the California National Primate Research Center in the University of California, Davis, CA. The Institutional Animal Care and Use Committee of the University of California, Davis (UC Davis), approved all animal protocols in advance of any procedures. A total of 10 rhesus macaques were dosed i.m. on day 0 with PBS (n = 2), VRP expressing SEAP at a dose of 1 × 108 (n = 4) and 75 µg CNE formulated SAM RNA expressing SEAP (n = 4). Blood was drawn on days 3, 7, and 14 and SEAP activity was measured using a chemiluminescent assay (Phospha-Light system; Applied Biosystems, Foster City, CA) according to the manufacturer's instructions.
SUPPLEMENTARY MATERIAL Figure S1. In vitro potency of CNE-formulated RNA before and after 30 minutes of treatment with 6.4 mAU of RNase A per µg of RNA. Figure S2. Ten rhesus macaques were dosed intramuscularly on day 0 with PBS (n = 2, solid line—triangle), 1 × 108 IU of VRP expressing SEAP (n = 4, dashed line—open circle), or 75 µg SAM RNA expressing SEAP formulated with CNE (n = 4, solid line—closed circle). Table S1. Particle size and zeta potential of CNE complexed with nucleic acids.
Acknowledgments
We thank the RNA Vaccine Platform Team at Novartis Vaccines and Diagnostics and, in particular, Nisha Chander, Mildred Ugozzoli, Madhura Shidhore, and Simona Mangiavacchi for coordinating the delivery of formulations for the animal studies; Vania Kenanova, Alicia Carlson, and Farid SaniSarraf for conducting the bioluminescence studies in mice; James Monroe for serology support; Marianna Taccone for cell infiltration work; May Liu and Keith Mansfield at NIBR for assisting with pathology; LAS in Cambridge for conducing mouse studies; Rino Rappouli and Dong Yu for the helpful discussions; Peter Barry at University of California Davis for conducing SEAP experiments in NHP; Anthony Cristillo, Sharon Orndorff, and Ranajit Pal at Advanced Biosciences Laboratories for conducting CMV experiments in NHP; and Joe Beirao at Josman LLC for conducting the HIV experiments in rabbits. Funding for the HIV studies was provided by HIV Vaccine Research and Design Grant 5P01AI066287. Additional funding was provided in part from the Defense Advanced Research Project Agency under agreement HR0011-12-3-001. The authors are Novartis shareholders and employees of Novartis Vaccines and Diagnostics.
Supplementary Material
In vitro potency of CNE-formulated RNA before and after 30 minutes of treatment with 6.4 mAU of RNase A per µg of RNA.
Ten rhesus macaques were dosed intramuscularly on day 0 with PBS (n = 2, solid line—triangle), 1 × 108 IU of VRP expressing SEAP (n = 4, dashed line—open circle), or 75 µg SAM RNA expressing SEAP formulated with CNE (n = 4, solid line—closed circle).
Particle size and zeta potential of CNE complexed with nucleic acids.
References
- Rappuoli R, Mandl CW, Black S, De Gregorio E. Vaccines for the twenty-first century society. Nat Rev Immunol. 2011;11:865–872. doi: 10.1038/nri3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutzler MA, Weiner DB. DNA vaccines: ready for prime time. Nat Rev Genet. 2008;9:776–788. doi: 10.1038/nrg2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardesai NY, Weiner DB. Electroporation delivery of DNA vaccines: prospects for success. Curr Opin Immunol. 2011;23:421–429. doi: 10.1016/j.coi.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geall AJ, Mandl CW, Ulmer JB. RNA: The new revolution in nucleic acid vaccines. Seminars in Immunol. 2013;25:152–159. doi: 10.1016/j.smim.2013.05.001. [DOI] [PubMed] [Google Scholar]
- Geall AJ, Verma A, Otten GR, Shaw CA, Hekele A, Banerjee K, et al. Nonviral delivery of self-amplifying RNA vaccines. Proc Natl Acad Sci USA. 2012;109:14604–14609. doi: 10.1073/pnas.1209367109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petsch B, Schnee M, Vogel AB, Lange E, Hoffmann B, Voss D, et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat Biotechnol. 2012;30:1210–1216. doi: 10.1038/nbt.2436. [DOI] [PubMed] [Google Scholar]
- Luo D, Saltzman WM. Synthetic DNA delivery systems. Nat Biotechnol. 2000;18:33–37. doi: 10.1038/71889. [DOI] [PubMed] [Google Scholar]
- Perri S, Greer CE, Thudium K, Doe B, Legg H, Liu H, et al. An alphavirus replicon particle chimera derived from venezuelan equine encephalitis and sindbis viruses is a potent gene-based vaccine delivery vector. J Virol. 2003;77:10394–10403. doi: 10.1128/JVI.77.19.10394-10403.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner JO, Dryga SA, Kamrud KI. Alphavirus vectors and vaccination. Rev Med Virol. 2002;12:279–296. doi: 10.1002/rmv.360. [DOI] [PubMed] [Google Scholar]
- Barnett SW, Burke B, Sun Y, Kan E, Legg H, Lian Y, et al. Antibody-mediated protection against mucosal simian-human immunodeficiency virus challenge of macaques immunized with alphavirus replicon particles and boosted with trimeric envelope glycoprotein in MF59 adjuvant. J Virol. 2010;84:5975–5985. doi: 10.1128/JVI.02533-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein DI, Reap EA, Katen K, Watson A, Smith K, Norberg P, et al. Randomized, double-blind, Phase 1 trial of an alphavirus replicon vaccine for cytomegalovirus in CMV seronegative adult volunteers. Vaccine. 2009;28:484–493. doi: 10.1016/j.vaccine.2009.09.135. [DOI] [PubMed] [Google Scholar]
- Atkins GJ, Fleeton MN, Sheahan BJ. Therapeutic and prophylactic applications of alphavirus vectors. Expert Rev Mol Med. 2008;10:e33. doi: 10.1017/S1462399408000859. [DOI] [PubMed] [Google Scholar]
- Robert-Guroff M. Replicating and non-replicating viral vectors for vaccine development. Curr Opin Biotechnol. 2007;18:546–556. doi: 10.1016/j.copbio.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smerdou C, Liljeström P. Non-viral amplification systems for gene transfer: vectors based on alphaviruses. Curr Opin Mol Ther. 1999;1:244–251. [PubMed] [Google Scholar]
- Zimmer G. RNA replicons - a new approach for influenza virus immunoprophylaxis. Viruses. 2010;2:413–434. doi: 10.3390/v2020413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hagan DT, Ott GS, Nest GV, Rappuoli R, Giudice GD. The history of MF59(®) adjuvant: a phoenix that arose from the ashes. Expert Rev Vaccines. 2013;12:13–30. doi: 10.1586/erv.12.140. [DOI] [PubMed] [Google Scholar]
- Swanson KA, Settembre EC, Shaw CA, Dey AK, Rappuoli R, Mandl CW, et al. Structural basis for immunization with postfusion respiratory syncytial virus fusion F glycoprotein (RSV F) to elicit high neutralizing antibody titers. Proc Natl Acad Sci USA. 2011;108:9619–9624. doi: 10.1073/pnas.1106536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MA. DNA vaccines: an historical perspective and view to the future. Immunol Rev. 2011;239:62–84. doi: 10.1111/j.1600-065X.2010.00980.x. [DOI] [PubMed] [Google Scholar]
- Doria-Rose NA, Learn GH, Rodrigo AG, Nickle DC, Li F, Mahalanabis M, et al. Human immunodeficiency virus type 1 subtype B ancestral envelope protein is functional and elicits neutralizing antibodies in rabbits similar to those elicited by a circulating subtype B envelope. J Virol. 2005;79:11214–11224. doi: 10.1128/JVI.79.17.11214-11224.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritto E, Mosca F, De Gregorio E. Mechanism of action of licensed vaccine adjuvants. Vaccine. 2009;27:3331–3334. doi: 10.1016/j.vaccine.2009.01.084. [DOI] [PubMed] [Google Scholar]
- Seubert A, Monaci E, Pizza M, O'Hagan DT, Wack A. The adjuvants aluminum hydroxide and MF59 induce monocyte and granulocyte chemoattractants and enhance monocyte differentiation toward dendritic cells. J Immunol. 2008;180:5402–5412. doi: 10.4049/jimmunol.180.8.5402. [DOI] [PubMed] [Google Scholar]
- Calabro S, Tortoli M, Baudner BC, Pacitto A, Cortese M, O'Hagan DT, et al. Vaccine adjuvants alum and MF59 induce rapid recruitment of neutrophils and monocytes that participate in antigen transport to draining lymph nodes. Vaccine. 2011;29:1812–1823. doi: 10.1016/j.vaccine.2010.12.090. [DOI] [PubMed] [Google Scholar]
- O'Hagan DT, Ott GS, De Gregorio E, Seubert A. The mechanism of action of MF59 - an innately attractive adjuvant formulation. Vaccine. 2012;30:4341–4348. doi: 10.1016/j.vaccine.2011.09.061. [DOI] [PubMed] [Google Scholar]
- Hippalgaonkar K, Majumdar S, Kansara V. Injectable lipid emulsions-advancements, opportunities and challenges. AAPS PharmSciTech. 2010;11:1526–1540. doi: 10.1208/s12249-010-9526-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WJ, Kim JK, Choi SH, Park JS, Ahn WS, Kim CK. Low toxicity of cationic lipid-based emulsion for gene transfer. Biomaterials. 2004;25:5893–5903. doi: 10.1016/j.biomaterials.2004.01.031. [DOI] [PubMed] [Google Scholar]
- Ott G, Singh M, Kazzaz J, Briones M, Soenawan E, Ugozzoli M, et al. A cationic sub-micron emulsion (MF59/DOTAP) is an effective delivery system for DNA vaccines. J Controlled Release. 2002;79:1–5. doi: 10.1016/s0168-3659(01)00545-4. [DOI] [PubMed] [Google Scholar]
- Roman VR, Jensen KJ, Jensen SS, Leo-Hansen C, Jespersen S, Te DD, et al. Therapeutic vaccination using cationic liposome-adjuvanted HIV type 1 peptides representing HLA-supertype-restricted subdominant T cell epitopes: safety, immunogenicity, and feasibility in Guinea-Bissau. AIDS Res and Human Retroviruses. 2013;29:1504–1512. doi: 10.1089/AID.2013.0076. [DOI] [PubMed] [Google Scholar]
- Spearman P, Lally MA, Elizaga M, Montefiori D, Tomaras GD, McElrath MJ, et al. A trimeric, V2-deleted HIV-1 envelope glycoprotein vaccine elicits potent neutralizing antibodies but limited breadth of neutralization in human volunteers. J Infect Dis. 2011;203:1165–1173. doi: 10.1093/infdis/jiq175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LR, Wloch MK, Ye M, Reyes LR, Boutsaboualoy S, Dunne CE, et al. Phase 1 clinical trials of the safety and immunogenicity of adjuvanted plasmid DNA vaccines encoding influenza A virus H5 hemagglutinin. Vaccine. 2010;28:2565–2572. doi: 10.1016/j.vaccine.2010.01.029. [DOI] [PubMed] [Google Scholar]
- Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release. 2006;114:100–109. doi: 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Xu L, Anchordoquy T. Drug delivery trends in clinical trials and translational medicine: challenges and opportunities in the delivery of nucleic acid-based therapeutics. J Pharmaceut Sci. 2011;100:38–52. doi: 10.1002/jps.22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson DX, Ljungberg K, Kakoulidou M, Liljeström P. Intradermal electroporation of naked replicon RNA elicits strong immune responses. PLoS One. 2012;7:e29732. doi: 10.1371/journal.pone.0029732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths PD, Stanton A, McCarrell E, Smith C, Osman M, Harber M, et al. Cytomegalovirus glycoprotein-B vaccine with MF59 adjuvant in transplant recipients: a phase 2 randomised placebo-controlled trial. Lancet. 2011;377:1256–1263. doi: 10.1016/S0140-6736(11)60136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel K, Martinez J, Yue Y, Lacey SF, Wang Z, Strelow L, et al. Vaccine-induced control of viral shedding following rhesus cytomegalovirus challenge in rhesus macaques. J Virol. 2011;85:2878–2890. doi: 10.1128/JVI.00883-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kormann MS, Hasenpusch G, Aneja MK, Nica G, Flemmer AW, Herber-Jonat S, et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat Biotechnol. 2011;29:154–157. doi: 10.1038/nbt.1733. [DOI] [PubMed] [Google Scholar]
- Wolff JA, Malone RW, Williams P, Chong W, Acsadi G, Jani A, et al. Direct gene transfer into mouse muscle in vivo. Science. 1990;247:1465–1468. doi: 10.1126/science.1690918. [DOI] [PubMed] [Google Scholar]
- Pollard C, Rejman J, De Haes W, Verrier B, Van Gulck E, Naessens T, et al. Type I IFN counteracts the induction of antigen-specific immune responses by lipid-based delivery of mRNA vaccines. Mol Ther. 2013;21:251–259. doi: 10.1038/mt.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotin-Mleczek M, Duchardt KM, Lorenz C, Pfeiffer R, Ojkić-Zrna S, Probst J, et al. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J Immunother. 2011;34:1–15. doi: 10.1097/CJI.0b013e3181f7dbe8. [DOI] [PubMed] [Google Scholar]
- Kallen KJ, Heidenreich R, Schnee M, Petsch B, Schlake T, Thess A, et al. A novel, disruptive vaccination technology: Self-adjuvanted RNActive ((R)) vaccines. Human Vaccines & Immunotherapeutics. 2013;9:2263–2276. doi: 10.4161/hv.25181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheel B, Teufel R, Probst J, Carralot JP, Geginat J, Radsak M, et al. Toll-like receptor-dependent activation of several human blood cell types by protamine-condensed mRNA. Eur J Immunol. 2005;35:1557–1566. doi: 10.1002/eji.200425656. [DOI] [PubMed] [Google Scholar]
- Weide B, Pascolo S, Scheel B, Derhovanessian E, Pflugfelder A, Eigentler TK, et al. Direct injection of protamine-protected mRNA: results of a phase ½ vaccination trial in metastatic melanoma patients. J Immunother. 2009;32:498–507. doi: 10.1097/CJI.0b013e3181a00068. [DOI] [PubMed] [Google Scholar]
- Hoerr I, Obst R, Rammensee HG, Jung G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur J Immunol. 2000;30:1–7. doi: 10.1002/1521-4141(200001)30:1<1::AID-IMMU1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Mooij P, Bogers WM, Oostermeijer H, Koornstra W, Ten Haaft PJ, Verstrepen BE, et al. Evidence for viral virulence as a predominant factor limiting human immunodeficiency virus vaccine efficacy. J Virol. 2000;74:4017–4027. doi: 10.1128/jvi.74.9.4017-4027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montefiori DC. Measuring HIV neutralization in a luciferase reporter gene assay. Methods Mol Biol. 2009;485:395–405. doi: 10.1007/978-1-59745-170-3_26. [DOI] [PubMed] [Google Scholar]
- Loomis RJ, Lilja AE, Monroe J, Balabanis KA, Brito LA, Palladino G, et al. Vectored co-delivery of human cytomegalovirus gH and gL proteins elicits potent complement-independent neutralizing antibodies. Vaccine. 2013;31:919–926. doi: 10.1016/j.vaccine.2012.12.009. [DOI] [PubMed] [Google Scholar]
- Srivastava IK, Stamatatos L, Legg H, Kan E, Fong A, Coates SR, et al. Purification and characterization of oligomeric envelope glycoprotein from a primary R5 subtype B human immunodeficiency virus. J Virol. 2002;76:2835–2847. doi: 10.1128/JVI.76.6.2835-2847.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
In vitro potency of CNE-formulated RNA before and after 30 minutes of treatment with 6.4 mAU of RNase A per µg of RNA.
Ten rhesus macaques were dosed intramuscularly on day 0 with PBS (n = 2, solid line—triangle), 1 × 108 IU of VRP expressing SEAP (n = 4, dashed line—open circle), or 75 µg SAM RNA expressing SEAP formulated with CNE (n = 4, solid line—closed circle).
Particle size and zeta potential of CNE complexed with nucleic acids.