

Abstract
The high transduction efficiency of lentiviral vectors in a wide variety of cells makes them an ideal tool for forward genetics screenings addressing issues of cancer research. Although molecular targeted therapies have provided significant advances in tumor treatment, relapses often occur by the expansion of tumor cell clones carrying mutations that confer resistance. Identification of the culprits of anticancer drug resistance is fundamental for the achievement of long-term response. Here, we developed a new lentiviral vector-based insertional mutagenesis screening to identify genes that confer resistance to clinically relevant targeted anticancer therapies. By applying this genome-wide approach to cell lines representing two subtypes of HER2+ breast cancer, we identified 62 candidate lapatinib resistance genes. We validated the top ranking genes, i.e., PIK3CA and PIK3CB, by showing that their forced expression confers resistance to lapatinib in vitro and found that their mutation/overexpression is associated to poor prognosis in human breast tumors. Then, we successfully applied this approach to the identification of erlotinib resistance genes in pancreatic cancer, thus showing the intrinsic versatility of the approach. The acquired knowledge can help identifying combinations of targeted drugs to overcome the occurrence of resistance, thus opening new horizons for more effective treatment of tumors.
Introduction
The analysis of recurrent genetic lesions in human tumors allowed the rational design of novel targeted therapies and currently assists in selecting anticancer drug regimens according to the mutational profile of each patient.1 However, despite providing significant rates of response, targeted therapies rarely result in disease eradication due to the emergence of drug-resistant clones that cause tumor relapse. The likelihood of response of each cancer to treatment with specific drugs is strongly influenced by the mutational landscape of its genome,2 which can render the targeted protein resistant to inhibition, reactivate downstream the targeted pathway, or engage alternative pathways that bypass the blocks provided by the therapeutic compound.3 The identification of genes and molecular networks whose deregulation causes unresponsiveness to therapy is urgently required for better stratification of patients toward more effective personalized treatments and to design combinations of existing and novel drugs capable to overcome the resistance to a single compound.4
A number of strategies have been devised in order to identify the culprits of drug resistance5 including association between the genomic mutation landscape and the sensitivity/resistance profiles of clinical cases, in vivo and in vitro induction of spontaneous resistance upon chronic drug administration/exposure and functional screens with cDNA, siRNA and shRNA libraries. The main disadvantages of these strategies are the difficulty to distinguish driver mutations among the plethora of bystander lesions and the limitation of screenings for anticancer drug resistance by overexpressing or knocking down only the subset of known genes included in the libraries. In order to identify driver genetic lesions, cancer genomic studies utilize statistical approaches that usually require large collection of human samples and sequencing efforts. On the other hand, insertional mutagenesis is a forward genetic approach that has been used for the functional identification of novel genes involved in the pathogenesis of human cancers.6,7 Retroviruses or transposons may activate cellular oncogenes or inactivate tumor suppressor genes near the integration site and induce cancer formation in mice. Identification of genomic regions recurrently targeted by integrations (Common Integration Sites (CIS)) in tumors induced by insertional mutagens has allowed the discovery of new cancer driver genes.6,7 Recently, we have developed new insertional mutagens based on lentiviral vectors (LVs) and used them to identify novel genes involved in hepatocellular carcinogenesis.8 LVs represent an appealing tool for forward genetic studies since they have high transduction efficiency, wide tissue tropism, and their mutagenic properties can be modified by changing the vector design.7,9,10 Since not only cell transformation, but most selectable phenotypes can be studied with insertional mutagenesis, we took advantage of the recently validated LV-based insertional mutagens to build up a new experimental platform to identify the culprits of drug resistance to targeted anti-cancer therapies in different tissue types.
Breast cancer is the most commonly diagnosed cancer and a leading cause of death in women worldwide.11 Recently, targeted therapies have been developed to treat different subtypes of the disease characterized by specific genotypes.12 In particular, HER2-directed monoclonal antibodies and small molecules have been used to treat HER2+ breast carcinomas, which represent 30% of human mammary tumors.13 Despite providing significant clinical responses, intrinsically resistant tumors exist, and even when an initial regression is observed, the incidence of tumor relapse is extremely high.14 Therefore, we decided to apply LV-based insertional mutagenesis screening to identify genes whose deregulation is involved in resistance to lapatinib, a HER2 and EGFR inhibitor recently approved for the treatment of metastatic HER2+ breast cancer.15
As targets for insertional mutagenesis, we used the BT474 and SKBR3 cell lines. The BT474 cell line expresses the estrogen and progesterone receptors (ER and PR respectively) and represents a luminal B subtype breast tumor, while the SKBR3 cell line is ER- PR- and represents the typical HER2+ subtype.16 Since both cell lines highly overexpress the HER2 receptor, they are commonly employed in research for studies on HER2-targeting agents, including trastuzumab (a monoclonal antibody) and lapatinib (a tyrosine kinase inhibitor), to which they display substantial sensitivity.17
By applying LV-based insertional mutagenesis to these two breast cancer cell lines, we identified 62 genes putatively involved in lapatinib resistance. We showed that forced expression of the top ranking CIS genes establishes resistance to the drug and found that those genes are frequently mutated or overexpressed in human breast tumors characterized by poor prognosis. Then, as a proof of principle of the broad applicability of this LV-based platform to different types of cancers and drugs, we successfully applied it to another clinically relevant context, such as the resistance to erlotinib in the highly lethal pancreatic adenocarcinoma. Our screening identified three candidate erlotinib resistance genes in the HPAC pancreatic cancer cell line. Overall, this study describes and validates an efficient and versatile strategy for the identification of anticancer drug resistance genes of potentially high clinical relevance.
Results
LV-based insertional mutagenesis induces resistance to lapatinib in a dose-dependent fashion in breast cancer cell lines
In order to induce drug resistance, we designed an LV construct containing the strong spleen focus-forming virus (SF) enhancer/promoter in the long terminal repeats (LTR) and no transgene inside (LV.SF.LTR, Figure 1a), a vector configuration similar to the one proved to be highly genotoxic in sensitive in vivo assays.10,18 The rationale of our in vitro screening is to infect a bulk population of drug-sensitive cells with LV.SF.LTR to generate drug-resistant clones by insertional mutagenesis. Upon drug exposure, drug-resistant clones are positively selected and expand, while sensitive clones die and are no more represented in the bulk population. After drug selection, DNA is extracted from the cell population enriched in LV-induced resistant cells, and LV insertions are retrieved and mapped. Statistical analysis identifies CIS in the resistant populations that are not found in cell cultures not exposed to the drugs. These loci represent novel candidate anticancer drug resistance genes (Figure 1a).
Figure 1.

Lentiviral vector (LV)-transduction induces resistance to different lapatinib concentrations in a LV dose-dependent fashion in two different breast cancer cell lines. (a) Schematic representation of the project rationale: (1) initially drug-sensitive cells are transduced with a highly mutagenic LV (depicted above) with strong SF enhancer/promoter elements in the LTR and no transgene inside. SA, splice acceptor; SD, splice donor; SF, spleen focus forming virus enhancer/promoter sequences. (2) Insertional mutagenesis induces the emergence of drug resistant clones (in red) with traceable mutations (the LV integrations) near or within genes whose deregulation confers resistance to a targeted anticancer drug of interest. (3) Upon drug selection, resistant cell clones grow and are strongly enriched in the bulk population. Following steps include (4) DNA collection from a bulk population of resistant clones, (5) amplification of vector-genome junctions by LAM-PCR and (6) deep sequencing of PCR products. (7) Bioinformatics and (8) statistical analysis finally allow mapping of the integration sites and the identification of commonly targeted genes in this selected population (CIS), which mark novel candidate culprits of drug resistance. (b, c) Representative ×50 magnifications at the end of selection for untransduced and LV-transduced (multiplicity of infection (MOI): 100) SKBR3 cells and untransduced and LV-transduced (MOI: 50) BT474 cells. Yellow dashed lines mark the borders of representative resistant clones. Scale is indicated in red. (d–g) Total number of colonies (d and f) and cells (e and g) counted at the end of selection with two different drug concentrations tested. The experimental groups are shown in the legends. The standard deviation is shown for each group. Asterisks indicate statistical significance by one-way Anova analysis with Bonferroni's multiple comparison test correction, with one, two or three asterisks if the P value is below 0.05, 0.01, or 0.001, respectively.
Two HER2+ breast cancer cell lines, SKBR3 and BT474, were infected with the LV.SF.LTR vector at multiplicity of infection (MOI) 1, 10, and 100 (SKBR3 cells) or MOI 0.5, 5, and 50 (BT474 cells). Two weeks later, both LV-infected and untransduced cells were seeded at a density of 107 cells/plate and supplemented with medium containing different concentrations of lapatinib (0.5, 1.0, or 2.0 μmol/l) or vehicle (“dimethyl sulfoxide (DMSO)-only” samples).
Lapatinib treatment induced major cell death, but the few surviving cells grew and formed colonies (Figure 1b,c; Supplementary Figure S1a). Lapatinib treatment was carried on until counting the number of colonies becomes feasible (see Materials and Methods). LV transduction induced a significant increase in the number of resistant cells and resistant colonies with respect to treatment-matched untransduced cells in both cell lines (P value ranging from <0.001 to <0.05 by one-way analysis of variance with Bonferroni's multiple comparison test correction). Moreover, the number of resistant cells and resistant colonies increased with the LV dose, at different lapatinib concentrations tested (Figure 1d–g; Supplementary Figure S1b–d). These data suggest that LV-mediated insertional mutagenesis induced lapatinib resistance in a LV dose-dependent fashion.
Integration analysis reveals candidate lapatinib resistance genes
Lapatinib-resistant cell colonies were pooled and harvested. DNA was extracted and subjected to linear amplification-mediated PCR (LAM-PCR) in order to retrieve LV/cellular genome junctions representative of the vector integration sites (Supplementary Figure S2a). Together with lapatinib-selected samples, we performed LAM-PCR also on samples collected before lapatinib selection (“PRE”) and on DMSO-only samples. Altogether, we generated 36 samples that were subjected to LAM-PCR with two different restriction enzymes (72 total LAM-PCRs, see Supplementary Table S1 and Supplementary Figure S2b).
LAM-PCR products were tagged with barcoded fusion primers and sequenced in four MiSeq (Illumina) runs. Sequences were analyzed by a dedicated informatics pipeline and mapped on the human genome by the Burrows-Wheeler Aligner (BWA). Overall, we mapped more than 8 million LAM-PCR products, corresponding to 39,365 unique integration sites, of which 24,306 were retrieved from SKBR3 samples (12,382 in lapatinib-selected samples and 11,924 in controls) and 15,059 from BT474 samples (9,007 in lapatinib-selected samples and 6,052 in controls). The LV integrations were broadly distributed throughout the genome (Supplementary Figure S3) and the deepness of integration retrieval allowed us to have a representative picture of the integration events in the analyzed samples (see Materials and Methods).
CIS were defined as previously described19 but applying additional cut-offs of stringency to avoid false positive CIS in large datasets of integrations (see Materials and Methods). Comparison with the list of CIS identified in PRE and DMSO-only samples corrected for biases due to the aberrant karyotype of the cell lines and LV integration site preferences, and excluded targeted genes promoting cell proliferation and fitness independently from drug exposure (Supplementary Table S2; Supplementary Figure S4a–d). By subtracting these CIS from the CIS list found in lapatinib-selected samples (Supplementary Table S3), we identified 27 CIS in SKBR3 cells and 35 CIS in BT474 cells, which represent candidate lapatinib resistance genes (Table 1; Supplementary Table S4; Supplementary Table S5; Supplementary Table S6).
Table 1. List of the top ranking common integration sites (CIS) corresponding to novel candidate lapatinib resistance genes.
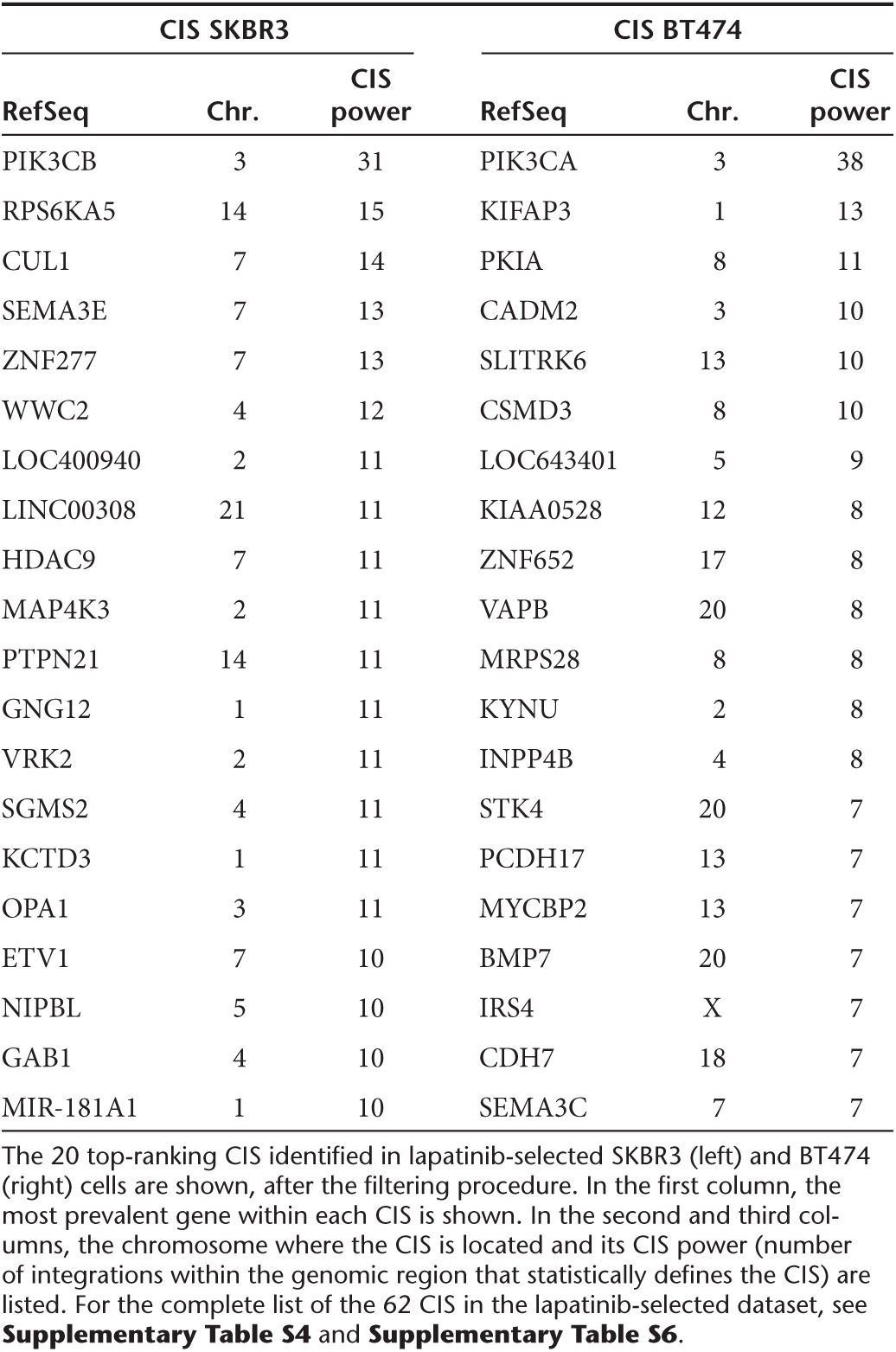
The top-ranking CIS in BT474 cells was PIK3CA on chromosome 3, targeted by 38 independent integrations occurring in a 65 Kb window (Figure 2a). All the integrations were located in the first intron of the gene, upstream of the first coding exon. Moreover, 36/38 integrations were orientated in sense with the transcription of the PIK3CA gene, which is very unlikely to happen by chance. Indeed, these findings strongly suggest active selection of clones carrying insertion of the SF promoter upstream of the PIK3CA gene, which could generate high levels of chimeric transcripts encoding full-length protein by promoter insertion, a well-known mechanism of insertional mutagenesis.8,18 PIK3CA encodes for the alpha isoform of the p110 catalytic subunit of PI3K, and BT474 cells carry a K111N-mutated version of PIK3CA, which has transforming potential20 and was never associated before to lapatinib resistance.
Figure 2.

Representative CIS in the lapatinib-selected dataset. The UCSC Genome Browser snapshots are shown for four representative CIS. (a) PIK3CA on chromosome 3 was targeted by 38 integrations in a 65 Kb window. All the integrations were located in the first intron of the gene, upstream of the first coding exon. Thirty-six of 38 integrations were orientated in sense with the transcription of the gene (which is shown by a black arrow). A green arrow sketches the marked orientation preference of integrations. (b) PIK3CB on chromosome 3 was targeted in SKBR3 cells by 31 integrations clustered in a 147 Kb window upstream of the gene with marked orientation preference (green arrow). C) LINC00308 on chromosome 21, identified as a CIS in SKBR3 cells, is a noncoding RNA gene targeted by integrations without orientation preference, sketched by green and red arrows pointing in opposite directions. (d) INPP4B on chromosome 4, identified as a CIS in BT474 cells, was targeted by intragenic insertions without orientation preference. In each panel, the genomic region showed in the snapshot is highlighted on top of the chromosome outline as a red vertical bar. Integrations are shown as colored vertical bars in each dataset (from the top: BT474 DMSO/PRE controls, BT474 lapatinb-selected cells, SKBR3 DMSO/PRE controls and SKBR3 lapatinib-selected cells). For the integrations in selected samples, red, blue and green bars indicate that they have been retrieved from cell population transduced at high, intermediate and low multiplicity of infection (MOI), respectively. For SKBR3, high MOI = 100, intermediate MOI = 10, low MOI = 1: for BT474 high MOI = 50, intermediate MOI = 5, low MOI = 0.5. A purple line underscores the dataset in which this CIS was identified. The succession of introns (lines) and exons (filled squares) of the gene(s) is shown in blue on the bottom of the panel.
Notably, the top ranking CIS in SKBR3 cells was PIK3CB, another isoform of the p110 catalytic subunit of PI3K, targeted by 31 integrations clustered in a 147 Kb window upstream of the gene with marked orientation preference (27/31 integrations are in sense with PIK3CB transcription), which might again be indicative of a promoter insertion mechanism of mutagenesis (Figure 2b). PIK3CB was not previously linked to resistance to targeted drugs, including lapatinib.
Additionally, we identified CIS genes targeted by LV integrations with a pattern of orientation and distribution that is suggestive of an enhancer-mediated mechanism of insertional mutagenesis, such as MAP4K3 and CADM2 (both with six integrations occurring in antisense to gene transcription and five in sense, Supplementary Figure S5a,b). We also found CIS associated to noncoding RNAs, such as LINC00308, MIR181A1, and LOC647107 (Figure 2c; Supplementary Figure S5c; Supplementary Table S4; Supplementary Table S6) and other CIS genes in the PI3K pathway such as INPP4B, MAP4K3, GAB1, and RPS6KA5 (Figure 2d; Supplementary Figure S5a; Supplementary Table S4; Supplementary Table S6).
Validation of PIK3CA and PIK3CB as lapatinib resistance genes
In order to validate the two top-ranking CIS discovered by our study as bona fide lapatinib resistance genes, we tested the prosurvival effect of their forced expression in cells cultured in the presence of lapatinib. We generated LVs with self-inactivating LTRs (a design associated with low risk of insertional mutagenesis)18 in which the expression of the putative lapatinib resistance genes (K111N-mutant PIK3CA or wild-type PIK3CB) is driven by the SF enhancer/promoter in an internal position (Figure 3a), and used them to transduce BT474 and SKBR3 cells. The BT474 cells transduced with K111N-mutant PIK3CA displayed a significant survival advantage upon lapatinib treatment as compared to mock treated cells and wild-type PIK3CA-overexpressing cells (P < 0.001 at 1 week by unpaired t-test, Figure 3b,c and Supplementary Figure S6a,c; P < 0.01 at 24 hours, P < 0.05 at 72 hours by unpaired t-test). Similarly, PIK3CB-transduced SKBR3 cells displayed a significant survival advantage upon lapatinib treatment compared to mock-treated cells (P < 0.001 at 2 weeks by unpaired t-test, Figure 3d,e; P < 0.01 at 24 and 72 hours by unpaired t-test, Supplementary Figure S6b). These results were confirmed by measuring the lapatinib dose–response curve of K111N PIK3CA overexpressing BT474 cells and PIK3CB-ovrexpressing SKBR3 cells (Supplementary Figure S6d). These data indicate that the genes identified in our screening are bona fide lapatinib resistance genes.
Figure 3.
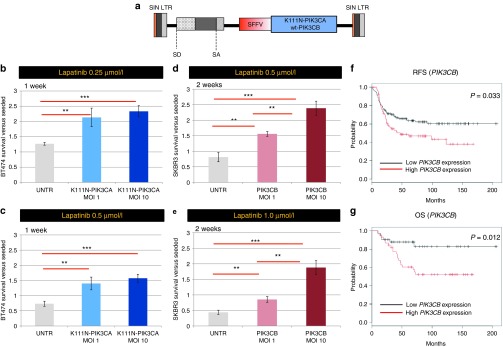
Validation of K111N-mutant PIK3CA and wild-type PIK3CB as novel lapatinib resistance genes. (a) The structure of the SIN lentiviral vector (LV) used for validation experiments is shown. Acronyms as in Figure 1a legends. (b–e) The fold change difference toward initially plated cells is plotted in the graph for BT474 cells (b, c) and SKBR3 cells (d, e) transduced at two different vector doses (multiplicity of infection (MOI): 1 and MOI: 10) in order to overexpress the K111N-mutant PIK3CA gene (b, c) or the wild-type PIK3CB gene (d, e), and cultured for 1 week at 0.25 and 0.5 μmol/l lapatinib (b, c) or for 2 weeks at 0.5 and 1.0 μmol/l lapatinib (d, e) together with untransduced cells (UNTR). The standard deviation is shown for each group. Asterisks indicate statistical significance by unpaired t-test, with one, two, or three asterisks if the P value is below 0.05, 0.01, or 0.001, respectively. (f, g) The relapse-free survival (RFS) and overall survival (OS) Kaplan–Meier plots are shown for human HER2+ breast cancer patients taken from http://kmplot.com, split in two cohorts according to PIK3CB expression (red and black lines corresponding to high- and low-expressers, respectively). The logrank P values is shown.
We then performed data-mining on human breast cancer datasets to validate the clinical relevance of the top 2 ranking lapatinib resistance genes. PIK3CA is the most frequently mutated gene in human breast cancer together with TP53 (International Cancer Genome Consortium Database: http://dcc.icgc.org and refs. 21,22). PIK3CA activating mutations were recently found to be associated to a worse prognosis23 and to trastuzumab and lapatinib resistance in breast cancer patients,24,25 thus validating the clinical relevance of our screening strategy. Since overexpression of wild-type PIK3CB was also able to induce lapatinib resistance in our study, we interrogated its expression profile in human breast cancers (see Materials and Methods). Remarkably, we found that increased expression of PIK3CB is associated to a significantly decreased overall survival (OS) and relapse-free survival (RFS, P value = 0.033 and 0.012 by Log Rank test, respectively) in patients affected by HER2+ breast cancer (Figure 3f,g).
LV-based insertional mutagenesis identifies erlotinib resistance genes in pancreatic cancer cell line
In order to demonstrate the broad applicability of the LV platform for insertional mutagenesis studies aimed at identifying novel drug resistance genes, we performed a screening in a different tumor type and with a different drug. HPAC, a well-characterized pancreatic adenocarcinoma cell line, was used to identify the culprits of resistance to erlotinib, an EGFR inhibitor used in the clinical practice for pancreatic cancer.26,27 HPAC cells were infected at MOI 75 with the LV.SF.LTR vector. Two weeks later, both LV-infected and untransduced cells were seeded at a density of 4 × 106 cells/plate and supplemented with medium containing different concentrations of erlotinib (50 or 100 μmol/l) or vehicle (“DMSO-only”). The treatment with the vector induced a significant increase in the total number of resistant cells and resistant colonies (P value ranging from <0.01 to <0.05 by unpaired t-test, Figure 4a,b; Supplementary Figure S7a). Resistant colonies were pooled and harvested, and their genomic DNA subjected to LAM-PCR in order to retrieve vector integration sites. A MiSeq sequencing run was performed on a library of 16 LAM-PCR products from 8 samples (see Supplementary Table S1). We mapped around 1.25 million LAM-PCR products, corresponding to 2,610 unique integration sites, of which 1,733 in erlotinib-selected samples and 877 in DMSO-only/PRE controls.
Figure 4.
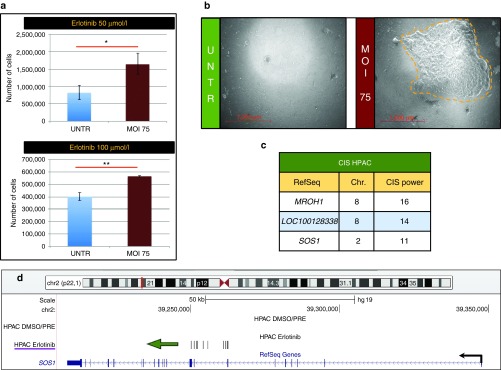
Lentiviral vector (LV)-based insertional mutagenesis unravelled erlotinib resistance genes in the HPAC pancreatic cancer cell line. (a) Total number of cells counted at the end of selection at each drug concentration tested. The experimental groups are shown in the legends. The standard deviation is shown for each group. Asterisks indicate statistical significance by unpaired t-test, with one, two, or three asterisks if the P value is below 0.05, 0.01, or 0.001, respectively. (b) Representative ×50 magnifications at the end of selection for untransduced and LV-transduced (MOI: 75) HPAC cells. Yellow dashed lines marks the borders of a representative resistant clone. Scale is indicated in red. (c) The three CIS identified in erlotinib-selected HPAC cells are shown, after the filtering procedure. In the first column, the most prevalent gene within each CIS is shown. In the second and third columns, the chromosome where the CIS is located and its CIS power (number of integrations within the fixed genomic region which statistically defines the CIS) are listed. (d) SOS1 on chromosome 2 was targeted in erlotinib-selected HPAC cells by 11 integrations clustered in a 12 Kb window upstream of the gene and all of them were orientated in sense with the transcription of the gene (which is shown by a black arrow). A green arrow pointing in the direction of gene transcription outlines the marked orientation preference of integrations.
The CIS were defined and filtered as described above. We retrieved from the erlotinib-selected dataset 3 CIS, which represent candidate erlotinib resistance genes (Figure 4c; Supplementary Figure S7b–d). Among them, SOS1 on chromosome 2 carried 11 integrations in a 12 Kb window within intron 8 (Figure 4d). Moreover, 11/11 integrations were orientated in sense with the transcription of the gene, suggesting that promoter insertion inducing overexpression of a truncated hyperactive protein may be the result of LV integration8 and be the culprit of erlotinib resistance.
Discussion
Targeted therapies represent a new frontier of cancer treatment, but their specificity is offset by the occurrence of pre-existing or acquired resistance. Understanding the mechanisms that dictate anticancer drug resistance is currently an unmet need that can have a significant impact on the clinics. Previous forward genetics studies aimed at unraveling the molecular bases of anticancer drug resistance have used γ-retroviruses or transposons to identify genes potentially endowing resistance towards cytotoxic chemotherapy or hormonal antagonists.28,29,30 However, when studying cancer cells, the low in vitro efficiency of transposon-based insertional mutagenesis systems strongly limited the discovery of novel culprits that induce resistance to anticancer drugs. In this study, we developed and validated a new screening strategy based on LV insertional mutagenesis that allows identifying genes conferring resistance to targeted anticancer drugs with high efficiency.
This LV-based platform offers several advantages. Differently from low-throughput single-cell-derived clonal studies, by working with bulk cultures exposed to increasing LV loads, we could screen a large number (5 × 106–109) of insertional mutagenesis events per Petri dish, which allowed identifying 65 candidate drug resistance genes. The finding that the yield of resistant cells and resistant clones significantly increases upon incrementing the LV dose further confirms that we are mainly scoring vector-induced events of drug resistance. All the identified CIS are constituted by integrations retrieved by at least two biological replicates of transduction and at least two different experimental conditions (MOI of infection and/or different drug concentration for the selection, Supplementary Table S6), thus strengthening the solidity of our findings.
LVs are able to transduce at high level a vast number of cell types9 and have a broad integration pattern throughout the genome (Supplementary Figure S3), with a bias toward gene dense regions and for expressed genes,31 two features which may facilitate the mechanisms of mutagenesis underlying the development of drug resistance. Preferential integration of LVs within transcriptional units can allow different mechanisms of gene deregulation such as enhancer insertion, promoter insertion, aberrant splicing, and loss of function.7,10 The platform is applicable to different clinically relevant contexts. Indeed, as a proof-of-concept study, we investigated the mechanisms of resistance to a clinically relevant drug, lapatinib, in a highly frequent and lethal cancer, i.e., HER2+ breast cancer. Then, we successfully applied the screen to a different tumor model (pancreatic adenocarcinoma cells) and drug (erlotinib). The versatility and scalability of the platform could make it suitable to generate comparative data by studying the genes that confer resistance to the same drug in different cell lines, or the genes that confer resistance to different drugs in the same cell line. Moreover, it could also be deployed to study mechanisms of resistance to different drug combinations in order to provide further steps of therapy optimization. There are also some limitations to be recognized in our strategy, such as the possibility to screen only cell-autonomous mechanisms in the in vitro setting, and the bias toward gain-of-function mutations typical of different insertional mutagens.6,7 Additionally, the analysis of bulk populations of resistant clones does not allow measuring quantitative deregulation of RNA and protein in specific resistant clones that may require a postscreening validation. By deep sequencing of LAM-PCR products from selected samples as well as from control samples collected before the occurring of selection (“PRE”) or cultured in parallel without the drug (“DMSO-only”), we could eliminate the effect of some biases that could hamper our analysis. By filtering out CIS detected in control samples, we eliminated the effect of LV integration site preferences and the false positive CIS due to the aberrant karyotype of the cell lines. Indeed, several of the filtered CIS map on chromosomal regions known to be amplified in SKBR3 and BT474 cells and thus resulting overtargeted when compared to the nonamplified genome (Supplementary Table S3; Supplementary Figure S4a).32 Moreover, by the DMSO-only controls we could also discard those genes that may provide a proliferative advantage both in the absence and presence of the drug (such as the well-known oncogenes MET, MYC, and MECOM Supplementary Figure S4b–d). Although we recognize that these genes may also be relevant for inducing drug resistance and warrant further investigation, here we conservatively preferred to focus on genes that were selected exclusively due to lapatinib selective pressure.
Remarkably, the top-ranking CIS in both cell lines are two closely related genes, PIK3CA and PIK3CB, which encode for two isoforms (α and β) of the p110 catalytic subunit of PI3K. Forced expression of these genes in cell lines validated them as lapatinib resistance genes. Moreover, our integration dataset is strongly enriched in genes from the PI3K cascade. The PIK3CA oncogene is the most frequently mutated gene in human breast cancer together with TP53 (International Cancer Genome Consortium Database: http://dcc.icgc.org and Refs [21,22]). Remarkably, recent studies have shown that PIK3CA mutations are significantly associated to resistance to lapatinib or trastuzumab (a monoclonal antibody targeting HER2).23,24,25,33 These data validate the clinical relevance of the findings obtained by our experimental platform.
In our screening, PIK3CA and PIK3CB were cell line-specific and mutually exclusive CIS in BT474 and SKBR3 cells, respectively. PIK3CA is the strongest CIS in BT474 cells, which harbor a K111N mutation in the N-terminal domain of the protein not previously associated to resistance. This mutation has been classified at intermediate level for its overall capacity to promote cell proliferation, growth factor independency, morphogenesis potential, focus formation and invasivity.20 We demonstrated that overexpression of the K111N-mutated PIK3CA is able to confer resistance to lapatinib in vitro, while overexpression of wild-type PIK3CA is not. Accordingly, PIK3CA was not found as a CIS in SKBR3 cells that carry wild-type alleles of the gene, highlighting how the pre-existing mutational landscape of the selected cell model can dictate the culprits of drug resistance after insertional mutagenesis. Collectively, in our study, only the concurrent mutation and overexpression of PIK3CA is able to provide resistance to lapatinib. On the other hand, we retrieved a SKBR3 cell-specific CIS in proximity of PIK3CB, a gene which has never been linked to lapatinib resistance before but which is emerging as an important player in breast cancer and a novel therapeutic target.34,35 PIK3CB overexpression was associated with worse breast tumor prognosis, higher grade and distant metastasis.36 Moreover, it was correlated to HER2 positivity and ER/PR negativity, in agreement with SKBR3 receptor status. It has been shown that differently from p110α, p110β is oncogenic when overexpressed in the wild-type state.37 Furthermore, when PTEN expression among different HER2-amplified cell lines was assessed, SKBR3 cells showed a low level of PTEN protein compared to other cell lines (e.g., BT474).38 Recently, it has been demonstrated that PTEN-deficient tumors rely on p110β signaling for growth in both cell-based and in vivo settings.35,39 Our data may thus uncover a relevant role for PIK3CB in inducing lapatinib resistance in this specific molecular subtype of breast cancer. Importantly, by mining gene expression and clinical data, we found that patients carrying HER2+ breast cancer characterized by high PIK3CB expression have a decreased RFS and OS compared to patients with low PIK3CB expression. These findings show that our screening was able to identify novel lapatinib resistance genes with potentially high clinical relevance. Moreover, given the relevance of p110β activity in PTEN-null tumors, we may speculate that its classification as a drug resistance gene may extend beyond the tissues and drugs that we have investigated. Overall, our findings indicate that hyperactivation of PI3K represents a converging node of lapatinib resistance. These results may corroborate the rationale of combining administration of lapatinib and PI3K inhibitors, which is currently being tested in a phase I/II clinical trial (PIKHER2) (http://clinicaltrials.gov/show/NCT01589861). Moreover, our findings prompt to explore the use of inhibitors specific for the alpha or beta subunits in tumors carrying the specific mutations or upregulation.
Regarding candidate erlotinib resistance genes identified in HPAC cells, one of the most intriguing finding was the presence of a CIS within SOS1, which encodes a guanine nucleotide exchange factor for RAS (whose mutations are detected in more than 90% of human pancreatic cancers).40 Interestingly, as we previously reported for LV-induced murine hepatocellular carcinomas,8 integrations in SOS1 were all in intron 8 and in the same orientation of gene transcription (Figure 4d). Since we showed that the LV-truncated SOS1 protein was responsible for hepatocarcinogenesis, we can speculate that also the observed drug resistance may be mediated by overexpression of a truncated and hyperactive SOS1 protein.
Several newly identified candidate lapatinib and erlotinib resistance genes are intertwined in cellular pathways that converge to transduce HER2/EGFR signaling (Figure 5). Among them, CUL1 (CIS power 14), a novel marker of poor prognosis and a potential therapeutic target in human breast cancer,41 is a core component of multiple E3 ubiquitin-protein ligase complexes, which mediate the ubiquitination of proteins involved in cell cycle progression, signal transduction and transcription. IRS4 (CIS power 7) was shown to potentiate PI3K activity.42 INPP4B (CIS power 8) is a tumor suppressor gene that inhibits PI3K signaling and whose loss of heterozygosity in breast cancer patients is correlated with lower overall survival.43 RPS6KA5 (CIS power 15, also known as MSK1) is a serine-threonine kinase that was previously reported to be involved in drug resistance in mouse models of breast cancer.44 Upon phosphorylation by ERK, MSK1 activates different transcription factors, among which ETV1 (CIS power 10), one of the major effectors of HER2-driven mammary tumorigenesis.45,46,47 This convergence may indicate that the selected tumor cells are still dependent on the pathways inhibited by the drug and that drug resistance occurs by downstream or parallel activation of the same signaling axis.
Figure 5.

Several newly identified lapatinib/erlotinib resistance genes are intertwined in the transduction pathway of HER2/EGFR. In the graph, the HER2/EGFR transduction pathway is outlined. Black arrows and red lines indicate positive and negative regulation, respectively. Within pink boxes, monoclonal antibodies and small molecule tyrosine kinase inhibitors used in the clinics to treat patients with aberrant activation of the HER2/EGFR pathway are shown. A yellow star marks genes that have been identified as novel lapatinib/erlotinib resistance genes in our screening.
An advantage of insertional mutagenesis over other functional studies, such as shRNA and siRNA screens, is that the genome wide distribution of LVs and other mutagens allow them to hit even noncanonical or nonannotated genes, which are not usually included in library-based approaches.7 Intriguingly, we identified not only protein-coding genes as culprits of drug resistance, but also microRNAs (miRNAs) and long noncoding RNAs (Figure 2c; Supplementary Figure S5c and Supplementary Table S4). For example, we found that the gene encoding for miR-181a1 is a CIS in BT474 cells. Notably, miR-181a1 was previously linked to drug resistance in HER2+ breast cancers, and corresponding anti-miRs are under development for therapeutic purposes.48
In summary, by LV-based insertional mutagenesis in breast and pancreatic cancer cell lines, we identified 65 candidate drug resistance genes whose deregulation confers resistance to lapatinib or erlotinib, respectively. Among them, we identified previously known anticancer drug resistance genes that validated our approach and new candidate drug resistance genes that include protein-coding and noncoding RNAs. Importantly, we found that our mutagenesis platform allowed the identification of lapatinib resistance genes whose mutation or expression impact the survival of breast cancer patients, thus indicating that our approach discovered clinically relevant genes.
Given the wide cell tropism of LVs, we envision, as future prospect, an expansion of this insertional mutagenesis screenings to a variety of primary and metastatic tumors, and also in the in vivo setting. Besides LVs that offer some advantages, other insertional mutagens could be exploited for this type of study, especially γ- or α-retroviruses that, with their different integration site preferences could provide complementary results for drug resistance genes identification. Integration of “omics” and insertional mutagenesis data could unravel novel interactions between the pre-existing mutational landscape of tumor cells and specific genes whose alteration is required to establish resistance to a given anticancer drug. These data may help designing new drug combinations that target both the mutated/amplified oncogenes and the drug resistance genes, potentially leading to sustained therapeutic responses.
Materials and Methods
Cell lines and chemicals. The BT474 and SKBR3 human breast cancer cell lines and the HPAC pancreatic adenocarcinoma cell line were purchased from the American Type Culture Collection (ATCC). They were cultured in RPMI-1640 medium supplemented with fetal bovine serum (10%), glutamine, and antibiotics (penicillin/streptomycin) at 37 °C with 5% CO2 in a humidified incubator. Lapatinib and erlotinib were purchased from Sequoia Biosciences (St Louis, MO) and were dissolved in DMSO to obtain 200 mmol/l stock solutions, which were then diluted 1:100 in phosphate buffer to obtain working solutions to be added to the cell media at different concentrations.
Vector production. The transfer plasmid for the production of LV.SF.LTR was cloned as follows. The GFP.PRE cassette was eliminated from the LV.SF.LTR.GFP.PRE plasmid18 by removing a 1269 bp KpnI-AgeI fragment encoding GFP.PRE. Then, DNA ends were blunted and intramolecular religation was performed. The transfer plasmids for the production of validation vectors were cloned as follows. The GFP.PRE cassette was eliminated from the backbone of SIN.LV.SF.GFP.PRE plasmid18 by removing a 736 bp AgeI-SalI fragment encoding GFP.PRE. The K111N-mutant and wild-type PIK3CA cDNAs were obtained by PCR (with DNA from BT474 and SKBR3 cells, respectively) using oligos that added at the two ends of the ORF the required restriction enzyme target sequences. The PIK3CB cDNA was obtained by PCR with DNA from SKBR3 cells. Then, PCR products were AgeI-SalI double-digested and ligated within the backbone construct. We produced concentrated LV stocks, pseudotyped with the VSV-G envelope, by transient co-transfection of four plasmids in 293T cells and titering on 293T cells as described.49
Selection protocol. 107 BT474 and SKBR3 cells were transduced with LV.SF.LTR in a 150 mm diameter Petri dish (volume of infection 20 ml, polybrene 8 μg/ml) at different vector doses (MOI: 0.5, 5, and 50 for BT474 cells and MOI: 1, 10, and 100 for SKBR3 cells). Three biological replicates of infection were performed. Two weeks after transduction, after the collection of a cell pellet for DNA extraction (“PRE” samples), 107 transduced and untransduced cells were seeded in 150 mm diameter Petri dishes. The following day, lapatinib selection was started by supplementing growth medium with different concentrations of lapatinib (0.5, 1, or 2 μmol/l) or vehicle only (“DMSO-only”) and renewing the treatment two to three times a week for 30 days (SKBR3 cells) or 60 days (BT474 cells). DMSO-only controls were kept in culture for the same period of time, and passaged with low passage ratios (1:3) upon reaching confluence. At the end of selection protocol, a cell pellet was collected from the whole dish where resistant clones have emerged upon lapatinib treatment, and from DMSO-only controls. To evaluate the development of lapatinib resistance, two counts were performed at the end of selection: total cell count and colony count. Total count of viable cells was performed with the aid of both Bürker chamber and the Countess automated cell counter (Life Technologies, Paisley, UK) upon Trypan Blue staining. Colony count was performed with the microscope at ×5 magnification: clones were counted on 10 nonconsecutive 4 cm2 areas across each plate (overall corresponding to around one fourth of the total surface), and these data were used to estimate the total number of colonies within the whole plate.
For erlotinib selection, 4 × 106 HPAC cells were initially transduced at MOI 75 with LV.SF.LTR in a 100 mm diameter Petri dish (volume of infection 10 ml, polybrene 8 µg/ml). Three biological replicates of infection were performed. Two weeks after transduction, after the collection of a cell pellet for DNA extraction, 4 × 106 transduced and untransduced cells were seeded in 100 mm diameter Petri dishes. The following day, erlotinib selection was started by supplementing growth medium with different concentrations of erlotinib (50 or 100 μmol/l) or vehicle only (“DMSO-only”) and renewing the treatment two to three times a week for 60 days. Samples at the end of the selection were collected as described above.
Vector copy number (VCN) analysis. Genomic DNA was extracted in a PCR-dedicated room from frozen cell pellets using the Qiagen blood and cell culture DNA Kits (Qiagen, Germantown, MD). Q-PCR analysis was performed as described50 with probes complementary to human genomic telomerase and a common LV sequence in the ψ-signal region. VCN was determined as the ratio between the relative amounts of LV versus total DNA (number of diploid genomes) evaluated by telomerase. A standard curve was made using dilutions from a clone of the cell line CEM (human T-cell lymphoblastic-like cell line) with a known LV VCN. Reactions were carried out according to manufacturer's instructions and analyzed using the ABI Prism 7900 HT Sequence Detection System (Applera, Life Technologies). Sequences of primers and probes are available upon request.
LAM-PCR procedure. LAM-PCR was performed on all drug-selected samples, DMSO-treated cells and PRE-selection samples as described.51 A clone of the cell line CEM (human T-cell lymphoblastic-like cell line) whose integrations are known was used as positive control for the reaction. Briefly, 200 ng was used as template for LAM-PCR for every sample, irrespective of the integration load. LAM-PCR was initiated with two rounds of 50-cycle linear PCR and overnight magnetic beads capture. Then, after second-strand synthesis, restriction digestions using the enzymes Tsp509 I or HpyCH4 IV were performed. Following steps included enzyme-specific linker-cassette ligation, denaturation and nested exponential PCR. LAM-PCR primers for LV were previously described.51 LAM-PCR amplicons were separated on Spreadex gels (Elchrom Scientific, Cham, Switzerland) to evaluate PCR efficiency and the bands pattern for each sample. Products of the second exponential amplification were tagged with barcoded primers specific for the Illumina platform, and were then pooled for MiSeq sequencing. See Supplementary Figure S2a for a general outline of the procedure.
Bioinformatics analysis of vector integrations. The output of MiSeq runs was analyzed by a bioinformatic pipeline developed within our laboratory, based on the BWA. CIS were defined according to the statistical definition of CIS developed by other studies19 and additionally we applied a stringent statistical cut-off that accounts for the increased size of the integration datasets in order to avoid false-positive CIS. In detail, a frequency distribution was built by ranking all the genomic regions recurrently targeted at least two times in 30 Kb, three times in 50 Kb, four times in 100 Kb, and five times in 200 Kb, according to their CIS power (number of integrations within the fixed genomic region). Then, the lower 75% of recurrently targeted regions was discarded and the upper quartile was considered to be composed of bona fide CIS. This was done separately for the lapatinib selected dataset and the control dataset, composed of DMSO-only and PRE samples. The stringency of these criteria is comparable to different approaches used in previous studies that analyzed a similar number of integrations. The closest gene to each CIS integration was annotated as a CIS-associated gene, and the most prevalent gene within each CIS is indicated in Table 1 and Supplementary Table S4. To filter the CIS generated by vector integration biases or those conferring an advantage independently from lapatinib selective pressure, we removed the CIS from the lapatinib-selected dataset with a matching CIS in the control dataset (DMSO-only and PRE datasets).
Validation experiments. We transduced 106 SKBR3 and BT474 cells with SIN LV vectors encoding PIK3CB, K111N-mutated PIK3CA, and wild-type PIK3CA, in a 100 mm diameter Petri dish (volume of infection 10 ml, polybrene 8 μg/ml) at two vector doses (MOI: 1 and MOI: 10). Then, for “long-term” validation experiments (1–2 weeks) we plated 2.5 × 105 transduced and untransduced cells in six-well plates and supplemented them with 0.5 or 1 μmol/l lapatinib for 2 weeks and 0.25 or 0.5 μmol/l lapatinib for 1 week, for SKBR3 and BT474 cells respectively. Three biological replicates of treatment were performed. At the end of treatment, total cells were counted with the Countess automated cell counter (Life Technologies) upon Trypan Blue staining. We then normalized the total number of cells at the end of treatment for the total number of cells plated at the beginning of the experiment, in order to plot the cell viability. For “short-term” experiments (24–72 hours), we plated 2 × 104 transduced and untransduced cells in 96-well plates and supplemented them with 1 μmol/l lapatinib for 24 and 72 hours. For each time point, three biological replicates of treatment were performed. At the end of treatment, cells were counted with the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI) protocol, in which the quantity of formazan product (as measured by the absorbance at 490 nm) is directly proportional to the number of living cells in culture. Briefly, at each time point 20 μl of the CellTiter reagent were added to each well of the assay and the plate was incubated at 37 °C for 2 hours before recording the absorbance at 490 nm using a 96-well plate reader (Victor X4; PerkinElmer, Waltham, MA). A time-zero reading was performed 6 hours after plating in order to normalize the absorbance levels at the end of treatment for the real starting absorbance. The ratio between the signal at the defined timepoint and the signal at 6 hours is indicated in the graphs and represent the cell viability.
The experiment described in Supplementary Figure S6d was performed as it follows. BT474 cells were transduced with the LV encoding for K111N-PIK3CA (MOI: 10) or GFP (MOI: 10) or mock-transduced and SKBR3 cells with two different doses on the LV encoding PIK3CB (MOI: 10 and 50) or mock-transduced. 200,000 cells were seeded in Multiwell 6 plates in biological triplicate. The day after seeding, the cells were treated with media containing 10, 40, and 160 nmol/l of lapatinib or matched-doses of vehicle (DMSO). Three days after the treatment, the media was replaced with new media containing the same dose of drug. At day 6, each cell population was counted. The survival was calculated as the ratio between each drug treated population and the average of the survival of the vehicle treated population. To design the survival curve, lapatinib nmol/l doses were converted in LOG10 scale and plotted with the above mentioned survival on the y-axis.
Overall survival and relapse-free survival analyses in breast cancer patients. For Kaplan–Meier plots, we used the Kaplan–Meier plotter (http://kmplot.com), an online tool capable to assess the effect of 22,277 genes on survival of 2,977 breast cancer patients.52 The analysis was restricted to HER2+ subtypes and excluded gene-biased arrays. For the survival analysis, we considered the expression level provided by the JetSet best probeset.53 Using the selected parameters, the analysis ran on 207 and 88 patients for RFS and OS, respectively. To analyze the prognostic value of PIK3CB overexpression, the patients were split into two groups according to PIK3CB expression selecting the “auto select best cutoff” option. The two patient cohorts were compared by a Kaplan–Meier survival plot and logrank P values was calculated.
Saturation and estimation of IS sampling. We can think at an amplicon library as a pool of individuals, grouped in species according to the different integration sites they represent. The sequencer draws amplicons from the library, without reintroduction, providing reads in 1 to 1 correspondence with the subsampled fraction of the library that experienced the sequencing process. To estimate if the sequencing efforts provide an exhaustive representation of the amplicon library and our integration site richness used the methods devised by Anne Chao et al. in “Sufficient Sampling for Asymptotic Minimum Species Richness Estimators” (2009, http://www.esajournals.org/doi/abs/10.1890/07-2147.1). The first step is the introduction of a non-parametric estimator for the number of classes in a population (A Chao – “Nonparametric Estimation of the Number of Classes in a Population” – 1984), referred as Chao1:
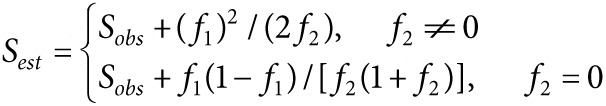 |
Here Sest and Sobs are the number of different species estimated for the environment and observed in the sample respectively while fr is the count of species represented by r individuals in the sample.
Under the hypotheses that the sample is large, the sampling is unbiased and the species have the same “catchability”, it should be considered as a universal law, valid under all types of species abundance distribution. We exploited this formula to estimate the total number of ISs in the amplicon library and thus the percentage of ISs detected by sequencing.
The second step is the derivation of a relationship between the increasing of sampling efforts and the additional species richness achieved in the sample, which comes from the estimated relative abundance (or discovery probability) of any species in the frequency class r, given by IJ Good as
where n is the number of individuals in the original sample.
As a consequence of the special case r = 0 (relative abundance of/probability to encountering each of the undetected species), the total relative abundance of the undetected species can be estimated by:
A probabilistic approach then lead to an estimate for m, the additional number of individuals to be taken required to reach the asymptotic richness:
where x* is the solution of the folowing equation:
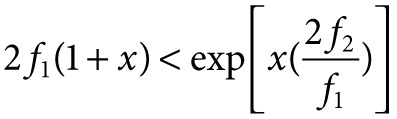 |
which always exists unique for x > 0.
To be clearer, it is expected that a sample of size n + m contains all species.
Conversely, letting g to be the desired percentage of Sest that someone may like to observe (gSest = Sobs desired), the required size of the sample can be estimated as n + mg, where
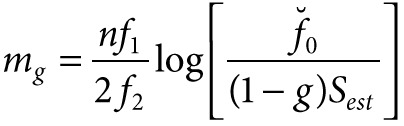 |
Exploiting (1), (2), and (3) on our data, we produced the following the table of results:
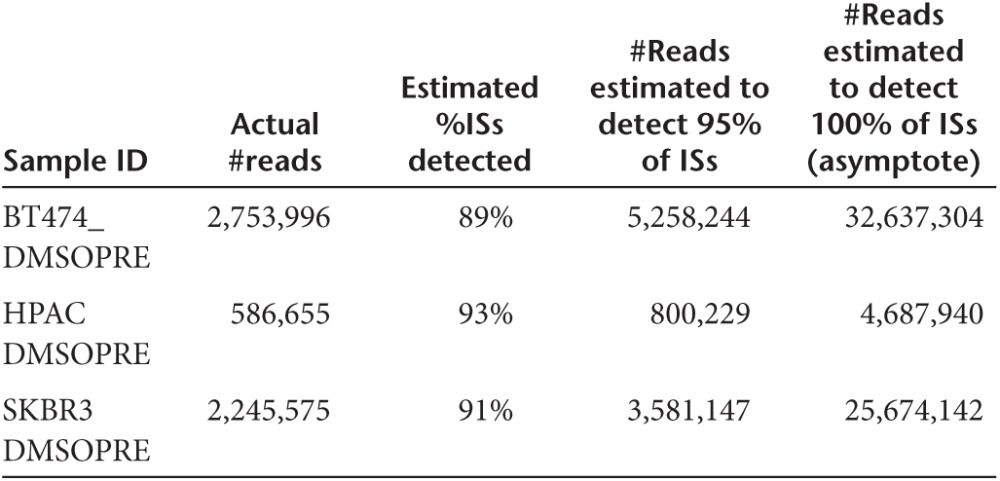 |
Roughly speaking, a double depth would be required to switch from 89–93% to 95% of ISs detected in each sample and over 10-fold to reach the asymptote (100% of ISs detected).
For the purpose of our work, we consider that the observation of 89–93% of ISs per sample is sufficient, not significantly different from 95% and that an increase of sequencing effort will not have a significant impact on the results presented in the manuscript.
SUPPLEMENTARY MATERIAL Figure S1. LV-based insertional mutagenesis induces resistance to different doses of lapatinib. Figure S2. LAM-PCR for the retrieval of LV integration sites. Figure S3. Genome-wide distribution of LV integrations in the different experimental datasets compared to gene density. Figure S4. Integrations from control samples are enriched in chromosomal amplified regions and clustered at genes that may induce drug-independent proliferative advantage. Figure S5. Representative CIS identified exclusively in lapatinib-selected cells. Figure S6. Overexpression of K111N-mutated PIK3CA and wild type PIK3CB promote the survival of breast cancer cell lines. Figure S7. LV-based insertional mutagenesis induces erlotinib resistance in a pancreatic cell line and allows the identification of erlotinib resistance genes. Table S1. DNA samples that have undergone LV integration analysis. Table S2. CIS genes identified in control samples. Table S3. CIS genes identified in lapatinib-selected samples before filtering the control CIS. Table S4. CIS genes identified in lapatinib-selected cells and filtered for the CIS identified in control samples. Table S5. Percentage of integration within CIS. Table S6. Details of the integrations falling within CIS.
Acknowledgments
We thank Daniela Cesana, Monica Volpin, Clara Alsinet Armengol, and Chi Wong for critical discussion of data, and Massimiliano Marini, Erika Tenderini, and Giulio Spinozzi for help with integration studies. This work was supported by grants from the Association for International Cancer Research (AICR 09-0784 to E.M.), Telethon Foundation (TGT11D1 to E.M.), European Union (Clinigene NoE LSHB-CT-2006–018933 to E.M. and PERSIST to L.N.), Italian Ministries of Health (GR-2007–684057 to E.M. and ONC-34/07 to L.N.), Associazione Italiana per la Ricerca sul Cancro (AIRC) 5x1000 (AIRC 5x1000 to L.N.). Funding for open access charge: Telethon Foundation. M.R. and S.A. designed and performed experiments and wrote the manuscript. A.C., F.B., and S.B. performed vector integration site and statistical analyses. P.G. performed experiments. L.N. provided intellectual input and edited the manuscript. E.M. revised the manuscript and supervised the project. The authors have declared that no conflict of interest exists.
Supplementary Material
References
- Stratton MR. Exploring the genomes of cancer cells: progress and promise. Science. 2011;331:1553–1558. doi: 10.1126/science.1204040. [DOI] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- Lieu CH, Tan AC, Leong S, Diamond JR, Eckhardt SG. From bench to bedside: lessons learned in translating preclinical studies in cancer drug development. J Natl Cancer Inst. 2013;105:1441–1456. doi: 10.1093/jnci/djt209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Jänne PA. Circumventing cancer drug resistance in the era of personalized medicine. Cancer Discov. 2012;2:214–226. doi: 10.1158/2159-8290.CD-12-0012. [DOI] [PubMed] [Google Scholar]
- Kool J, Berns A. High-throughput insertional mutagenesis screens in mice to identify oncogenic networks. Nat Rev Cancer. 2009;9:389–399. doi: 10.1038/nrc2647. [DOI] [PubMed] [Google Scholar]
- Ranzani M, Annunziato S, Adams DJ, Montini E. Cancer gene discovery: exploiting insertional mutagenesis. Mol Cancer Res. 2013;11:1141–1158. doi: 10.1158/1541-7786.MCR-13-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranzani M, Cesana D, Bartholomae CC, Sanvito F, Pala M, Benedicenti F, et al. Lentiviral vector-based insertional mutagenesis identifies genes associated with liver cancer. Nat Methods. 2013;10:155–161. doi: 10.1038/nmeth.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blömer U, Gallay P, Ory D, Mulligan R, Gage FH, et al. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- Cesana D, Ranzani M, Volpin M, Bartholomae C, Duros C, Artus A, et al. Uncovering and dissecting the genotoxicity of self-inactivating lentiviral vectors in vivo. Mol Ther. 2014;22:774–785. doi: 10.1038/mt.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R, DeSantis C, Virgo K, Stein K, Mariotto A, Smith T, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62:220–241. doi: 10.3322/caac.21149. [DOI] [PubMed] [Google Scholar]
- Lin SX, Chen J, Mazumdar M, Poirier D, Wang C, Azzi A, et al. Molecular therapy of breast cancer: progress and future directions. Nat Rev Endocrinol. 2010;6:485–493. doi: 10.1038/nrendo.2010.92. [DOI] [PubMed] [Google Scholar]
- Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2012;9:16–32. doi: 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3:269–280. doi: 10.1038/ncponc0509. [DOI] [PubMed] [Google Scholar]
- Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733–2743. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- Lacroix M, Leclercq G. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res Treat. 2004;83:249–289. doi: 10.1023/B:BREA.0000014042.54925.cc. [DOI] [PubMed] [Google Scholar]
- Hegde PS, Rusnak D, Bertiaux M, Alligood K, Strum J, Gagnon R, et al. Delineation of molecular mechanisms of sensitivity to lapatinib in breast cancer cell lines using global gene expression profiles. Mol Cancer Ther. 2007;6:1629–1640. doi: 10.1158/1535-7163.MCT-05-0399. [DOI] [PubMed] [Google Scholar]
- Montini E, Cesana D, Schmidt M, Sanvito F, Bartholomae CC, Ranzani M, et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Invest. 2009;119:964–975. doi: 10.1172/JCI37630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel U, Deichmann A, Bartholomae C, Schwarzwaelder K, Glimm H, Howe S, et al. Real-time definition of non-randomness in the distribution of genomic events. PLoS One. 2007;2:e570. doi: 10.1371/journal.pone.0000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu G, Dziubinski M, Yang Z, Ethier SP, Wu G. Comprehensive analysis of oncogenic effects of PIK3CA mutations in human mammary epithelial cells. Breast Cancer Res Treat. 2008;112:217–227. doi: 10.1007/s10549-007-9847-6. [DOI] [PubMed] [Google Scholar]
- Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, et al. Oslo Breast Cancer Consortium (OSBREAC) The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cizkova M, Dujaric ME, Lehmann-Che J, Scott V, Tembo O, Asselain B, et al. Outcome impact of PIK3CA mutations in HER2-positive breast cancer patients treated with trastuzumab. Br J Cancer. 2013;108:1807–1809. doi: 10.1038/bjc.2013.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497:108–112. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhang Q, Zhang J, Sun S, Guo H, Jia Z, et al. PI3K pathway activation results in low efficacy of both trastuzumab and lapatinib. BMC Cancer. 2011;11:248. doi: 10.1186/1471-2407-11-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. National Cancer Institute of Canada Clinical Trials Group Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- Wong HH, Lemoine NR. Pancreatic cancer: molecular pathogenesis and new therapeutic targets. Nat Rev Gastroenterol Hepatol. 2009;6:412–422. doi: 10.1038/nrgastro.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Agthoven T, Veldscholte J, Smid M, van Agthoven TL, Vreede L, Broertjes M, et al. Functional identification of genes causing estrogen independence of human breast cancer cells. Breast Cancer Res Treat. 2009;114:23–30. doi: 10.1007/s10549-008-9969-5. [DOI] [PubMed] [Google Scholar]
- Dorssers LC, van Agthoven T, Dekker A, van Agthoven TL, Kok EM. Induction of antiestrogen resistance in human breast cancer cells by random insertional mutagenesis using defective retroviruses: identification of bcar-1, a common integration site. Mol Endocrinol. 1993;7:870–878. doi: 10.1210/mend.7.7.8413311. [DOI] [PubMed] [Google Scholar]
- Chen L, Stuart L, Ohsumi TK, Burgess S, Varshney GK, Dastur A, et al. Transposon activation mutagenesis as a screening tool for identifying resistance to cancer therapeutics. BMC Cancer. 2013;13:93. doi: 10.1186/1471-2407-13-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- Jönsson G, Staaf J, Olsson E, Heidenblad M, Vallon-Christersson J, Osoegawa K, et al. High-resolution genomic profiles of breast cancer cell lines assessed by tiling BAC array comparative genomic hybridization. Genes Chromosomes Cancer. 2007;46:543–558. doi: 10.1002/gcc.20438. [DOI] [PubMed] [Google Scholar]
- Hanker AB, Pfefferle AD, Balko JM, Kuba MG, Young CD, Sánchez V, et al. Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors and induces resistance to combinations of anti-HER2 therapies. Proc Natl Acad Sci USA. 2013;110:14372–14377. doi: 10.1073/pnas.1303204110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–779. doi: 10.1038/nature07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J, Liu Q, Xie S, Carlson C, Von T, Vogel K, et al. Functional characterization of an isoform-selective inhibitor of PI3K-p110β as a potential anticancer agent. Cancer Discov. 2012;2:425–433. doi: 10.1158/2159-8290.CD-12-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho S, Milanezi F, Costa JL, Amendoeira I, Schmitt F. PIKing the right isoform: the emergent role of the p110beta subunit in breast cancer. Virchows Arch. 2010;456:235–243. doi: 10.1007/s00428-010-0881-0. [DOI] [PubMed] [Google Scholar]
- Dbouk HA, Backer JM. A beta version of life: p110β takes center stage. Oncotarget. 2010;1:729–733. doi: 10.18632/oncotarget.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien NA, Browne BC, Chow L, Wang Y, Ginther C, Arboleda J, et al. Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol Cancer Ther. 2010;9:1489–1502. doi: 10.1158/1535-7163.MCT-09-1171. [DOI] [PubMed] [Google Scholar]
- Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, et al. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci USA. 2008;105:13057–13062. doi: 10.1073/pnas.0802655105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Yong HM, Chen FF, Mei PJ, Liu H, Li C, et al. Cullin1 is a novel marker of poor prognosis and a potential therapeutic target in human breast cancer. Ann Oncol. 2013;24:2016–2022. doi: 10.1093/annonc/mdt147. [DOI] [PubMed] [Google Scholar]
- Hoxhaj G, Dissanayake K, MacKintosh C. Effect of IRS4 levels on PI 3-kinase signalling. PLoS One. 2013;8:e73327. doi: 10.1371/journal.pone.0073327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115–125. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliabadi HM, Maranchuk R, Kucharski C, Mahdipoor P, Hugh J, Uludağ H. Effective response of doxorubicin-sensitive and -resistant breast cancer cells to combinational siRNA therapy. J Control Release. 2013;172:219–228. doi: 10.1016/j.jconrel.2013.08.012. [DOI] [PubMed] [Google Scholar]
- Janknecht R. Regulation of the ER81 transcription factor and its coactivators by mitogen- and stress-activated protein kinase 1 (MSK1) Oncogene. 2003;22:746–755. doi: 10.1038/sj.onc.1206185. [DOI] [PubMed] [Google Scholar]
- Shepherd TG, Kockeritz L, Szrajber MR, Muller WJ, Hassell JA. The pea3 subfamily ets genes are required for HER2/Neu-mediated mammary oncogenesis. Curr Biol. 2001;11:1739–1748. doi: 10.1016/s0960-9822(01)00536-x. [DOI] [PubMed] [Google Scholar]
- Goel A, Janknecht R. Concerted activation of ETS protein ER81 by p160 coactivators, the acetyltransferase p300 and the receptor tyrosine kinase HER2/Neu. J Biol Chem. 2004;279:14909–14916. doi: 10.1074/jbc.M400036200. [DOI] [PubMed] [Google Scholar]
- Miller TE, Ghoshal K, Ramaswamy B, Roy S, Datta J, Shapiro CL, et al. MicroRNA-221/222 confers tamoxifen resistance in breast cancer by targeting p27Kip1. J Biol Chem. 2008;283:29897–29903. doi: 10.1074/jbc.M804612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follenzi A, Ailles LE, Bakovic S, Geuna M, Naldini L. Gene transfer by lentiviral vectors is limited by nuclear translocation and rescued by HIV-1 pol sequences. Nat Genet. 2000;25:217–222. doi: 10.1038/76095. [DOI] [PubMed] [Google Scholar]
- Montini E, Cesana D, Schmidt M, Sanvito F, Ponzoni M, Bartholomae C, et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol. 2006;24:687–696. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Schwarzwaelder K, Bartholomae C, Zaoui K, Ball C, Pilz I, et al. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR) Nat Methods. 2007;4:1051–1057. doi: 10.1038/nmeth1103. [DOI] [PubMed] [Google Scholar]
- Györffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123:725–731. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- Li Q, Birkbak NJ, Gyorffy B, Szallasi Z, Eklund AC. Jetset: selecting the optimal microarray probe set to represent a gene. BMC Bioinformatics. 2011;12:474. doi: 10.1186/1471-2105-12-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.