

Abstract
Block of human ether-à-go-go–related gene 1 (hERG1) K+ channels by many drugs delays cardiac repolarization, prolongs QT interval, and is associated with an increased risk of cardiac arrhythmia. Preferential block of hERG1 channels in an inactivated state has been assumed because inactivation deficient mutant channels can exhibit dramatically reduced drug sensitivity. Here we reexamine the link between inactivation gating and potency of channel block using concatenated hERG1 tetramers containing a variable number (0–4) of subunits harboring a point mutation (S620T or S631A) that disrupts inactivation. Concatenated hERG1 tetramers containing four wild-type subunits exhibited high-affinity block by cisapride, dofetilide, and MK-499, similar to wild-type channels formed from hERG1 monomers. A single S620T subunit within a tetramer was sufficient to fully disrupt inactivation gating, whereas S631A suppressed inactivation as a graded function of the number of mutant subunits present in a concatenated tetramer. Drug potency was positively correlated to the number of S620T subunits contained within a tetramer but unrelated to mutation-induced disruption of channel inactivation. Introduction of a second point mutation (Y652W) into S620T hERG1 partially rescued drug sensitivity. The potency of cisapride was not altered for tetramers containing 0 to 3 S631A subunits, whereas the potency of dofetilide was a graded function of the number of S631A subunits contained within a tetramer. Together these findings indicate that S620T or S631A substitutions can allosterically disrupt drug binding by a mechanism that is independent of their effects on inactivation gating.
Introduction
Human ether-à-go-go–related gene 1 (hERG1) subunits coassemble to form a voltage-gated K+ channel that conducts the rapid delayed rectifier K+ current (Sanguinetti et al., 1995; Trudeau et al., 1995). Mutations in the hERG1 gene KCNH2 are a major cause of congenital long QT syndrome (Curran et al., 1995). Loss of function mutations in KCNH2 reduce rapid delayed rectifier K+ current, delay ventricular repolarization, lengthen the QT interval of the body surface electrocardiogram, and increase the risk of lethal cardiac arrhythmia (Keating and Sanguinetti, 2001). Prolongation of the action potential of cardiomyocytes can also be caused by block of hERG1 channels as an unintended side effect of many medications, and such activity is a major safety concern in drug development (Fenichel et al., 2004). Elucidation of the molecular mechanisms responsible for high-affinity block of hERG1 channels has facilitated efforts to avoid drug-induced arrhythmia.
The structural basis of the high-affinity ligand binding site for potent hERG1 blockers was revealed by a site-directed mutagenesis approach. Two aromatic residues (Y652, F656) located in each of the four S6 segments that line the central cavity of the hERG1 channel are critical for interactions with a variety of structurally diverse compounds (Lees-Miller et al., 2000; Mitcheson et al., 2000; Fernandez et al., 2004). These two aromatic residues are conserved in hERG1 and ether-à-go-go (eag) channel subunits, but noninactivating eag channels are much less sensitive to most hERG1 blockers (Ficker et al., 1998). In addition, most inactivation-deficient hERG1 mutant channels (e.g., S620T, S631A, G628C/S631C) exhibit significantly reduced sensitivity to high-affinity drugs (Suessbrich et al., 1997; Wang, et al. 1997a; Ficker et al., 1998; Numaguchi et al., 2000; Perrin et al., 2008). Together these findings strongly suggest that high-affinity blockers preferentially bind to the inactivated state of hERG1 channels. Contrary to this hypothesis was the observation that the IC50 for block of wild-type and inactivation-deficient mutant channels is similar for some low-potency compounds such as quinidine (Lees-Miller et al., 2000; Perrin et al., 2008), erythromycin, and perhexilin (Perrin et al., 2008). Moreover, drug potency of high-affinity ligands is not always proportionate to the extent of channel inactivation. For example, G628C/S631C (GCSC) mutant channels do not inactivate at all, yet these channels have a significantly higher drug sensitivity to methanesulfonanilides (e.g., MK-499 [N-[(4R)-1′-[(2R)-6-cyano-1,2,3,4-tetrahydronaphthalen-2-yl]-4-hydroxyspiro[3,4-dihydrochromene-2,4′-piperidine]-6-yl]methanesulfonamide] and E-4031 [N-[4-[1-[2-(6-methylpyridin-2-yl)ethyl]piperidine-4-carbonyl]phenyl]methanesulfonamide]) than other inactivation-deficient mutant channels (e.g., S620T or S631A hERG1) (Wang, et al., 1997b; Mitcheson et al., 2000). Finally, point mutations that enhance inactivation (e.g., G648A, T623A, and F656A) were found to exhibit reduced sensitivity to block by MK-499 (Mitcheson et al., 2000). How inactivation gating of hERG1 modulates block of the channel by drugs is not well understood, but presumably the subtle conformational change in the selectivity filter that mediates inactivation of hERG1 (Stansfeld et al., 2008) is allosterically coupled to movement of key residues (Tyr652 or Phe656) in the S6 segments that interact with a drug located within the central cavity of the channel. There is little evidence for this hypothesis other than the finding that repositioning (by site-directed mutagenesis) of aromatic residues in the S6 of Drosophila eag introduced high-affinity block by cisapride in the normally insensitive channel (Chen et al., 2002).
We recently characterized the biophysical properties of concatenated hERG1 tetramers containing a variable number of wild-type and S620T or S631A mutant subunits with defined stoichiometry and positioning (Wu et al., 2014a). A single S620T subunit disrupted gating of the tetramer to the same extent as that observed for S620T homotetramers, indicating that hERG1 inactivation is mediated by an all-or-none cooperative interaction between all four subunits. In contrast, suppression of inactivation by S631A subunits was a graded function of the number of S631A subunits present in a concatenated tetramer. Here, we ask whether disruption of inactivation gating by the presence of 1–4 S620T or S631A subunits in a concatenated hERG1 tetramer is associated with a reduction in the potency for drug block as would be expected if there was a 1:1 correlation between inactivation and drug block. We also explored the relationship between channel inactivation and drug response with channels formed by coassembly of S620T monomers that contained an additional mutation of specific residues within the S6 segment that were previously shown to be critical for drug block. Our findings provide evidence for a new interpretation of how mutation-induced disruption of inactivation can reduce the potency of high-affinity hERG1 channel blockers.
Materials and Methods
Molecular Biology.
hERG1 cDNA (KCNH2, isoform 1a, NCBI Reference Sequence NM_000238) was cloned into the pSP64 oocyte expression vector, and point mutations were introduced using the QuickChange mutagenesis kit (Agilent Technologies, Santa Clara, CA). Concatenated hERG1 tetramers containing a variable number of S620T or S631A subunits were constructed as previously described (Wu et al., 2014a). Concatenated tetramers formed by four wild-type (WT), S620T (ST), or S631A (SA) mutant subunits are designated as WT4, ST4, and SA4, respectively. Heterotypic tetramers were named to indicate the relative positioning of the WT, ST, or SA mutant subunits. For example, the ST1/WT1/ST2 concatemer contained a WT subunit in the second position and S620T subunits in the first, third, and fourth position of the tetramer. All constructs were confirmed by DNA sequencing. cRNA encoding hERG1 monomers or concatenated tetramers were prepared by linearization of the plasmid with EcoRI and in vitro transcription using the mMessage mMachine SP6 kit (Ambion, Austin, TX).
Isolation and Two-Electrode Voltage-Clamp of Oocytes.
Oocytes were enzymatically isolated from ovarian lobes excised from Xenopus laevis using procedures as described previously (Abbruzzese et al., 2010) and approved by the University of Utah Institutional Animal Care and Use Committee. Single oocytes were injected with 5–30 ng of hERG1 cRNA and incubated for 1–4 days before use in voltage-clamp experiments.
Whole cell hERG1 currents were recorded using agarose-cushion microelectrodes and standard two-electrode voltage-clamp techniques (Goldin, 1991; Stühmer, 1992). Oocytes were voltage clamped to a holding potential of −80 mV, and 4-second pulses to a variable test potential (Vt), usually 0 or −30 mV, were applied once every 20 seconds. For some channel types, a current-voltage (Itest–Vt) relationship was determined by applying 4-second pulses to a Vt that ranged from −70 to +40 mV applied in 10-mV increments. Peak outward currents at each test potential (Itest) were measured, normalized to the largest current measured (at +40 mV), and plotted as a function of Vt. A Dell personal computer (Round Rock, TX), GeneClamp 500 amplifier (Molecular Devices, Inc., Sunnyvale, CA), Digidata 1322A data acquisition system (Molecular Devices, Inc.), and pCLAMP (ver. 8.2) software (Molecular Devices, Inc.) were used for data acquisition. pCLAMP and ORIGIN (ver. 8.6) software (OriginLab, Northhampton, MA) were used for offline analysis of digitized data.
Solutions and Drugs.
For two-electrode voltage clamp experiments, the extracellular solution contained (in mM): 96 NaCl, 2 KCl, 1 CaCl2, 5 HEPES, and 1 MgCl2, pH 7.6. Cisapride and dofetilide were purchased from Tocris Bioscience (Bristol, UK), and MK-499 was generously supplied by Merck & Co. (West Point, PA). All compounds were dissolved in dimethyl sulfoxide to make 10 mM stock solutions. Drug solutions at concentrations of 0.03–100 µM were made by diluting stock solutions with extracellular solution just before use in each experiment.
Data Analysis.
Relative block of hERG1 channels was calculated as (Icontrol − Idrug)/Icontrol, where Icontrol and Idrug are the magnitudes of current measured at the end of a 4-second depolarizing pulse under control conditions and after steady-state block in the presence of drug. Normalized current magnitude was plotted as a function of [drug] and the data fitted with the logistic equation:
where [D] is the concentration of drug, IC50 is the concentration of drug that caused a half-maximal inhibition, and nH is the Hill coefficient.
The voltage dependence of inactivation was estimated using a two-pulse protocol. Current was first activated by a 2-second pulse to +40 (or +80) mV, then a tail current (Itail) was elicited with a 1.5-second pulse to a variable return potential (Vret) that was applied in 10-mV increments ranging from +30 (or +70) to −140 mV. Normalized conductance (g/gmax) was estimated by calculating Itail/(Vret − Erev), where Erev is the reversal potential of Itail. The resulting relationship was fitted with a Boltzmann function:
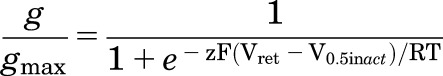 |
where V0.5inact is the half-point for inactivation, z is the equivalent charge, F is Faraday’s constant, R is the universal gas constant, and T is the absolute temperature. For some experiments, plots of IC50 versus g/gmax, or the number of mutant subunits/tetramer, were analyzed by linear regression, and the adjusted R2 value was used to estimate the degree of relationship between the two variables.
Data are presented as mean ± S.E.M. (n = number of individual oocytes). Where appropriate, data were analyzed with a one-way or two-way analysis of variance followed by a Tukey's multiple comparison test. A P < 0.05 was considered significant.
Results
Effects of Subunit Concatenation on the Potency of hERG1 Channel Blockers.
Concatenation of subunits can affect the pharmacology of the resulting tetrameric channels (Wu et al., 2014b, 2015). Therefore, we compared the concentration-response relationships for cisapride, dofetilide, and MK-499 on the block of concatenated WT hERG1 (WT4) channels and channels formed naturally by coassembly of single WT subunits (WTmonomer). Cisapride and dofetilide inhibited the two channel types with equal potency (Fig. 1, A and B), whereas WT4 channels were 2.6 times less sensitive to MK-499 than WTmonomer channels (Fig. 1C).
Fig. 1.

Concentration-dependent block of WT hERG1 and concatenated tetrameric hERG1 channels expressed in Xenopus oocytes by cisapride, dofetilide, and MK-499. (A) Concentration-effect relationship for cisapride-mediated block of concatenated WT hERG1 tetramers (WT4) and WT hERG1 channels formed naturally by coassembly of monomers (WTmonomer). The IC50 values determined by fitting data to a logistic equation (smooth curve) were 188 ± 20 nM for WT4 channels (n = 5) and 174 ± 21 nM for WTmonomer channels (n = 4). P = 0.99, two-way analysis of variance (ANOVA). (B) Concentration-effect relationship for dofetilide-mediated block of WT4 channels (IC50 = 131 ± 21 nM, n = 5) and WTmonomer channels (IC50 = 150 ± 12 nM, n = 5). P = 0.23, two-way ANOVA. (C) Concentration-effect relationship for MK-499–mediated block of WT4 channels (IC50 = 167 ± 25 nM, n = 4) and WTmonomer channels (IC50 = 64 ± 9 nM, n = 4). P < 0.0001, two-way ANOVA. For all compounds, channel inhibition was quantified by measuring currents at the end of 4-second pulses applied once every 20 seconds to a Vt of 0 mV.
The Number of S620T Mutant Subunits Contained within a Concatenated Tetramer Determines Sensitivity of Channel to hERG1 Blockers.
We previously reported (Wu et al., 2014a) that the presence of a single S620T mutant subunit in a concatenated hERG1 tetramer (ST1/WT3 tetramer) attenuates inactivation to the same extent as concatemers containing four S620T subunits (ST4 tetramer). If the lack of inactivation gating was the main determinant of reduced potency of drug block of S620T hERG1 channels, then we would expect that tetrameric channels containing one or more S620T subunits would have a similarly reduced sensitivity to cisapride, dofetilide, and MK-499. To assess the potency of these compounds on concatenated channels, repetitive 4-second test pulses to 0 mV were applied and extent of current block was assessed when the block reached a steady-state level. Representative current traces for WT4, ST1/WT3, ST4, and STmonomer channels in the absence and presence of 0.3 µM cisapride are shown in Fig. 2A. In addition, ST1/WT3 channel currents exhibited a tonic block and a time-dependent block that developed during each 4-second test pulse. Current magnitude was measured at the end of the 4-second pulses. The [cisapride]-response relationships for the six different concatenated hERG1 channels and STmonomer channels were fitted with a logistic equation to estimate the IC50 values (Fig. 2B; n = 4–6). ST4 channels (IC50 = 7.33 ± 0.92 µM) and STmonomer channels (IC50 = 6.75 ± 1.0 µM) were 35-fold less sensitive to block by cisapride compared with WT4 channels (IC50 = 188 ± 21 nM), whereas ST1/WT3 channels (IC50 = 588 ± 71 nM) were 3.1-fold less sensitive to block compared with WT4 channels. The IC50 for block of currents by cisapride was positively correlated to the number (0 to 4) of S620T subunits contained in a tetrameric hERG1 channel (Fig. 2C). Inactivation of WT4 channels reduced g/gmax to a value of 0.09, whereas channels containing S620T subunits exhibit almost no inactivation at 0 mV and g/gmax > 0.95 (Fig. 2D). To quantify the relationship between inactivation and cisapride potency, g/gmax measured at 0 mV was plotted as a function of the IC50 values for cisapride, also determined at 0 mV (Fig. 2E). There was no correlation between inactivation of these hERG1 channels and cisapride potency. Together these data indicate that the potency of cisapride block of hERG1 is dependent on the number of S620T subunits and not the extent of channel inactivation.
Fig. 2.

Potency for cisapride block of hERG1 channels is dependent on the number of S620T subunits contained within a concatenated tetramer. (A) Representative current traces for WT4, ST1/WT3, ST4, and STmonomer hERG1 channels, recorded before and after treatment of oocytes with 0.3 µM cisapride. Channels were activated with a 4-second pulse to 0 mV, and tail currents were elicited at −70 mV. (B) [Cisapride]-response relationships for inhibition of Itest measured at 0 mV for indicated concatenated hERG1 channels (n = 4–6). Data were fitted with a logistic equation (smooth curves). (C) IC50 values plotted as a function of the number of S620T subunits contained within a concatenated tetramer. IC50 for ST1/WT1/ST1/WT1 channels is shown as hatched bar; IC50 for STmonomer channels is shown as crosshatched bar (at far right). P < 0.0001, one-way analysis of variance; *P < 0.001, **P < 0.0001 compared with ST4 channels. (D) Plot of g/gmax as a function of Vret. Curves represent previously published average data for the indicated concatenated tetramers (Wu et al., 2014a). Vertical dotted line was used to determine g/gmax at 0 mV for each channel type. (E) Plot of IC50 values versus g/gmax at 0 mV for channels as indicated by key (B).
The role of inactivation gating as a determinant of high-affinity hERG1 block was next evaluated with two methanesulfonanilides, dofetilide and MK-499. Representative traces of WT4, ST1/WT3, ST4, and STmonomer channel currents measured before and after the application of 0.3 µM dofetilide are illustrated in Fig. 3A. ST1/WT3 channels were much less sensitive to dofetilide than WT4 channels but still significantly more sensitive than ST4 and STmonomer channels. The [dofetilide]-response relationships for the six different concatenated hERG1 channels and STmonomer channels were fitted with a logistic equation to estimate the IC50 values (Fig. 3B). The IC50 values for dofetilide block of the four concatenated heterotetrameric channels were similar (∼5 µM), 30–40 times greater than the IC50 for WT4 channels (0.13 µM) and 3 or 5 times less than ST4 or STmonomer channels, respectively. The IC50 for block of currents by dofetilide is plotted as a function of the number (0 to 4) of S620T subunits contained in a tetramer (Fig. 3C) or relative rectification at 0 mV (Fig. 3D). The same experimental approach and methods of analysis used for cisapride and dofetilide were repeated for MK-499 (Fig. 4, A–D). There was no significant correlation between the number of S620T subunits incorporated in a tetramer and the IC50 for dofetilide or MK-499. High-affinity block of hERG1 channels by dofetilide and MK-499 was more dependent on inactivation than cisapride; however, for all drugs the IC50 for block was reduced for concatenated channels with one or more WT subunit.
Fig. 3.

Dofetilide block of concatenated hERG1 channels containing a variable number of S620T subunits. (A) Representative current traces for WT4, ST1/WT3, ST4, and STmonomer hERG1 channels, recorded before and after treatment of oocytes with 0.3 µM dofetilide. Channels were activated with 4-second pulse to 0 mV, and tail currents were elicited at −70 mV. (B) [Dofetilide]-response relationships for inhibition of Itest measured at 0 mV for indicated concatenated hERG1 channels (n = 4–5). Data were fitted with a logistic equation (smooth curves). (C) IC50 values plotted as a function of the number of S620T subunits contained within a concatenated tetramer. IC50 for ST1/WT1/ST1/WT1 channels is shown as hatched bar; IC50 for STmonomer channels is shown as crosshatched bar (at far right). P < 0.0001, one-way analysis of variance; *P < 0.05, **P < 0.01, #P < 0.001 compared with ST4 channels. (D) Plot of IC50 values versus g/gmax at 0 mV for channels as indicated by key in (B).
Fig. 4.

MK-499 block of concatenated hERG1 channels containing a variable number of S620T subunits. (A) Representative current traces of WT4, ST1/WT3, ST4, and STmonomer channels recorded before and after treatment of oocytes with 0.3 µM MK-499. Channels were activated with 4-second pulse to 0 mV, and tail currents were elicited at −70 mV. (B) [MK-499]–response relationships for inhibition of Itest measured at 0 mV for indicated concatenated hERG1 channels (n = 3–5). Data were fitted with a logistic equation (smooth curves). (C) IC50 values plotted as a function of the number of S620T subunits contained within a concatenated tetramer. IC50 for ST1/WT1/ST1/WT1 channels is shown as hatched bar; IC50 for STmonomer channels is shown as crosshatched bar (at far right). P < 0.0001, one-way analysis of variance; *P < 0.0001 compared with ST4 channels. (D) Plot of IC50 values versus g/gmax at 0 mV for channels as indicated by key in (B).
Response of Concatenated Channels Containing a Variable Number of S631A Subunits to hERG1 Blockers.
Ser631 is located just outside the selectivity filter of the hERG1 subunit. Mutation of this residue to Ala (S631A) shifts V0.5inact to more positive potentials (reduces inactivation) as a graded function of the number of S631A subunits present in a concatenated tetramer, consistent with highly cooperative, but not fully concerted (i.e., all-or-none), subunit interactions (Wu et al., 2014a). We tested the effects of cisapride and dofetilide on concatenated channels containing 1, 2, 3, or 4 S631A subunits. The IC50 for cisapride was similar for WT4 channels (188 ± 20 nM, n = 5) and channels containing 1, 2, or 3 S631A subunits/tetramer (Fig. 5, A and B). Only SA4 channels exhibited a reduced sensitivity to cisapride with an IC50 of 0.77 ± 0.2 µM (n = 5). We previously reported the voltage dependence of inactivation for tetramers containing 1–4 S631A subunits, and these relationships are presented in Fig. 5C. The dotted vertical line in this figure was used to estimate the g/gmax at 0 mV for the different concatenated tetramers. The relationship between g/gmax at 0 mV and the IC50 for cisapride, also determined at 0 mV, is plotted in Fig. 5D. Despite an increase in g/gmax from 0.09 for WT4 channels to 0.73 for SA1/WT1/SA2 channels, there was no change in the IC50 for cisapride. The [dofetilide]-response relationships for WT4 and the four different concatenated tetramers containing S631A subunits are plotted in Fig. 5E. The IC50 values determined from these plots were highly correlated (adjusted R2 = 0.95) with the number of S631A subunits (from 0 to 4) contained in a tetramer (Fig. 5F). As shown in Fig. 5G, the IC50 values for dofetilide block of tetramers containing 1 or more S631A were also highly correlated with g/gmax at 0 mV (adjusted R2 = 0.98); however, when WT4 channels were included in the analysis, the correlation was reduced (adjusted R2 = 0.76). Thus, the potency of dofetilide is more closely related to the number of S631A subunits contained within a concatenated tetramer than with the extent of channel inactivation.
Fig. 5.

Cisapride and dofetilide block of concatenated hERG1 channels containing a variable number of S631A subunits. (A) [Cisapride]-response relationships for inhibition of Itest measured at 0 mV for indicated concatenated hERG1 channels (n = 3–5). Data were fitted with a logistic equation (smooth curves). (B) IC50 values for cisapride plotted as a function of the number of S631A subunits contained within a concatenated tetramer. (C) Plot of g/gmax as a function of Vret. Curves represent previously published data for the indicated concatenated tetramers (Wu et al., 2014a). Vertical dotted line was used to determine g/gmax at 0 mV for each channel type. (D) Plot of IC50 values versus g/gmax at 0 mV for indicated channels. Symbols are the same as indicated in (A). (E) [Dofetilide]-response relationships for inhibition of Itest measured at 0 mV for indicated concatenated hERG1 channels (n = 3–5). Data were fitted with a logistic equation (smooth curves). (F) IC50 values plotted as a function of the number of S631A subunits contained within a concatenated tetramer. The relationship was analyzed by linear regression (dashed line; adjusted R2 = 0.95). *P < 0.005 compared with SA4 channels. (G) Plot of IC50 values versus g/gmax at 0 mV for indicated channels. Dashed line represents fit to all data points (adjusted R2 = 0.76); dotted line represents fit to mutant concatenated channels (adjusted R2 = 0.98). Symbols are the same as indicated in (E).
Effect of Cisapride on G628C/S631C hERG1 Concatenated Tetrameric Channels.
The combination of two point mutations, one in the selectivity filter (G628C) and another just outside this structure (S631C), eliminates inactivation in hERG1 channels (Smith et al., 1996). Two concatenated tetramers incorporating G628C/S631C mutant subunits were constructed (GCSC1/WT3 and GCSC4) and used to further investigate the role of inactivation in the potency of block by cisapride. Figure 6A illustrates the effect of 0.3 µM cisapride on currents activated during 4-second pulses to 0 mV followed by repolarization to −70 mV to elicit tail currents. GCSC1/WT3 and GCSC4 hERG1 concatemers do not inactivate (Wu et al., 2014a), and both were equally sensitive to block by cisapride with IC50 values of 0.51 µM for GCSC1/WT3 channels and 0.49 µM for GCSC4 channels (Fig. 6B), only a 2.5-fold increase of IC50 compared with WT4 channels. Thus, when inactivation gating of hERG1 channels was removed with the G628C/S631C double mutations, the decrease in sensitivity to cisapride was far less than that observed when inactivation was eliminated with the S620T mutation. This finding suggests that inactivation per se is not the primary determinant of altered drug sensitivity in S620T channels.
Fig. 6.
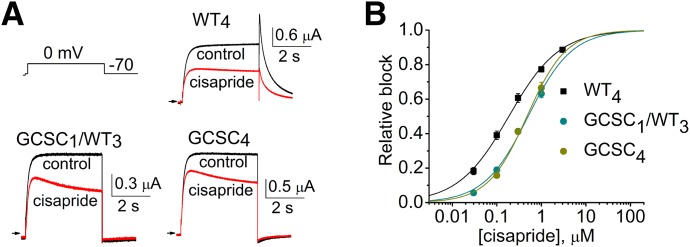
hERG1 concatemers containing one or four G628C/S631C subunits have equal sensitivity to cisapride. (A) Representative current traces recorded at 0 mV for WT4, GCSC1/ WT3 and GCSC4 hERG1 channels in the absence and presence of 0.3 µM cisapride. (B) [Cisapride]-effect relationship for inhibition of Itest measured at the end of the 4-second pulse to 0 mV for the indicated concatenated hERG1 channels. Data were fitted with a logistic equation (smooth curves) to determine IC50 for WT4 channels (188 ± 20 nM; data from Fig. 1A), GCSC1/WT3 channels (511 ± 48 nM, n = 3), and GCSC4 channels (487 ± 55 nM, n = 3).
Second Site Mutation (Y652W) Partially Restores Blocker Sensitivity of S620T hERG1 Channels.
The putative binding site for most drugs that block hERG1 channels, including cisapride (Mitcheson et al., 2000), dofetilide (Kamiya et al., 2006), and MK-499 (Mitcheson et al., 2000) is located in the central cavity. Site-directed mutagenesis assays indicate that these drugs primarily interact with a few residues that face toward the central cavity, including two aromatic residues in the S6 segment (Tyr652 and Phe656) and three residues located between the pore helix and the selectivity filter (Thr623, Ser624, and Val625). We hypothesized that a S620T mutation in the pore helix might reduce the affinity for interaction with one or more of these key residues and that this action rather than simply a lack of inactivation gating might be the cause of the reduced drug potency. To test this idea, we determined if introduction of a second mutation into S620T subunits could partially rescue drug sensitivity that was reduced by S620T. In addition, we also determined if drug potency could be rescued by “repositioning” the single Tyr and Phe residues in the S6 segment that are the most important molecular determinants of blocker potency (Mitcheson and Perry, 2003; Fernandez et al., 2004). Specifically, Tyr652 or Phe656 were mutated to Ala and the preceding residue was mutated to Tyr or Phe to yield M651Y+Y652A and I655F+F656A channels. In essence, each double mutation moves a key aromatic residue to the preceding position in the S6 segment, a procedure that we previously found could introduce sensitivity to cisapride block of otherwise insensitive eag channels (Chen et al., 2002).
For these experiments, channels formed by natural coassembly of single subunits expressed in oocytes were studied. Itest-Vt relationships for all the different mutant channels were determined by applying 4-second pulses to a Vt that was varied from −70 to +40 mV. Representative current traces at a Vt of −20 and +20 mV for WT, S620T, and S620T+Y652W hERG1 channels are illustrated in Fig. 7A. For WT hERG1 channel, Itest at −20 mV was about 2.5-fold larger than Itest at +20 mV, reflecting strong channel inactivation at the depolarized potential. In contrast, for S620T and S620T+Y652W channels Itest was much larger at a Vt of +20 mV than at −20 mV, a consequence of attenuated channel inactivation. S620T+Y652W channel current at +20 mV exhibited a time-dependent decay that presumably indicates that these channels retained an ability to partially inactivate. The normalized Itest-Vt relationships for mutant channels containing the S620T mutation alone or combined with one or two additional mutations are plotted in Fig. 7, B and C. The Itest-Vt relationship exhibited outward rectification with no region of negative slope conductance, indicating that inactivation was highly disrupted in all 10 of the mutant channels. The effect of the three drugs on channel currents was assessed by applying repetitive pulses to 0 mV once every 20 seconds. The percent inhibition of Itest of WT and the 10 mutant hERG1 channels by 0.3 µM cisapride, dofetilide, and MK-499 are summarized in Fig. 7, D–F, respectively. Y652W was the only mutation that partially restored sensitivity of S620T channels to block by all three drugs. However, as shown in Fig. 7A, S620T+Y652W channels also appeared to inactivate, albeit only slightly compared with WT channels. The voltage dependence of inactivation of S620T+Y652W channels is quantitatively compared with WT, Y652W, and S620T channels in Fig. 7G. Although the voltage dependence of g/gmax for WT and Y652W channels was similar, rectification of S620T+Y652W channels was less attenuated than that observed for S620T channels.
Fig. 7.

Drug sensitivity of S620T hERG1 mutant channels harboring additional mutation(s) in the central cavity. (A) Representative current traces recorded at −20 mV and +20 mV for WT, S620T, and S620T+Y652W channels. (B and C) Itest-Vt relationships for oocytes injected with cRNA encoding monomeric mutant subunits as indicated (n = 4–8). Currents were normalized to peak Itest at +40 mV. (D–F) Inhibition of Itest at 0 mV expressed as % inhibition at steady-state ([Icontrol − Idrug]/Icontrol × 100) by 0.3 µM cisapride (D), dofetilide (E), and MK-499 (F). n = 3–5 for all drugs. (G) Voltage dependence of inactivation for WT, Y652W, S620T, and S620T+Y652W channel currents (n = 4–6).
To better quantify the relationship between drug potency and channel inactivation of these four channels, we determined the concentration-dependent inhibition of Itest by all three drugs (Fig. 8, A–C) and then plotted the calculated IC50 values as a function of the relative channel conductance (g/gmax) at 0 mV (Fig. 8, D–F). The value of g/gmax at 0 mV was chosen because the relative block of Itest was also determined at a Vt of 0 mV. As before (Figs. 2E, 3D, and 4D), there was no correlation between the extent of current rectification and drug potency, suggesting that inactivation is not the primary determinant of hERG1 channel sensitivity to block by cisapride, dofetilide, or MK-499. Finally, we also determined the IC50 for block of S620T and S620T+Y652W channels by all three drugs in oocytes that were repetitively pulsed to a Vt of −30 mV, a potential where both of the mutant channels do not inactivate (i.e., g/gmax = 1.0). The plots of relative block of Itest at −30 mV as a function of the concentration of cisapride, dofetilide, and MK-499 are shown in Fig. 9, A–C, respectively. Although S620T channels were still less sensitive to block than S620T+Y652W channels when assessed at −30 mV, the fold increase in IC50 was less at −30 than 0 mV for cisapride and dofetilide. In contrast, the fold increase in IC50 at the two test potentials was not appreciably changed for MK-499 (Fig. 9D). Thus, even when the extent of inactivation is quantitatively accounted for in our analysis, mutating Tyr652 to the more bulky and hydrophobic Trp partially restores the sensitivity of S620T hERG1 channels to block by the three drugs examined in this study.
Fig. 8.

Relationship between inactivation and IC50 for block determined at a test potential of 0 mV for WT, Y652W, S620T and S620T+Y652W mutant hERG1 channels. (A–C) Concentration-response relationship for block of Itest at 0 mV for WT (▪), Y652W (red circle), S620T (green circle), and S620T/Y652W (purple circle) channels by cisapride (n = 4–6), dofetilide (n = 4–5), and MK-499 (n = 4–5) as indicated. Data were fitted with a logistic equation (smooth curves). (D–F) IC50 values, determined from plots shown in (A–C), are plotted as a function of g/gmax at 0 mV for cisapride (D), dofetilide (E), and MK-499 (F).
Fig. 9.

Block of S620T and S620T+Y652W mutant hERG1 channels at a test potential of −30 mV. (A–C) Concentration-response relationships for block of Itest at −30 mV for S620T (▾) and S620T+Y652W (▴) mutant hERG1 channels by cisapride (n = 3–4), dofetilide (n = 4), and MK-499 (n = 3) as indicated. Data were fitted with a logistic equation (smooth curves). (D) Comparison of the fold increase in IC50 value for block of S620T channels compared with block of S620T+Y652W channels when Itest was measured at 0 mV or −30 mV for individual drugs as indicated.
Discussion
Based on numerous studies, it is generally believed that hERG1 channels are preferentially blocked when in an inactivated state. The most compelling evidence for this state-dependent block is the finding that most inactivation-deficient mutant channels exhibit dramatically reduced drug sensitivity (Suessbrich et al., 1997; Wang et al., 1997a; Ficker et al., 1998; Numaguchi et al., 2000; Perrin et al., 2008). However, there is not a simple correlation between point mutation-induced reduction or elimination of inactivation and the potency of hERG1 blockers. For example, the low-affinity blockers quinidine, perhexiline, and erythromycin inhibit WT and inactivation-deficient N588K hERG1 channels with equal potency (Perrin et al., 2008).
The study of channels formed by concatenation of multiple hERG1 subunits has provided unique insights into the mechanisms of drug action. Concatenated hERG1 dimers were used to show that cisapride interacts with Tyr652 residues on adjacent subunits, whereas terfenadine interacts with Tyr652 residues on diagonal, but not on adjacent, subunits (Imai et al., 2009). We previously used concatenated hERG1 tetramers to show that modification of hERG1 channel gating requires occupancy of all four available ligand binding sites to achieve maximal efficacy by the agonists PD-118057 [2-[[4-[2-(3,4-dichlorophenyl)ethyl]phenyl]amino]benzoic acid], ICA-105574 [3-nitro-N-(4-phenoxyphenyl)-benzamide] (Wu et al., 2014b), and RPR-260243 [(3R,4R)-4-[3-(6-methoxyquinolin-4-yl)-3-oxopropyl]-1-[3-(2,3,5-trifluorophenyl)prop-2-ynyl]piperidine-3-carboxylic acid] (Wu et al., 2015). In the present study, we further explored the link between inactivation gating and potency of block by cisapride, dofetilide, and MK-499 using concatenated hERG1 tetramers containing a variable number of mutant subunits harboring the S620T or S631A mutation known to disrupt inactivation without significantly changing ion selectivity (as occurs with the double mutation G628C/S631C). Previous studies have shown that concatenation of voltage-gated K channel subunits can result in unanticipated coassembly of multiple units (McCormack et al., 1992; Hurst et al., 1995; Sack et al., 2008). Improper assembly is less problematic when concatenated subunits are nearly identical to one another (Hurst et al., 1992; Sack, et al., 2008). As discussed previously (Wu et al., 2014a,b), the formation of multimerized hERG1 concatemers is unlikely.
A single S620T subunit in a concatenated tetramer is sufficient to disrupt inactivation gating to the same extent as that observed for concatenated S620T tetramers (Wu et al., 2014a). However, despite equivalent disruption of inactivation gating, the sensitivity of ST4 channels to block by dofetilide, MK-499 and especially cisapride was much less than ST1/WT3 channels. These and other findings indicate that Ser620 mutations have effects on drug efficacy that are not fully explained by high-affinity binding to an inactivated state of the channel. For example, whereas S620C and S620T mutations are equally effective at eliminating inactivation, S620C did not affect the potency of channel block by cocaine, whereas block of S620T channels was reduced by 21-fold (Guo et al., 2006). In contrast to S620T, the suppression of inactivation induced by S631A was a graded function of the number of mutant subunits contained within a concatenated tetramer. The blocking potency of cisapride was unaffected by the presence of 1 to 3 S631A subunits and was only increased (4-fold) in SA4 channels, suggesting that only a single WT subunit is sufficient for high-affinity block, irrespective of the ability of the channel to inactivate. The potency of dofetilide was a graded function of the number of S631A mutant subunits with a maximum 10-fold reduction in IC50 observed for SA4 tetramers, a finding that is consistent with binding affinity being dependent on either the extent of inactivation or the number of mutant subunits contained within the concatenated tetramer. However, our findings with dofetilide block of S620T hERG1 tetramers would argue that inactivation is less important than the presumed effect of S631A subunits on binding affinity.
Mutations that cause nearly equivalent reduction or elimination of inactivation gating of hERG1 channels do not reduce the potency of blockers equally. In general, S620T channels are more resistant to block than S631A, N588K, or G628C/S631C mutant channels. One possible explanation for some of these findings is that whereas S620T completely eliminates inactivation, the point mutations S631A or N588K retain the ability to inactivate, albeit only at highly positive membrane voltages [V0.5inact is shifted by >100 mV (Zou et al., 1998; Cordeiro et al., 2005)]. Thus, as suggested previously (Perrin et al., 2008), whereas S631A and N588K channels only rarely enter an inactivated state at moderate potentials (e.g., 0 mV), the much higher affinity for inactivated state block would naturally result in a lower IC50 for block of these channels compared with S620T channels. Perrin et al. (2008) suggested that block of S620T hERG1 channels by dofetilide provided an estimate of the open state binding affinity, and Markov modeling was used to calculate affinities for the open and inactivated state that differed by 73-fold. However, we found that ST1/WT3 channels were more sensitive than ST4 channels to block by dofetilide (and MK-499 and cisapride) despite the fact that inactivation was absent in both mutant channel types. Moreover, GCSC1/WT3 and GCSC4 channels do not inactivate, yet were equally sensitive to block by cisapride and only 2.7 times less sensitive than WT4 channels. Interestingly, we found that ST1/WT3 channels were 3.1 times less sensitive to cisapride than WT4 channels, and Perrin et al. (2008) reported that when expressed in Chinese hamster ovary cells, N588K channels were 2.7-fold less sensitive to cisapride than WT hERG1 channels. Together these findings suggest that (1) block of G628C/S631C or ST1/WT3 inactivation-removed channels might provide a better estimate of drug binding affinity to the open state of hERG1 channels than that estimated from block of S620T homotetrameric channels and (2) inactivation state-dependent binding only accounts for ∼3-fold increase in sensitivity to channel block by cisapride.
Molecular dynamics simulations of a homology model suggest that inactivation of hERG1 channels results when the carbonyl of Phe627 in the selectivity filter rotates away from the ion conduction axis and that this is prevented by the S620T mutation (Stansfeld et al., 2008). We presume that each S620T mutation affects drug affinity by an intrasubunit effect, because increasing the number of S620T subunits contained within a concatenated tetramer produced a progressive reduction in the potency of cisapride, dofetilide, and MK-499. As suggested previously (Perrin et al., 2008), the S620T mutation might induce a conformational change in one or more of the critical drug binding residues located at the base of the pore helix or nearby S6 residues that face toward the central cavity (Mitcheson et al., 2000). We probed for this possibility by introducing into S620T channels a second site mutation of a residue located at the base of the pore helix (Ser624, Thr623, Val625) or in the S6 segment (Tyr652, Phe656). Only one second site mutation (Y652W) appreciably enhanced drug sensitivity of S620T channels. Substitution of Tyr with Trp could facilitate hydrophobic interactions between the channel and a drug located in the central cavity; however, the Y652W mutation alone did not enhance drug potency. Based on these findings, we propose that S620T, in addition to disrupting the molecular machinery of inactivation, may also allosterically alter the positon of the side chain of Tyr652 in a manner that reduces high affinity drug binding.
In summary, our findings suggest that although the S620T and S631A mutations reduce drug sensitivity in part by eliminating inactivation, these mutations also apparently cause an allosteric disruption of drug binding by an independent mechanism. Thus, the subtle reconfiguration of channel structure associated with inactivation gating facilitates, but is not a strict requirement for, high-affinity block of hERG1 channels.
Abbreviations
- E-4031
N-[4-[1-[2-(6-methylpyridin-2-yl)ethyl]piperidine-4-carbonyl]phenyl]methanesulfonamide
- eag
ether-à-go-go
- GCSC
G628C/S631C
- ICA-105574
3-nitro-N-(4-phenoxyphenyl)benzamide
- MK-499
N-[(4R)-1′-[(2R)-6-cyano-1,2,3,4-tetrahydronaphthalen-2-yl]-4-hydroxyspiro[3,4-dihydrochromene-2,4′-piperidine]-6-yl]methanesulfonamide
- PD-118057
2-[[4-[2-(3,4-dichlorophenyl)ethyl]phenyl]amino]benzoic acid
- RPR-260243
(3R,4R)-4-[3-(6-methoxyquinolin-4-yl)-3-oxopropyl]-1-[3-(2,3,5-trifluorophenyl)prop-2-ynyl]piperidine-3-carboxylic acid
- ST
S620T
- SA
S631A
- WT
wild-type
Authorship Contributions
Participated in research design: Wu, Gardner, Sanguinetti.
Conducted experiments: Wu, Gardner.
Performed data analysis: Wu, Sanguinetti.
Wrote or contributed to the writing of the manuscript: Wu, Sanguinetti.
Footnotes
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grant R01-HL055236] and the Nora Eccles Harrison presidential endowed chair in cardiology (to M.C.S.).
References
- Abbruzzese J, Sachse FB, Tristani-Firouzi M, Sanguinetti MC. (2010) Modification of hERG1 channel gating by Cd2+. J Gen Physiol 136:203–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Seebohm G, Sanguinetti MC. (2002) Position of aromatic residues in the S6 domain, not inactivation, dictates cisapride sensitivity of HERG and eag potassium channels. Proc Natl Acad Sci USA 99:12461–12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro JM, Brugada R, Wu YS, Hong K, Dumaine R. (2005) Modulation of I(Kr) inactivation by mutation N588K in KCNH2: a link to arrhythmogenesis in short QT syndrome. Cardiovasc Res 67:498–509. [DOI] [PubMed] [Google Scholar]
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. (1995) A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80:795–803. [DOI] [PubMed] [Google Scholar]
- Fenichel RR, Malik M, Antzelevitch C, Sanguinetti M, Roden DM, Priori SG, Ruskin JN, Lipicky RJ, Cantilena LR, Independent Academic Task Force (2004) Drug-induced torsades de pointes and implications for drug development. J Cardiovasc Electrophysiol 15:475–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez D, Ghanta A, Kauffman GW, Sanguinetti MC. (2004) Physicochemical features of the HERG channel drug binding site. J Biol Chem 279:10120–10127. [DOI] [PubMed] [Google Scholar]
- Ficker E, Jarolimek W, Kiehn J, Baumann A, Brown AM. (1998) Molecular determinants of dofetilide block of HERG K+ channels. Circ Res 82:386–395. [DOI] [PubMed] [Google Scholar]
- Goldin AL. (1991) Expression of ion channels by injection of mRNA into Xenopus oocytes. Methods Cell Biol 36:487–509. [DOI] [PubMed] [Google Scholar]
- Guo J, Gang H, Zhang S. (2006) Molecular determinants of cocaine block of human ether-á-go-go-related gene potassium channels. J Pharmacol Exp Ther 317:865–874. [DOI] [PubMed] [Google Scholar]
- Hurst RS, Kavanaugh MP, Yakel J, Adelman JP, North RA. (1992) Cooperative interactions among subunits of a voltage-dependent potassium channel. Evidence from expression of concatenated cDNAs. J Biol Chem 267:23742–23745. [PubMed] [Google Scholar]
- Hurst RS, North RA, Adelman JP. (1995) Potassium channel assembly from concatenated subunits: effects of proline substitutions in S4 segments. Receptors Channels 3:263–272. [PubMed] [Google Scholar]
- Imai YN, Ryu S, Oiki S. (2009) Docking model of drug binding to the human ether-à-go-go potassium channel guided by tandem dimer mutant patch-clamp data: a synergic approach. J Med Chem 52:1630–1638. [DOI] [PubMed] [Google Scholar]
- Kamiya K, Niwa R, Mitcheson JS, Sanguinetti MC. (2006) Molecular determinants of HERG channel block. Mol Pharmacol 69:1709–1716. [DOI] [PubMed] [Google Scholar]
- Keating MT, Sanguinetti MC. (2001) Molecular and cellular mechanisms of cardiac arrhythmias. Cell 104:569–580. [DOI] [PubMed] [Google Scholar]
- Lees-Miller JP, Duan Y, Teng GQ, Duff HJ. (2000) Molecular determinant of high-affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: involvement of S6 sites. Mol Pharmacol 57:367–374. [PubMed] [Google Scholar]
- McCormack K, Lin L, Iverson LE, Tanouye MA, Sigworth FJ. (1992) Tandem linkage of Shaker K+ channel subunits does not ensure the stoichiometry of expressed channels. Biophys J 63:1406–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. (2000) A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci USA 97:12329–12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitcheson JS, Perry MD. (2003) Molecular determinants of high-affinity drug binding to HERG channels. Curr Opin Drug Discov Devel 6:667–674. [PubMed] [Google Scholar]
- Numaguchi H, Mullins FM, Johnson JP, Jr, Johns DC, Po SS, Yang IC, Tomaselli GF, Balser JR. (2000) Probing the interaction between inactivation gating and Dd-sotalol block of HERG. Circ Res 87:1012–1018. [DOI] [PubMed] [Google Scholar]
- Perrin MJ, Kuchel PW, Campbell TJ, Vandenberg JI. (2008) Drug binding to the inactivated state is necessary but not sufficient for high-affinity binding to human ether-à-go-go-related gene channels. Mol Pharmacol 74:1443–1452. [DOI] [PubMed] [Google Scholar]
- Sack JT, Shamotienko O, Dolly JO. (2008) How to validate a heteromeric ion channel drug target: assessing proper expression of concatenated subunits. J Gen Physiol 131:415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. (1995) A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81:299–307. [DOI] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T, Yellen G. (1996) The inward rectification mechanism of the HERG cardiac potassium channel. Nature 379:833–836. [DOI] [PubMed] [Google Scholar]
- Stansfeld PJ, Grottesi A, Sands ZA, Sansom MS, Gedeck P, Gosling M, Cox B, Stanfield PR, Mitcheson JS, Sutcliffe MJ. (2008) Insight into the mechanism of inactivation and pH sensitivity in potassium channels from molecular dynamics simulations. Biochemistry 47:7414–7422. [DOI] [PubMed] [Google Scholar]
- Stühmer W. (1992) Electrophysiological recording from Xenopus oocytes. Methods Enzymol 207:319–339. [DOI] [PubMed] [Google Scholar]
- Suessbrich H, Schönherr R, Heinemann SH, Lang F, Busch AE. (1997) Specific block of cloned Herg channels by clofilium and its tertiary analog LY97241. FEBS Lett 414:435–438. [DOI] [PubMed] [Google Scholar]
- Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. (1995) HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 269:92–95. [DOI] [PubMed] [Google Scholar]
- Wang S, Liu S, Morales MJ, Strauss HC, Rasmusson RL. (1997a) A quantitative analysis of the activation and inactivation kinetics of HERG expressed in Xenopus oocytes. J Physiol 502:45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Morales MJ, Liu S, Strauss HC, Rasmusson RL. (1997b) Modulation of HERG affinity for E-4031 by [K+]o and C-type inactivation. FEBS Lett 417:43–47. [DOI] [PubMed] [Google Scholar]
- Wu W, Gardner A, Sanguinetti MC. (2014a) Cooperative subunit interactions mediate fast C-type inactivation of hERG1 K+ channels. J Physiol 592:4465–4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Gardner A, Sanguinetti MC. (2015) Concatenated hERG1 tetramers reveal stoichiometry of altered channel gating by RPR-260243. Mol Pharmacol 87:401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Sachse FB, Gardner A, Sanguinetti MC. (2014b) Stoichiometry of altered hERG1 channel gating by small molecule activators. J Gen Physiol 143:499–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou A, Xu QP, Sanguinetti MC. (1998) A mutation in the pore region of HERG K+ channels expressed in Xenopus oocytes reduces rectification by shifting the voltage dependence of inactivation. J Physiol 509:129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]