

Abstract
Previously, we showed that vesicular stomatitis virus (VSV) engineered to express a cDNA library from human melanoma cells (ASMEL, Altered Self Melanoma Epitope Library) was an effective systemic therapy to treat subcutaneous (s.c.) murine B16 melanomas. Here, we show that intravenous treatment with the same ASMEL VSV-cDNA library was an effective treatment for established intra-cranial (i.c.) melanoma brain tumors. The optimal combination of antigens identified from the ASMEL which treated s.c. B16 tumors (VSV-N-RAS+VSV-CYTC-C+VSV-TYRP-1) was ineffective against i.c. B16 brain tumors. In contrast, combination of VSV-expressed antigens—VSV-HIF-2α+VSV-SOX-10+VSV-C-MYC+VSV-TYRP1—from ASMEL which was highly effective against i.c. B16 brain tumors, had no efficacy against the same tumors growing subcutaneously. Correspondingly, i.c. B16 tumors expressed a HIF-2αHi, SOX-10Hi, c-mycHi, TYRP1, N-RASloCytclo antigen profile, which differed significantly from the HIF-2αlo, SOX-10lo, c-myclo, TYRP1, N-RASHiCytcHi phenotype of s.c. B16 tumors, and was imposed upon the tumor cells by CD11b+ cells within the local brain tumor microenvironment. Combining T-cell costimulation with systemic VSV-cDNA treatment, long-term cures of mice with established i.c. tumors were achieved in about 75% of mice. Our data show that the anatomical location of a tumor profoundly affects the profile of antigens that it expresses.
Introduction
During our studies to develop effective oncolytic viruses for cancer therapy, we showed that vesicular stomatitis virus (VSV) not only acts as an oncolytic agent1,2 but also serves as a powerful immune adjuvant.2,3,4,5,6 Thus, therapy of intra-tumorally (i.t.) delivered VSV in the B16-ova model derived predominantly from immune bystander effects of the anti-viral innate immune response at the tumor site.2,3,4,5,6 In addition, viral oncolysis of B16 metastases in the tumor draining lymph node (TDLN) was significantly more effective at priming adaptive T-cell responses against Tumor Associated Antigens (TAA), and clearing tumor, than was direct i.t. virus.7 This reflected a potent VSV-mediated activation of APC in the TDLN for presentation of TAA released from metastases undergoing oncolysis.7 We,2,8 and others,9,10,11 have also enhanced priming of CD8+ T-cell responses against TAA by incorporating specific TAA into the oncolytic virus,2,8 thereby combining truly systemic delivery of oncolytic viruses with effective tumor vaccination to mobilize therapeutic anti-tumor T-cell responses. However, a major hurdle to cancer immunotherapy is that tumors readily evolve to escape from single antigen specific immune responses.12,13 If immunotherapies are developed to target a broad repertoire of TAAs, it will become increasingly difficult for tumors to lose expression of all of these TAA simultaneously, and/or for highly heterogeneous tumors to maintain sub-dominant clones which already lack expression of a subset of TAA, in order to escape a multi-targeted immune response.14 In this respect, we previously showed that by inducing “stressful death” of normal cells it was possible to generate autoimmune responses, which were also effective against tumor cells which share antigens with the normal tissue.15,16,17,18,19,20
Therefore, we combined the concepts of inflammatory killing of normal cells to treat tumors, with the use of VSV as both an oncolytic and adjuvant. We hypothesized that, by expressing a cDNA library of a normal/tumor tissue from systemically delivered, immunogenic VSV, it would be possible to display a very broad repertoire of TAA, which are also expressed on tumors of the same histological type and against which T-cell responses would mediate tumor rejection. Consistent with this, intravenous (i.v.) injection of VSV expressing a cDNA library from normal human prostate induced rejection of established murine prostate tumors, without detectable autoimmunity.21 In this system, VSV induced inflammatory signals in the TDLN, which activated anti-tumor T-cell responses2,7,9,22,23,24,25,26 against the repertoire of potential TAA encoded by the cDNA library.21 Similarly, a VSV-cDNA library from human melanoma cells (an Altered Self Melanoma Epitope Library (ASMEL) in the murine context) also treated subcutaneous (s.c.) murine B16 melanomas.27 In both prostate21 and melanoma27 models, re-stimulation of lymph nodes (LN)/splenocytes from mice cured of tumor with the VSV-cDNA library stimulated tumor specific, cytokine recall responses in vitro. Using this assay, we identified 3 VSV-cDNA viruses—VSV-N-RAS/VSV-CYT-C/VSV-TYRP-1—from the ASMEL melanoma VSV-cDNA library which, in combination, but not alone, induced s.c. B16 tumor rejection by priming a Th17 anti-tumor response.27
The goal of the current study was to investigate whether the ASMEL VSV-cDNA library strategy would show a more generalizable therapy against disseminated disease in other body sites. To test this, we treated mice with intra-cranial (i.c.) B16 tumors as opposed to s.c. with the ASMEL VSV-cDNA library. Here, we show that, in the presence of additional T-cell costimulation with IL-2Cx,28 intravenously delivered ASMEL cured over 75% of mice bearing 5 days established B16 tumors in the brain. However, to our surprise, the combination of antigens from the ASMEL which treated s.c. B16 tumors (VSV-N-RAS/VSV-CYT-C/VSV-TYRP-1), was ineffective against i.c. B16 brain tumors; in contrast, a different combination of VSV-expressed antigens—VSV-Hypoxia-Inducible Factor 2α (HIF-2α)/VSV-SOX-10/VSV-C-MYC/VSV-TYRP1—identified from the ASMEL, which was highly effective against i.c. B16 brain tumors, had no efficacy against s.c. B16 tumors. Our data show that the site of tumor growth, even of tumors of the same histological type, profoundly affects the profile of potential immunogens expressed by the tumor cells. This may either be through the direct imposition of a niche-specific phenotype upon the tumor cells by the local tumor/immune environment or by selection of a subset of pre-existing clones from the heterogeneous tumor mass, which express different antigen repertoires associated with optimal growth in different niches. Therefore, the design of therapeutic strategies must take into account not only the specific histological type of tumor being treated, but also the antigenic/immunogenic identity imposed by the local tumor microenvironment, which may differ radically between tissue/organ sites.
Results
VSV expressing a foreign model TAA treats i.c. tumors
Systemic (i.v.) delivery of VSV-expressing OVA, a model TAA of B16-ova tumors, significantly prolonged survival of mice with established intra-cranial B16-ova tumors (P = 0.0259 compared to VSV-GFP), but was unable to generate any cures (Figure 1a). As we reported previously for s.c. B16-ova tumors which escaped OVA-targeted immunotherapy,29 i.c. tumors which developed following i.v. VSV-ova had lost expression of the target OVA antigen (data not shown).
Figure 1.
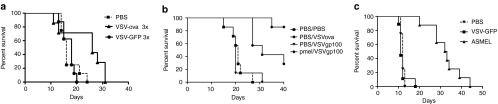
Systemic VSV expressing a tumor antigen treats brain tumors. (a) C57BL/6 mice bearing 5d established intra-cranial B16-ova tumors were treated intravenously with PBS (100 μl) or 5 × 106 pfu/100 μl of VSV-GFP or VSV-ova on days 6, 8, and 10. Survival with time (tumor <1.0 cm diameter) is shown. (b) C57BL/6 mice bearing 5d established i.c. tumors (7–8/grp) were treated with naive Pmel T cells (1 × 106 cells/100 μl) or PBS (100 μl) on day 6. Three doses of VSV-hgp100 or VSV-ova (5 × 106 pfu/100 μl) or PBS (100 μl) were given intravenously every other day starting 1 day after adoptive T-cell transfer. (c) C57BL/6 mice bearing 5d established intra-cranial B16-ova tumors were treated intravenously with 5 × 106 pfu of VSV-GFP or the ASMEL on days 6, 8, 10; 13, 15, 17; 20, 22, 24; and 27, 19, 31. Survival with time is shown.
VSV expressing a self-TAA is ineffective against i.c. tumors
In contrast to our ability to target OVA, a foreign TAA against which no tolerance exists in C57BL/6 mice, VSV-hgp100, targeting the endogenous self-TAA gp100, against which tolerance is intact in C57BL/6 mice, generated no significant therapy against i.c. B16-ova tumors in vivo (Figure 1b), and all the tumors recovered from these mice still retained unchanged levels of gp100 expression (data not shown). However, although adoptive transfer of naive Pmel hgp100 antigen-specific T cells had no therapeutic effect compared to PBS30,31 (data not shown) >80% of mice bearing i.c. B16-ova tumors were cured by combining i.v. VSV-hgp100 with adoptive transfer of naive PMEL antigen-specific T cells (Figure 1b)—similar to our results treating s.c. B16 tumors.30,31 These data confirm that B16 melanoma in the brain (a common site of clinical metastases) is amenable to immunotherapy, as we have shown for s.c. disease.
ASMEL VSV-cDNA library treats melanoma in the brain
We therefore hypothesized that it would be possible to treat i.c. tumors by targeting multiple different self-TAA on the tumor cells using a cDNA library, would raise multiple (weak) T-cell responses against several different self-antigens, which would, cumulatively, be sufficient to impact on tumor growth. Consistent with this, i.v. treatment with the ASMEL VSV-cDNA library, constructed from cDNA of human melanoma cells,27 significantly extended survival of mice bearing 5 days established i.c. B16-ova tumors (P < 0.0001 compared to VSV-GFP) (Figure 1c). Although the ASMEL VSV-cDNA did not generate any long-term cures, it targeted only self-antigens (OVA is not expressed in the ASMEL) and it was significantly more effective than VSV-mediated expression of either a single self-TAA (gp100, no therapy) (Figure 1b) or a non-tolerized, foreign TAA (VSV-ova) (Figure 1a) in two separate experiments.
ASMEL boosts a Th17 anti-tumor memory response
Previously we had shown that anti-tumor efficacy of VSV-cDNA libraries correlates with the ability of LN/splenocytes from VSV-cDNA library treated, tumor-cured mice to mount an IL-1721,27 or IFN-γ32 recall response in vitro upon re-stimulation with either tumor targets or the library itself. LN/splenocytes from C57BL/6 mice bearing i.c. B16-ova tumors and treated with the ASMEL, secreted IL-17, and IFN-γ in response to re-stimulation in vitro with the ASMEL (Lane 2) (Figure 2a,b). Interestingly, LN/splenocytes from mice which had no primary tumor, but were treated with ASMEL, secreted significantly reduced amounts of IL-17, relative to tumor-bearing mice, upon in vitro re-stimulation with the ASMEL (Lanes 2 versus 4) (Figure 2a), but did exhibit a similar memory IFN-γ recall response to the ASMEL (Lanes 2 versus 4) (Figure 2b). Consistent with this IFN-γ response being predominantly directed against VSV antigens, although tumor bearing mice treated with either the ASMEL VSV-cDNA library, or with VSV-ova, did not have an IL-17 recall response to VSV-ova (Lane 7) (Figure 2a), VSV-hgp100 (data not shown) or VSV-GFP (data not shown), they did have a Th1, IFN-γ memory response in all cases when they were re-stimulated with VSV (either the ASMEL or VSV-ova)(Lanes 6 and 7) (Figure 2b). This Th1, IFN-γ response was absent unless both in vivo treatment and re-stimulation, included VSV; hence, IFN-γ was not seen on re-stimulation of VSV-ova treated mice with cells (not VSV) not expressing OVA (Lane 10) (Figure 2b) or with re-stimulation with OVA peptide (not expressed by VSV) when in vivo treatment was with VSV-GFP (Lane 11) (Figure 2b). However, treatment with VSV-ova was able to generate a Th1, IFN-γ memory recall response against the foreign encoded OVA antigen, as demonstrated either by re-stimulation with OVA-expressing cells (Lanes 8 and 10) (Figure 2b) or with OVA peptide (Lanes 9 and 10) (Figure 2b). Hence, in the context of i.c. B16, a Th17 memory response indicated priming against ASMEL-encoded TAA, whilst a Th1, IFN-γ response corresponded to priming against the VSV adjuvant or an artificially dominant antigen, such as OVA.
Figure 2.

Systemic treatment with the ASMEL. (a,b) Pooled LN/splenocytes (1 × 106/well) from mice which either never had a tumor (Tumor; -) or had established i.c. tumors (Tumor; +) which had been treated with either the ASMEL (LIB; 9 i.v. injections), PBS (-), VSV-ova (VSV-o), or VSV-GFP (VSV-G) were re-stimulated in vitro with nothing (-), ~1 × 106 pfu of the parental ASMEL virus stock (LIB), freeze/thaw lysate of B16 cells (B16), ~106 pfu of VSV-ova (VSV-o), freeze thaw lysate of B16-ova cells (B16-ova) or with the ova-specific SIINFEKL peptide (SIIN). 24 hours later, the cultures were replenished with an additional 1 × 106 LN/splenocytes with a further round of virus infection/re-stimulation 24 after that. 48 hours following the final infection with virus, supernatants were assayed for (a) IL-17 or (b) IFN-γ by ELISA. (c) LN/splenocytes cultures (104/well) from C57BL/6 mice bearing i.c. B16 tumors and treated with the ASMEL were screened for secretion of IL-17 induced by infection with aliquots of ~1 × 104 pfu of the parental ASMEL virus stock in the presence of recombinant hsp70 (10 µg/ml). Aliquots which contained virus competent for inducing the IL-17 recall response were pooled and expanded in BHK cells (24–36 hours.). New LN/splenocytes cultures from ASMEL-treated mice were infected with serial dilutions of this expanded stock in the presence of recombinant hsp70, and assayed for IL-17 production. The highest dilution of the virus stock (~1 × 101 pfu) which induced IL-17 at levels significantly above background (>100 pg/ml) was amplified by passaging through BHK cells for 24–36 hours. Serial dilutions of this expanded stock were screened for their ability to induce IL-17. 10 µl aliquots of the highest dilution of the virus which induced IL-17 (1 × 102 pfu) were used as the starting point for limiting dilution cloning on BHK cells to identify the dilution at which a single virus particle generated cytopathic effect (+). Of 18 individual viruses screened from this experiment 5 viruses encoded part of the human HIF-2α gene,33 5 viruses encoded part of the human SOX-10 gene,34 3 viruses encoded part of the human TYRP1 gene,36 and 4 viruses encoded sequence of the human C-MYC gene.35
Identification of brain tumor antigens
We exploited the ability of LN/splenocytes (harvested from all the LN of the mice) from intra-cranial tumor-bearing ASMEL-treated mice to secrete IL-17 upon in vitro re-stimulation with the ASMEL (Lane 2) (Figure 2a) to identify individual VSV-cDNA viruses which encoded proteins that are the immunogenic targets of this IL-17 memory response. Using an assay previously validated to clone immunogenic proteins from VSV-cDNA libraries,21,32 including the ASMEL,27 we isolated the highest dilution of the ASMEL which was still active in stimulating an IL-17 recall response from LN/splenocytes (Figure 2c). Limiting dilution cloning from this highly diluted stock identified single VSV encoding 5′ cDNA sequences from HIF-2α,33 SOX-10,34 C-MYC,35 and TYRP-1.27,36
Anatomical location of tumor affects antigen expression
Consistent with our previous observations for the treatment of s.c. B16 tumors,27 re-stimulation of LN/splenocytes from tumor bearing, ASMEL-treated mice with any of these VSV-cDNA individually, in double or triple combinations, did not induce significant IL-17 (Figure 3a). However, when all four were combined, at the same total dose of virus, an IL-17 recall response was induced in vitro at levels approaching that induced by re-stimulation with the intact, un-fractionated ASMEL (Figure 3a).
Figure 3.
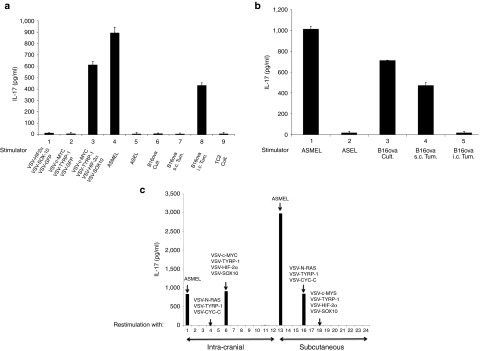
TAA expression is determined by anatomical location of the tumor. (a) LN/splenocytes cultures from mice treated for i.c. B16-ova tumors with the ASMEL were screened for IL-17 secretion following re-stimulation in vitro with a total of 107 pfu of combinations of the viruses selected from the screen of Figure 2c, including VSV-HIF-2α+VSV-SOX-10+VSV-GFP (Lane 1); VSV-C-MYC+VSV-TYRP-1+VSV-GFP (Lane 2); VSV-C-MYC+VSV-TYRP-1+VSV-HIF-2α+VSV-SOX-10 (Lane 3); the melanoma derived ASMEL (Lane 4) or the control ASEL VSV-cDNA library from human prostate cDNA (Lane 5).21,32 In addition, re-stimulation was also performed with freeze-thaw lysates from long-term in vitro cultured B16-ova cells (Lane 6); B16-ova cells freshly resected (within 48 hours of explant) from s.c. tumors (Lane 7); B16-ova cells freshly resected from three pooled i.c. tumors (Lane 8) or from long-term in vitro cultured TC2 murine prostate cells (Lane 9). (b) LN/splenocytes cultures from mice treated for s.c. B16-ova tumors with the ASMEL were screened for IL-17 secretion following re-stimulation in vitro with 107 pfu of the melanoma derived ASMEL (Lane 1) or the control ASEL VSV-cDNA library from human prostate cDNA (Lane 2);21,32 or with freeze-thaw lysates from long-term in vitro cultured B16-ova cells (Lane 3); B16-ova cells freshly resected from s.c. tumors (Lane 4); or B16-ova cells freshly resected from three pooled i.c. tumors (Lane 5). (c) LN/splenocytes cultures from mice treated for either i.c. (Lanes 1–12) or s.c. (Lanes 13–24) B16-ova tumors with the ASMEL were screened for IL-17 secretion following re-stimulation in vitro with 1 × 107 pfu of the melanoma derived ASMEL (Lane 1 and 13); PBS (Lanes 2 and 14); VSV-GFP (Lanes 3 and 15); or with a total of 1 × 107 pfu of different combinations of either three viruses including VSV-N-RAS+VSV-TYRP-1+VSV-CYT-C (Lanes 4 and 16) or VSV-SOX10+VSV-HIF-2α+VSV-C-MYC (Lanes 5 and 17); of four viruses including VSV-cMYC+VSV-TYRP-1+VSV-HIF-2α+VSV-SOX10 (Lanes 6 and 18); or of combinations of two different viruses: VSV-CYT-C+VSV-N-RAS (Lanes 7 and 19); VSV-CYT-C+VSV-TYRP-1 (Lanes 8 and 20); VSV-N-RAS+VSV-TYRP-1 (Lanes 9 and 21); VSV-SOX-10+VSV-HIF-2α (Lanes 10 and 22); VSV-SOX-10+VSV-C-MYC (Lanes 11 and 23) or VSV-C-MYC+ VSV-HIF-2α (Lanes 12 and 24).
To our surprise, and despite the fact that the ASMEL generated significant therapy against i.c. B16-ova tumors, LN/splenocytes from C57BL/6 mice bearing intra-cranial B16-ova tumors, which had been treated with the ASMEL, did not secrete IL-17 (or IFN-γ) when re-stimulated in vitro with lysates of cultured B16-ova cells (Lane 3) (Figure 2a,b) (Lane 6) (Figure 3a). This was in sharp contrast to our previous findings where LN/splenocytes from mice in which s.c. tumors were cured by treatment with the ASMEL had a Th17 memory response against both the ASMEL and cultured tumor cells in both the B16 and B16-ova models (Figure 3b).27 However, the IL-17 recall response was effectively induced from LN/splenocytes of mice with i.c. B16-ova tumors, treated with the ASMEL, upon re-stimulation with tumor cells recovered directly from i.c.-grown B16-ova brain tumors (Lane 8) (Figure 3a). In contrast, LN/splenocytes from mice treated for s.c. B16-ova tumors with the ASMEL could not be re-stimulated with brain tumor-derived B16 targets (Lane 5) (Figure 3b).
These data suggested that B16-ova tumors growing in the brain expressed a distinct set of potentially immunogenic TAA, compared to B16-ova tumors growing s.c., against which a Th17 response was raised by the presence of the tumor and which was boosted by i.v. treatment with the ASMEL. Consistent with this, LN/splenocytes from mice bearing i.c. B16-ova tumors, treated with the ASMEL, secreted high levels of IL-17 upon re-stimulation with either the unfractionated ASMEL or the combination of (VSV-HIF-2α/VSV-SOX-10/VSV-c-myc/VSV-TYRP-1) but not with other combinations of VSV-cDNA, including (VSV-N-RAS/VSV-CYT-C/VSV-TYRP-1) (Figure 3c). In contrast, LN/splenocytes from mice bearing s.c. B16-ova tumors, treated with the ASMEL, also secreted high levels of IL-17 upon re-stimulation with the unfractionated ASMEL, but not with the combination of (VSV-HIF-2α/VSV-SOX-10/VSV-c-myc/VSV-TYRP-1); however, re-stimulation with (VSV-N-RAS/VSV-CYT-C/VSV-TYRP-1), which was therapeutically active against s.c. B16-ova tumors,27 generated high levels of IL-17 (Figure 3c).
Melanoma tumors in the brain express a specific profile of potential tumor antigens
Therefore, we tested B16-ova cells, recovered from s.c. or i.c. environments, for their profiles of antigen expression. B16-ova cells maintained in vitro, and the source for implantation of both s.c. and i.c. tumors, expressed low (CT GAPDH-gene <0) levels of both HIF-2α and c-MYC, and moderate levels of TYRP-1, N-RAS and CYT-C mRNA (Lanes 16–20) (Figure 4a). Consistent with N-RAS, CYT-C and TYRP-1 as immunogens expressed by s.c. B16-ova tumors,27 freshly explanted s.c. B16-ova tumors had a very similar HIF-2αlo, c-MYClo, profile of antigen expression, and a slightly higher expression of both N-RAS and CYT-C mRNA (Lanes 1–5) (Figure 4a). In contrast, and consistent with HIF-2α and c-MYC as immunogens of i.c. tumors, freshly explanted i.c. B16-ova tumors from the brain had a very different HIF-2αHi, c-MYCHi profile as well as significantly lower levels of expression of N-RAS and CYT-C mRNAs (Lanes 6–10) (Figure 4a). Expression levels of TYRP-1 were similar across B16-ova cells recovered from in vitro passage, s.c. or i.c. sites. Expression of SOX-10 between i.c. and s.c. B16-ova tumor explants closely mirrored changes in HIF-2α (data not shown). Finally, upon prolonged in vitro culture, B16-ova cells recovered from i.c. tumors gradually reverted from a HIF-2αHi, SOX-10Hi, c-MYCHi profile to a more in vitro/s.c.-like HIF-2αlo, SOX-10lo, c-myclo, profile, with a gradual increase of N-RAS expression (Lanes 11–15) (Figure 4a). The qRT-PCR data was validated at the protein expression level for HIF-2α using an enzyme-linked immunosorbent assay (ELISA) assay (Figure 4b) and by western blotting for N-RAS and C-MYC (Figure 4d,e). GL261 glioma cells, freshly resected from the brain, also expressed a HIF-2αHi phenotype, which was significantly different from the in vitro cultured cells from which those tumors were derived (Lanes 1 and 2) (Figure 4c). In addition, as for s.c. B16-ova tumors, freshly resected s.c. TC2 prostate tumors had a very similar level of HIF-2α as their in vitro cultured counterparts (Lanes 3 and 4) (Figure 4c).
Figure 4.
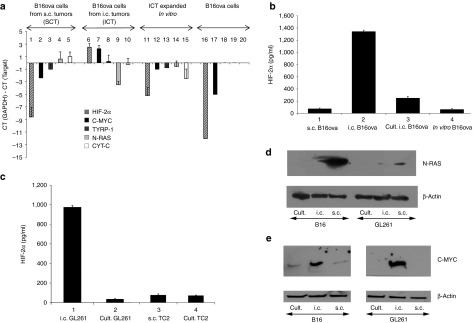
Intra-cranial and s.c. tumor phenotypes are distinct. (a) cDNA from B16-ova cells freshly resected from s.c. tumors (SCT) (Lanes 1–5), B16-ova cells freshly resected from three pooled i.c. tumors (ICT) (Lanes 6–10), B16-ova cells resected from three pooled i.c. tumors and maintained in culture for 3 weeks (Lanes 11–15) or long-term in vitro cultured B16-ova cells (Lanes 16–20) was screened by qRT-PCR for expression of HIF-2α (Lanes 1, 6, 11, 16), C-MYC (Lanes 2, 7, 12, 17), TYRP-1 (Lanes 3, 8, 13, 18), N-RAS (Lanes 4, 9, 14, 19) and CYT-C (Lanes 5, 10, 15, 20). The difference in cycle threshold (CT) for expression of the control GAPDH gene and the target gene (CT(GAPDH)-CT(Target gene)) is shown. Results are representative of three separate experiments. (b) HIF-2α protein expression from B16-ova cells freshly resected from s.c. tumors (Lane 1), B16-ova cells freshly resected from three pooled i.c. tumors (Lane 2), B16-ova cells resected from three pooled i.c. tumors and maintained in culture for 3 weeks (Lane 3) or long-term in vitro cultured B16-ova cells (Lane 4) was measured by ELISA with samples standardized for equal protein loading. (c) The experiment of b was repeated using glioma GL261 cells freshly resected from i.c. tumors (Lane 1) or long-term in vitro cultured GL261 cells (Lane 2); prostate TC2 cells freshly resected from s.c. tumors (Lane 3) or long-term in vitro cultured TC2 cells (Lane 4). (d) N-RAS or (e) C-MYC protein expression from long-term in vitro cultured B16-ova (Lane 1) or GL261 (Lane 4) cells, B16-ova (2) or GL261 (5) cells freshly resected from i.c. tumors, or B16-ova (3) or GL261 (6) cells freshly resected from s.c. tumors, was measured by Western Blot with samples standardized for equal protein loading using β-actin.
Intra-cranial CD11b+ cells mediate the HIF-2αHi phenotype
In vitro culture of B16-ova cells with cell free lysates of mouse brain did not change the HIF-2αlo phenotype of cultured B16-ova cells (Lanes 6–8) (Figure 5a). In contrast, coculture with cell-intact brain cell suspensions significantly increased levels of HIF-2α expression by qRT-PCR (Lanes 9–11) (Figure 5a), confirmed at the protein level by ELISA (data not shown). Whilst depletion of neither CD8+ T cells, NK cells nor neutrophils significantly prevented induction of the HIF-2αHi phenotype by brain cell suspensions (Figure 5b), depletion of CD11b+ cells completely abrogated this effect (Lanes 1, 2, and 6) (Figure 5b). Depletion of CD4+ T cells had a significant, but incomplete, effect on preventing the HIF-2αlo to HIF-2αHi high transition (Lanes 1,2,4) (Figure 5b). Finally, coculture of B16 cells with purified CD11b+ cells from mouse brains was able to mimic almost entirely the effects of brain cell suspensions on inducing the HIF-2αlo to HIF-2αHi high transition of B16-ova cells in culture (Figure 5c), an effect that was specific to brain derived CD11b+ cells as opposed to splenic CD11b+ cells (Figure 5c,d).
Figure 5.
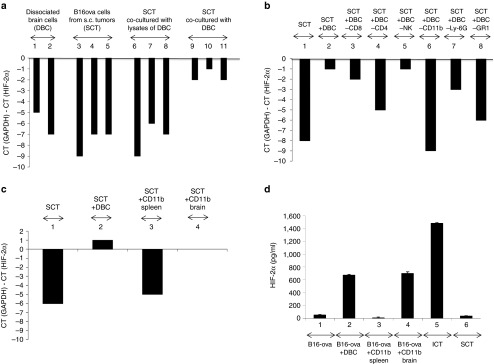
The i.c. phenotype is imposed by brain associated immune cells. (a) cDNA from two different brain-cell suspensions (DBC, dissociated intact brain cells) (Lanes 1,2); B16-ova cells from three different freshly resected s.c. tumors (SCT) (Lanes 3,4,5); cocultures of B16-ova cells from freshly resected s.c. tumors with freeze/thaw lysates of mouse brain cells (Lanes 6–8), or cocultures of B16-ova cells from freshly resected s.c. tumors with dissociated brain-cell suspensions (Lanes 9–11) (Materials and Methods section) were screened by qRT-PCR for expression of HIF-2α relative to GAPDH. The difference in cycle threshold for expression of the control GAPDH gene and HIF-2α (CT(GAPDH)-CT(HIF-2α)) is shown. (b) cDNA from B16-ova cells from a freshly resected s.c. tumor with no added brain-cell suspension (Lane 1); cocultured with dissociated brain-cell suspension (Lane 2); or cocultured with dissociated brain-cell suspensions depleted of CD8 (Lane 3); CD4 (Lane 4); NK (Lane 5); CD11b (Lane 6); Ly-6G+ neutrophils (using the IA8 depleting antibody) (Lane 7); or GR1+ cells (neutrophils, some DC, some monocytes, using the RB6-8C5 depleting antibody) (Lane 8); were screened by qRT-PCR for expression of HIF-2α relative to GAPDH. (c) cDNA from B16-ova cells from a freshly resected s.c. tumor were cocultured with no added cells (Lane 1); with dissociated brain-cell suspension (Lane 2); or with CD11b+ cells purified from spleens (Lane 3) or brains (Lane 4) of C57BL/6 mice were screened by qRT-PCR for expression of HIF-2α relative to GAPDH. (d) HIF-2α protein expression was measured by ELISA from B16-ova cells cocultured with no added cells (lane 1); with dissociated brain-cell suspension (Lane 2); with CD11b+ cells purified from spleens (Lane 3) or brains (Lane 4). HIF-2α protein expression was also measured from B16-ova cells freshly resected from an i.c. (Lane 5) or s.c. (Lane 6) tumor.
i.c. tumors are treated by different immunogens than s.c. tumors
Intravenous treatment of mice bearing established s.c. B16-ova tumors with a combination of VSV expressing HIF-2α, SOX-10, c-MYC, and TYRP1 was completely ineffective at controlling tumor growth, or overall survival (Figure 6a). However, as reported previously, a combination of VSV expressing N-RAS, CYT-C, and TYRP-1 delivered i.v. was highly effective against s.c. B16-ova tumors (P < 0.001 compared to controls) (Figure 6a). In contrast, for mice bearing i.c. B16-ova tumors, although the VSV-N-RAS, CYT-C, and TYRP-1 combination had no therapeutic effect on survival, the combination of VSV expressing HIF-2α, SOX-10, c-MYC, and TYRP1 generated highly significant survival benefits over controls (P < 0.0001) (Figure 6b), similar to those produced by the intact ASMEL (Figures 2a and 6b). The combination of (VSV-HIF-2α+VSV-TRP-1) had a significant, but very modest, benefit on survival compared to controls (P = 0.02) in one experiment, whilst (VSV-c-MYC+VSV-TYRP-1) was no better than controls (VSV-GFP or PBS) in treating i.c. B16-ova tumors (data not shown), showing that multiple combinations of immunogens are optimal for VSV-mediated immunotherapy of B16-ova (as reported previously).27 The combination of VSV expressing HIF-2α, SOX-10, c-MYC, and TYRP1 was at least as effective at treating i.c. B16 tumors (Figure 6c), as it was at treating i.c. B16-ova tumors (Figure 6b), confirming that the ova antigen was not a target immunogen in these experiments.
Figure 6.
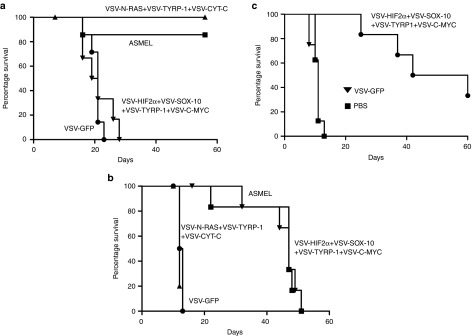
Differential immunotherapy for s.c. and i.c. tumors. C57BL/6 mice bearing 5d established (a) s.c. or (b) intra-cranial B16-ova tumors or (c) intra-cranial B16 tumors were treated intravenously with a total of 5 × 106 pfu of the ASMEL; (VSV-N-RAS+VSV-TYRP-1+VSV-CYT-C); (VSV-HIF2α+VSV-SOX-10+VSV-C-MYC+VSV-TYRP-1); VSV-GFP or PBS. Virus/PBS injections were administered on days 6, 8, 10; 13, 15, 17; 20, 22, 24; and 27, 29, 31. Survival with time is shown.
Additional T-cell stimulation improves therapy
As for the s.c. treatment model,27 6 i.v. injections of the unfractionated ASMEL, or of the VSV-HIF-2α/SOX-10/c-MYC/TYRP1 combination, instead of the 12 of Figure 6b, still gave significant (P < 0.01 compared to controls), but very moderate, survival benefits against B16-ova i.c. tumors (Figure 7a). However, in the presence of additional T-cell stimulation with IL-2Cx,28 this suboptimal dose of VSV-cDNA was converted into a highly effective immunotherapy, which cured over 75% of mice with i.c. B16-ova tumors (Figure 7a). LN/splenocytes from mice treated with VSV (either VSV-GFP or VSV-cDNA) and IL-2Cx had significantly enhanced Th1, IFN-γ memory recall responses against VSV (re-stimulation in vitro with a VSV-infected, non B16 cell line) (Figure 7b). As before (Figure 3c), mice treated with the VSV-cDNA combination alone did not have a Th1 IFN-γ response against i.c. or s.c. derived B16-ova cell targets (Figure 7c). However, treatment with VSV-cDNA (but not VSV-GFP) in the presence of IL-2Cx, induced weak, but significant, Th1 responses in all 5 mice tested against re-stimulating targets from B16-ova i.c. tumor growth; however, only 1 mouse had a detectable Th1 response against s.c. B16-ova tumor targets (Figure 7c). Interestingly, although treatment with VSV-cDNA alone did not generate any reactivity against glioma GL261 cells freshly explanted from the brain, IL-2Cx cotreatment uncovered IFN-γ recall responses of similar magnitude to those against i.c. B16-ova targets (Figure 7d), suggesting that some antigens are shared between the two models.
Figure 7.

T-cell costimulation enhances VSV-cDNA therapy of i.c. tumors. (a) C57BL/6 mice bearing 5d established intra-cranial B16-ova tumors were treated intravenously with PBS+PBS, or with a total of 5 × 106 pfu of (VSV-HIF2α+VSV-SOX-10+VSV-C-MYC+VSV-TYRP-1)+PBS; or with 5 × 106 pfu (VSV-HIF2α+VSV-SOX-10+VSV-C-MYC+VSV-TYRP-1)+IL-2Cx; or with 5 × 106 pfu of VSV-GFP + IL-2Cx, with virus on days 6, 8, 10, 13, 15, 17 and IL-2Cx on days 13, 15, 17. Survival with time is shown. (b–d) Pooled LN/splenocytes (106/well) from mice bearing i.c. B16-ova tumors treated with VSV-GFP+IL-2Cx (A1-A4); (VSV-HIF2α+VSV-SOX-10+VSV-C-MYC+VSV-TYRP-1)+PBS (VSV-cDNA+PBS, B1-B3); (VSV-HIF2α+VSV-SOX-10+VSV-CMYC+VSV-TYRP-1)+IL-2Cx (VSV-cDNA+IL-2Cx, C1-C5); or PBS+PBS (D1-D3) were re-stimulated in vitro with (b) freeze/thaw lysates of TC2 cells (white bars), or TC2 cells preinfected for 24 hours with VSV-GFP (MOI 0.1) (black bars); (c) freeze/thaw lysates of B16-ova cells freshly resected from s.c. (white bars) or i.c. (black bars) tumors; or (d) with freeze/thaw lysates of GL261 cells freshly resected from i.c. tumors (black bars) or cultured long-term in vitro (white bars). 24 hours later, the cultures were replenished with an additional 106 LN/splenocytes with a further round of re-stimulation 24 after that. 48 hours following the final re-stimulation, supernatants were assayed for IFN-γ by ELISA. (e,f) The same supernatants from b–d were also assayed for IL-17 secretion following re-stimulation as shown.
Finally, as in Figure 2a, treatment with neither VSV-GFP nor VSV-cDNA induced a Th17 response against the VSV either with, or without, IL-2Cx (Figure 7e). However, the VSV-cDNA combination generated a strong Th17 response against i.c.-derived, but not s.c.-derived, B16-ova, consistent with Figure 3a. Coadministration of IL-2Cx enhanced the magnitude of these Th17 responses against i.c.-derived B16-ova targets (Figure 7e). Interestingly, although the Th1 responses induced by cotreatment of VSV-cDNA with IL-2Cx were of comparable magnitude against i.c. B16 and i.c. GL261 targets (Figure 7b,d), the Th17 responses against i.c. B16 targets were generally higher than those against i.c.-derived GL261 glioma targets (Figure 7e,f). As for the B16 targets, LN/splenocytes from mice treated with VSV-cDNA either with, or without, IL-2Cx, did not generate recall responses to GL261 cells maintained in culture (Figure 7d,f).
Discussion
Although systemically delivered, VSV-mediated expression of the non-self, foreign OVA antigen primed therapeutic T-cell responses against tumor within the brain, i.v. treatment with VSV-hgp100, targeting a fully tolerized, endogenous self-TAA, gave no significant therapy against i.c. B16-ova tumors (Figure 1). However, combination with adoptive T-cell therapy could generate long-term cures, suggesting that targeting just a single weak self-TAA alone was challenging for priming/expanding therapeutic levels of anti-tumor T cells (Figure 1b). Therefore, we reasoned that an immunotherapy which targeted multiple different TAA on the tumor cells would raise multiple (probably weak) T-cell responses against a range of self-antigens, would, cumulatively, generate levels of anti-tumor T cells sufficient to impact on tumor growth. In addition, targeting multiple different antigens would limit the ability of the cells to evolve resistance against only one, or a few, T-cell responses. Consistent with this, we show here that i.v. treatment with the ASMEL VSV-cDNA library significantly extended survival of mice with i.c. B16-ova tumors, without targeting any foreign, non-self-antigens in the tumor cells (Figure 1c). Treatment with the ASMEL acted to boost a Th17 immune response, in the presence of i.c. B16-ova tumor (Figure 2a). Interestingly, neither OVA, hgp100 nor GFP, when encoded by VSV, could boost this Th17 memory response in vitro (Figure 2a), suggesting that a specific set of antigens encoded within the cDNA library, in the context of VSV as an adjuvant, are preferentially processed into, and presented by, a Th17 pathway of antigen presentation and that these proteins may be critical in generating anti-self/tumor responses. We also consistently observed that treatment with VSV (in the form of the ASMEL, VSV-ova, VSV-hgp100, or VSV-GFP) primed a Th1 IFN-γ recall response against VSV antigens (Figure 2b).
LN/splenocytes from ASMEL-treated mice with i.c. B16-ova tumors did not secrete IL-17 (or IFN-γ) when re-stimulated in vitro with lysates of cultured B16-ova cells, or B16-ova tumor cells freshly explanted from s.c. tumors (Figures 2a,b and 3a). This was surprising because LN/splenocytes from mice in which s.c. B16-ova tumors had been cured by treatment with the ASMEL had a significant Th17 memory response against both the ASMEL and cultured B16-ova cells (Figure 3b) (reported previously27). Moreover, LN/splenocytes of ASMEL-treated mice cured of s.c. B16-ova tumors did not generate a Th17 memory recall response to lysates of B16-ova tumor cells recovered from the intra-cranial site (Figure 3b). Therefore, these data suggested for the first time that neither cultured B16 cells, nor s.c. tumor B16-ova cells, expressed the same repertoire of antigens that were the targets of the in vivo Th17-cell responses that treated i.c. B16-ova tumors.
Using an in vitro assay,27,32 we identified the combination of VSV-HIF-2α, VSV-SOX-10, VSV-c-MYC, and VSV-TYRP-1 (Figure 2c) as the immunologically active components of the ASMEL which could stimulate the Th17 recall response from LN/splenocytes of i.c. B16-ova tumor cured, ASMEL-treated mice (Figure 3a). We are currently identifying the major epitopes of these proteins which elicit the Th17 responses in vivo and are testing whether these epitopes represent altered peptide ligands, compared to the corresponding mouse epitopes. Significantly, this combination of antigens was ineffective in stimulating the Th17 recall response from LN/splenocytes of mice cured of s.c. tumors by the ASMEL27 (Figure 3c). Consistent with these immunological data, both RNA and protein data showed that freshly resected i.c. B16-ova tumors expressed a HIF-2αHi, SOX-10Hi, c-MYCHi, TYRP1, N-RASlo, Cytclo phenotype, which was clearly distinct from the HIF-2αlo, SOX-10lo, c-myclo, TYRP1, N-RASHiCytcHi phenotype associated with s.c. B16-ova or in vitro cultured B16-ova tumors (Figure 4a,b). There are almost certainly additional proteins which are either up-, or downregulated in i.c., compared to s.c., tumors but which would not necessarily be identified by our immunological screen. We also showed (Figure 5) that the induction of the HIF-2αHi phenotype of B16-ova tumors growing intra-cranially was associated with the local cellular, rather than physical (such as hypoxic) microenvironment, principally mediated by brain-associated (but not splenic) CD11b+ cells. Such a cell-mediated mechanism of phenotype modulation is consistent with our observations that the HIF-2αHi, SOX-10Hi, c-MYCHi, TYRP1, N-RASloCytclo i.c.-associated B16-ova phenotype reverted with time in culture to the HIF-2αlo, SOX-10lo, c-MYClo, TYRP1, N-RASHiCytcHi phenotype associated with s.c. B16 or in vitro cultured B16 tumors (Figure 4a,b).
The therapeutic relevance of the location-dependent difference in antigenic phenotype of B16-ova tumors was shown by the fact that, whereas VSV-expressed HIF-2α, SOX-10, C-MYC and TYRP-1 were as potently immunogenic against established i.c. B16-ova tumors as the intact ASMEL (Figure 6b), these viruses were completely ineffective against s.c. B16-ova tumors (Figure 6a). In contrast, VSV-expressed N-RAS, CYT-C and TYRP-1 were highly effective against s.c. B16-ova tumors (Figure 6a)27 but gave no survival benefit compared to controls against i.c. B16-ova brain tumors (Figure 6b). The therapeutic effects observed on established i.c. B16-ova tumors were not dependent on the expression of the foreign antigen ova by the tumor cells (Figure 6c). Taken together, these data demonstrate that the anatomical location of tumor profoundly affects the profile of immunogenic antigens which are expressed and that a treatment which is highly effective against a tumor growing in one location, can be completely ineffective against a tumor of the same histological type, but growing elsewhere in the body.
Finally, we showed that by treating mice with the combination of VSV-HIF-2α, VSV-SOX-10, VSV-c-MYC and VSV-TYRP-1, in combination with additional T-cell costimulation in the form of IL-2Cx,28 long-term cures of mice with established intra-cranial tumors was achieved in about 75% of mice. (Figure 7a). None of these mice developed overt signs of autoimmune toxicity to the brain as judged by weight loss or behavioral abnormalities, although detailed histological analysis of the brains was not performed. The combination of VSV-TAA and T-cell costimulation induced increased (Figure 7b,c), and broader spectrum (Figure 7d), Th1-cell responses against i.c.-associated B16-ova TAA as well as VSV together with an increased magnitude of Th17 responses against i.c.-derived tumor targets (Figure 7e,f). Moreover, there was significant cross reactivity of the Th1 and Th17 responses induced by the melanoma-derived ASMEL between i.c.-derived B16 melanoma and GL261 glioma targets (Figure 7e,f). These intriguing results suggest that an intra-cranial (HIF-2αHi) phenotype (Figure 4), dependent upon the CD11b+ cells of the brain microenvironment (Figure 5), which we observed to be induced in tumors of at least two different histological types (Figure 4b,c), may render brain tumors vulnerable to common therapeutic strategies independent of tumor origin.
In addition, our data that both B16 melanoma and GL261 gliomas share a common HIF-2αHi, SOX-10Hi, c-mycHi phenotype suggest that it may be that the immunogenic profiles of tumors of different histologies, but growing in the same anatomical niche, may converge to some extent, raising the possibility of common location specific therapies for different tumor types.
Our data here are highly significant for the design of location-specific anti-tumor therapies. It seems likely that melanomas growing in other critical tissue/organ sites (such as the liver) may also have very different profiles of antigen/immunogen expression from those in the skin or the brain, as determined by the respective local tissue microenvironments. Therefore, it may not be unrealistic to consider tumors of the same histological type, but which are growing in different tissue locations, as sets of different “quasi-immunogenic species,” similar perhaps to distinct populations of viruses which evolve in vivo from a single initiating virus during infection. As a result, it may be that no single “one fits all” therapy can be used for these different “quasi-species.” In contrast, each location-specific quasi-species will need to be targeted separately by immune-, chemo- or radiation-therapies which exploit the specific profile of antigens expressed at the site dictated by the local microenvironment. Our findings also raise concerns about immunotherapies which rely on targeting only one, or a few, TAA. As well as the issue of antigen loss over time, our data suggest that metastatic disease at different anatomical locations may already have inherently different antigenic profiles. In addition, it may be that the immunogenic profiles of tumors of different histologies, but growing in the same anatomical niche, may converge to some extent, raising the possibility of common location specific therapies for different tumor types. With regard to the current rapid clinical progress in immunotherapy, it is perhaps noteworthy that the most promising agents to date (anti-CTLA4 and anti-PD1/PDL1 antibodies) do not rely on any specific, defined antigens as their targets, thereby potentially enhancing immune reactivity against a broad spectrum of TAA, as would be presented by a VSV-cDNA library, expressed on the quasi-species of tumors, irrespective of their location.
Our data also suggest that the cell intrinsic pathways that promote tumor growth—including pro-growth, anti-apoptotic, pro-angiogenic and anti-immunogenic factors—are very different depending upon the site of tumor growth. Thus, HIF-2α, SOX-10 and c-MYC are presumably critically important for tumor growth in the intra-cranial niche whereas N-RAS and CYT-C presumably provide alternative pathways for tumor growth subcutaneously. In this respect, it may be that the local microenvironment of a tumor directly imposes a niche-specific phenotype upon the tumor cells through, for example, the secretion of soluble factors. Alternatively, the location-dependent microenvironment may act upon the highly heterogeneous tumor population to select out a subset of pre-existing clones, which express different antigen repertoires associated with optimal growth in different niches (s.c. or intra-cranial). The HIF-2αHi, SOX-10Hi, c-MYCHi, TYRP1, N-RASloCytclo i.c.-associated B16-ova phenotype was transient, and reverted in culture to the HIF-2αlo, SOX-10lo, c-MYClo, TYRP1, N-RASHiCytcHi phenotype. This suggests that the imposition of the i.c. phenotype was effected through an epigenetic mechanism, rather than through selection of genetically stable sub-populations of tumor cells which then populate a tumor growing intra-cranially. However, it may still be that the s.c. in vitro dominant clone(s) of B16-ova, which are optimally adapted for growth s.c. or on plastic, are still carried as sub-populations in i.c. tumors and can then become re-selected for dominance in vitro. Characterization of these different mechanisms will be possible using genetically marked populations of B16-ova cells.32 Whatever the mechanisms, identifying niche-specific patterns of tumor antigen expression, through technologies such as gene profiling, or VSV-cDNA screening, will uncover new sets of potential targets for therapies against location-specific “quasi-species” of tumors.
In summary, we show here that a VSV-cDNA library generates immune re-activities against a range of weak self-tumor associated antigens which are cumulatively strong enough to treat intra-cranial melanomas. However, the immunogens, which are the targets of those T-cell responses, are very different in B16-ova tumors growing intra-cranially compared to the same B16-ova tumor cells growing subcutaneously. These findings have important implications for the design of tumor-type, but location-specific, therapies. In addition, they suggest that it may also be possible to design therapies which are themselves specific for tumors across histological types but growing in a common location.
Materials and Methods
Cell lines. Murine B16 cells (American Type Culture Collection, Manassas, Va.) were grown in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Carlsbad, CA) supplemented with 10% (v/v) fetal calf serum (FCS; Life Technologies) and L-glutamine (Life Technologies). B16-ova melanoma cells (H2-Kb) were derived from B16 cells transduced with a cDNA encoding the chicken ovalbumin gene.37 B16-ova cells were grown in the same medium as B16 cells but supplemented with 5 mg/ml G418 (Mediatech, Manassas, VA) to select for retention of the ova gene. All cell lines were routinely monitored and found to be free of Mycoplasma infection. TRAMP-C2 (TC2) cells are derived from a prostate tumor that arose in a TRAMP mouse (H-2kb)38 and were characterized by Dr Esteban Celis. TC2 cells grow in an androgen-independent manner and are routinely grown as tumors in C57BL/6 male mice.20
Mice. C57BL/6 mice (Thy 1.2+) were purchased from The Jackson Laboratory (Bar Harbor, ME) at 6–8 weeks of age. Pmel-1 transgenic mice (C57BL/6 background) express the Vα1/Vβ13 T-cell receptor that recognizes amino acids 25–33 of gp100 of pmel-17 presented by H2-Db MHC class I molecules.39 Pmel-1 breeding colonies were purchased from The Jackson Laboratory at 6–8 weeks of age.
Viruses. The ASMEL VSV-cDNA library was generated as described previously.21,27 Briefly, cDNA from two human melanoma cell lines, Mel624 and Mel888, was pooled, cloned into the pCMV.SPORT6 cloning vector (Invitrogen, Grand Island, NY) and amplified by PCR. The PCR amplified cDNA molecules were size fractionated to below 4 kbp for ligation into the parental VSV genomic plasmid pVSV-XN240 between the G and L genes. The complexity of the ASMEL cDNA library cloned into the VSV backbone plasmid between the Xho1-Nhe1-sites was 7.0 × 106 colony forming units. Virus was generated from BHK cells by cotransfection of pVSV-XN2-cDNA library DNA along with plasmids encoding viral genes as described in ref. 40. Virus was expanded by a single round of infection of BHK cells and purified by sucrose gradient centrifugation. VSV-GFP, VSV-ova, and VSV-hgp100 were described previously.30,40
In vivo studies. All procedures were approved by the Mayo Foundation Institutional Animal Care and Use Committee. To establish s.c. tumors, 5 × 105 B16-ova tumor cells in 100 μl of PBS were injected into the flanks of C57BL/6 mice (7–8 mice per treatment group unless stated otherwise). To establish i.c. brain tumors, 5 × 104 B16or B16-ova cells were injected stereotactically (1 mm anterior and 2 mm lateral to the bregma) using a syringe bearing a 26G needle 2.5 mm into the brains of C57BL/6 mice. Virus or PBS control (100 μl) was administered intravenously following tumor establishment (day 5 after cell implantation) and occurred every other day as dictated by the specific study.
Naive Pmel-1 T cells were isolated from the spleens and lymph nodes of pmel-1 transgenic mice. Single-cell suspensions were prepared by crushing tissues through a 100 μm filter and red blood cells were removed by incubation in ACK buffer (sterile distilled H2O containing 0.15 mol/l NH4Cl, 1.0 mmol/l KHCO3, and 0.1 mmol/l EDTA adjusted to pH 7.2–7.4) for 2 minutes. CD8+ T cells were isolated using the MACS CD8α (Ly-2) microbead magnetic cell sorting system (MiltenyiBiotec, Auburn, CA). For adoptive transfer experiments, mice were intravenously administered naive (1 × 106 total cells in 100 μl PBS) Pmel-1 cells after tumor establishment. Mice were examined daily for overall health as well as changes in whisker and coat pigmentation. For s.c. tumors, tumor sizes were measured three times weekly using calipers and were euthanized when tumor size was approximately 1.0 cm × 1.0 cm in two perpendicular directions.
IL-2Cx,28 a conjugate of murine IL-2 (2 µg/mouse) (Peprotech, Rocky Hill, NJ) preincubated in vitro with anti-mouse IL-2 Ab (10 µg per mouse) (BioXCell, West Lebanon, NH) at 4 °C for 18 hours, was injected intravenously in 100 µl PBS to enhance T-cell costimulation in vivo.
Quantitative RT-PCR. Tumors were immediately excised from euthanized mice and dissociated in vitro to achieve single-cell suspensions. RNA was extracted from cells using the QiagenRNeasy kit (Qiagen, Valencia, CA). cDNA was made from 1 μg total cellular RNA using the First Strand cDNA Synthesis Kit (Roche, Indianapolis, IN). A cDNA equivalent of 1 ng RNA was amplified by PCR with gene-specific primers to test for the expression of HIF2α (5′-ATGACAGCTGACAAGGAGAAGAA-3′ and 5′-TCTGACAGAAAGATCATGTCGC-3′) c-myc (5′-ATGCCCCTCAACGTTAGCTTCACC-3′ and 5′-CGGGGTTCGGGCTGCCGCTGTCT-3′), TYRP-1 (5′-ATGAGTGCTCCTAAACTCCTCTCTCT-3′and 5′-GTATTGTCTATTATGTCCAATAGG-3′), NRAS (5′-ATGACTGAGTACAAACTGGT-3′ and 5′-CTATTATTGATGGCAAATACA-3′) and cyt-c (5′-ATGATGGCGTCGGCGGTGCTG-3′ and 5′-AAGGGCTTGGTCCTGATGCGC-3′). Expression of mGAPDH was used as a positive control/reference gene for expression of genes being quantified by qRT-PCR. mGAPDH sense: TCATGACCACAGTCCATGCC, mGAPDH antisense: TCAGCTCTGGGATGACCTTG. qRT-PCR was carried out using a LightCycler480 SYBRGreen Master kit and a LightCycler480 instrument (Roche) according to the manufacturer's instructions. Typically, RNA was prepared from equal numbers of cells from each sample (usually 5,000 cells) and reverse transcribed as described above. PCR (primers at 0.5 µmol/l) was run with diluted cDNA samples (neat, 1:10, 1:100, 1:1,000). GAPDH amplification was used as a control for equal loading of target cDNAs. The threshold cycle (Ct) at which amplification of the target sequence was detected and used to compare the relative levels of mRNA between samples. Relative quantities of the target gene mRNA were normalized with Ct of GAPDH amplification.
In vitro splenic T-cell reactivation and ELISA for IFN-γ/IL-17/HIF-2α. Spleens and lymph nodes were immediately excised from euthanized mice and dissociated in vitro to achieve single-cell suspensions. Red blood cells were lysed with ACK lysis buffer for 2 minutes as described above. Cells were re-suspended at 1 × 106 cells/ml in Iscove's modified Dulbecco's medium (IMDM; Gibco, Grand Island, NY) + 5% FBS + 1% Pen-Strep + 40 μmol/l 2-ME. Supernatants were harvested from 1 × 106 LN/splenocytes previously stimulated with virus stocks as described in the text, with the H-2Kb-restricted peptide, ova257–264: SIINFEKL peptide (2.5 μg/ml) and/or with freeze thaw lysates from tumor cells in triplicate, every 24 hours for 3 days. 48 hours later, cell-free supernatants were collected and tested by ELISA for IL-17 (R&D Systems, Minneapolis, MN) or IFN-γ (BD Biosciences, San Jose, CA). HIF-2α protein was measured using a sandwich enzyme immunoassay as per the manufacturer's instructions (USCN Life Sciences, Houston, TX).
Virus re-stimulation of lymph nodes (LN)/splenocytes. Pooled LN/splenocytes (1 × 106/well) from previously treated mice were re-stimulated in vitro with nothing, ~1 × 106 pfu of virus stocks (ASMEL or VSV-ova), freeze/thaw lysates of tumor cells, or with the ova-specific SIINFEKL peptide (2.5 μg/ml). 24 hours later, the cultures were replenished with an additional 106 LN/splenocytes with a further round of virus infection/re-stimulation 24 after that. 48 hours following the final re-stimulation supernatants were assayed for IL-17 or IFN-γ by ELISA.
In vitro tumor-cell/brain-cell suspension cocultures. B16-ova cells (1 × 105/well) were cocultured with PBS, lysate from dissociated brain cells, freeze/thawed ×3 (equivalent of 1 × 106 cells per well), or with 1 × 106 cells/well from dissociated, but intact mouse brains. 24 and 48 hours later further PBS, freeze/thawed lysate or brain cell suspensions were added to the wells. 24 hours later, cultures were washed three times with PBS, cDNA prepared and screened by qRT-PCR for expression of HIF-2α relative to GAPDH. Brain-cell suspensions were depleted for immune cell types by 24 hours incubation with depleting antibodies. CD11b+ cells (1 × 105/well) were purified from brain-cell suspensions of multiple mouse brains using CD11b microbeads (Miltenyi Biotech, Auburn, CA) as directed by the manufacturer.
Western blotting. Proteins were resolved by SDS-PAGE using 10% Tris-glycine precast gels gel (Bio-rad, Hercules, CA). Following electrophoresis, proteins were transferred onto nitrocellulose membranes and blocked in PBS with 0.05% Tween-20 (PBS-T) and 5% milk. The membranes were probed using primary antibodies to c-myc (eBioscience, San Diego, CA) or N-RAS (Thermo Scientific, Waltham, MA) or β-actin (Sigma, St. Louis, MO) prepared in PBS-T with 3% milk. After several washes in PBS-T, the blots were incubated with respective secondary antibodies linked to horseradish peroxidase (GE healthcare, Piscataway, NJ) in PBS-T with 1% milk. The blots were washed in PBS-T, and proteins were detected using enhanced chemiluminescence substrate (Thermo Scientific).
Statistics. Survival data from the animal studies were analyzed by the log-rank test using GraphPad Prism 5 (GraphPad Software, La Jolla, CA). Two-sample, unequal variance Student's t-test analysis was applied for in vitro data. Statistical significance was determined at the level of P < 0.05.
Acknowledgments
This work was inspired by, and dedicated to, Shannon O'Hara. This work is supported by The Richard M Schulze Family Foundation, the Mayo Foundation, Cancer Research UK, the National Institute of Health (R01CA107082, R01CA130878, and R01CA132734) and a grant from Terry and Judith Paul. We thank Toni Higgins for expert secretarial assistance. The authors declare no conflict of interest.
References
- Barber GN. Vesicular stomatitis virus as an oncolytic vector. Viral Immunol. 2004;17:516–527. doi: 10.1089/vim.2004.17.516. [DOI] [PubMed] [Google Scholar]
- Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- Galivo F, Diaz RM, Thanarajasingam U, Jevremovic D, Wongthida P, Thompson J, et al. Interference of CD40L-mediated tumor immunotherapy by oncolytic vesicular stomatitis virus. Hum Gene Ther. 2010;21:439–450. doi: 10.1089/hum.2009.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galivo F, Diaz RM, Wongthida P, Thompson J, Kottke T, Barber G, et al. Single-cycle viral gene expression, rather than progressive replication and oncolysis, is required for VSV therapy of B16 melanoma. Gene Ther. 2010;17:158–170. doi: 10.1038/gt.2009.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wongthida P, Diaz RM, Galivo F, Kottke T, Thompson J, Melcher A, et al. VSV oncolytic virotherapy in the B16 model depends upon intact MyD88 signaling. Mol Ther. 2011;19:150–158. doi: 10.1038/mt.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wongthida P, Diaz RM, Galivo F, Kottke T, Thompson J, Pulido J, et al. Type III IFN interleukin-28 mediates the antitumor efficacy of oncolytic virus VSV in immune-competent mouse models of cancer. Cancer Res. 2010;70:4539–4549. doi: 10.1158/0008-5472.CAN-09-4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao J, Kottke T, Willmon C, Galivo F, Wongthida P, Diaz RM, et al. Purging metastases in lymphoid organs using a combination of antigen-nonspecific adoptive T cell therapy, oncolytic virotherapy and immunotherapy. Nat Med. 2008;14:37–44. doi: 10.1038/nm1681. [DOI] [PubMed] [Google Scholar]
- Wongthida P, Diaz RM, Pulido C, Rommelfanger D, Galivo F, Kaluza K, et al. Activating systemic T-cell immunity against self tumor antigens to support oncolytic virotherapy with vesicular stomatitis virus. Hum Gene Ther. 2011;22:1343–1353. doi: 10.1089/hum.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridle BW, Stephenson KB, Boudreau JE, Koshy S, Kazdhan N, Pullenayegum E, et al. Potentiating cancer immunotherapy using an oncolytic virus. Mol Ther. 2010;18:1430–1439. doi: 10.1038/mt.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelo-Branco P, Passer BJ, Buhrman JS, Antoszczyk S, Marinelli M, Zaupa C, et al. Oncolytic herpes simplex virus armed with xenogeneic homologue of prostatic acid phosphatase enhances antitumor efficacy in prostate cancer. Gene Ther. 2010;17:805–810. doi: 10.1038/gt.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil A, Martinez O, Chua MA, García-Sastre A. Recombinant Newcastle disease virus as a vaccine vector for cancer therapy. Mol Ther. 2008;16:1883–1890. doi: 10.1038/mt.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake CG, Jaffee E, Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. 2006;90:51–81. doi: 10.1016/S0065-2776(06)90002-9. [DOI] [PubMed] [Google Scholar]
- Poschke I, Mougiakakos D, Kiessling R. Camouflage and sabotage: tumor escape from the immune system. Cancer Immunol Immunother. 2011;60:1161–1171. doi: 10.1007/s00262-011-1012-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koos D, Josephs SF, Alexandrescu DT, Chan RC, Ramos F, Bogin V, et al. Tumor vaccines in 2010: need for integration. Cell Immunol. 2010;263:138–147. doi: 10.1016/j.cellimm.2010.03.019. [DOI] [PubMed] [Google Scholar]
- Ferrone S. Immunotherapy dispenses with tumor antigens. Nat Biotechnol. 2004;22:1096–1098. doi: 10.1038/nbt0904-1096. [DOI] [PubMed] [Google Scholar]
- Jevremovic D, Vile R. Stuhler G, Walden P. Cancer Immune Therapy. Wiley VCH: Germany; 2002. The immune system in cancer: if it isn't broken, Can we fix it; pp. 204–229. [Google Scholar]
- Kottke T, Pulido J, Thompson J, Sanchez-Perez L, Chong H, Calderwood SK, et al. Antitumor immunity can be uncoupled from autoimmunity following heat shock protein 70-mediated inflammatory killing of normal pancreas. Cancer Res. 2009;69:7767–7774. doi: 10.1158/0008-5472.CAN-09-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll DM. Inducing autoimmune disease to treat cancer. Proc Natl Acad Sci USA. 1999;96:5340–5342. doi: 10.1073/pnas.96.10.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmiani G. Tumor immunity as autoimmunity: tumor antigens include normal self proteins which stimulate anergic peripheral T cells. Immunol Today. 1993;14:536–538. doi: 10.1016/0167-5699(93)90183-L. [DOI] [PubMed] [Google Scholar]
- Kottke T, Sanchez-Perez L, Diaz RM, Thompson J, Chong H, Harrington K, et al. Induction of hsp70-mediated Th17 autoimmunity can be exploited as immunotherapy for metastatic prostate cancer. Cancer Res. 2007;67:11970–11979. doi: 10.1158/0008-5472.CAN-07-2259. [DOI] [PubMed] [Google Scholar]
- Kottke T, Errington F, Pulido J, Galivo F, Thompson J, Wongthida P, et al. Broad antigenic coverage induced by vaccination with virus-based cDNA libraries cures established tumors. Nat Med. 2011;17:854–859. doi: 10.1038/nm.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braxton CL, Puckett SH, Mizel SB, Lyles DS. Protection against lethal vaccinia virus challenge by using an attenuated matrix protein mutant vesicular stomatitis virus vaccine vector expressing poxvirus antigens. J Virol. 2010;84:3552–3561. doi: 10.1128/JVI.01572-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridle BW, Boudreau JE, Lichty BD, Brunellière J, Stephenson K, Koshy S, et al. Vesicular stomatitis virus as a novel cancer vaccine vector to prime antitumor immunity amenable to rapid boosting with adenovirus. Mol Ther. 2009;17:1814–1821. doi: 10.1038/mt.2009.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobleigh MA, Buonocore L, Uprichard SL, Rose JK, Robek MD. A vesicular stomatitis virus-based hepatitis B virus vaccine vector provides protection against challenge in a single dose. J Virol. 2010;84:7513–7522. doi: 10.1128/JVI.00200-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbert TW, Geisbert JB, Leung A, Daddario-DiCaprio KM, Hensley LE, Grolla A, et al. Single-injection vaccine protects nonhuman primates against infection with marburg virus and three species of ebola virus. J Virol. 2009;83:7296–7304. doi: 10.1128/JVI.00561-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JA, Buonocore L, Suguitan AL, Jr, Silaghi A, Kobasa D, Kobinger G, et al. Potent vesicular stomatitis virus-based avian influenza vaccines provide long-term sterilizing immunity against heterologous challenge. J Virol. 2010;84:4611–4618. doi: 10.1128/JVI.02637-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido J, Kottke T, Thompson J, Galivo F, Wongthida P, Diaz RM, et al. Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat Biotechnol. 2012;30:337–343. doi: 10.1038/nbt.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HI, Reyes-Vargas E, Delgado JC, Celis E. A potent vaccination strategy that circumvents lymphodepletion for effective antitumor adoptive T-cell therapy. Cancer Res. 2012;72:1986–1995. doi: 10.1158/0008-5472.CAN-11-3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaluza KM, Thompson JM, Kottke TJ, Flynn Gilmer HC, Knutson DL, Vile RG. Adoptive T cell therapy promotes the emergence of genomically altered tumor escape variants. Int J Cancer. 2012;131:844–854. doi: 10.1002/ijc.26447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommelfanger DM, Wongthida P, Diaz RM, Kaluza KM, Thompson JM, Kottke TJ, et al. Systemic combination virotherapy for melanoma with tumor antigen-expressing vesicular stomatitis virus and adoptive T-cell transfer. Cancer Res. 2012;72:4753–4764. doi: 10.1158/0008-5472.CAN-12-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaluza KM, Kottke T, Diaz RM, Rommelfanger D, Thompson J, Vile R. Adoptive transfer of cytotoxic T lymphocytes targeting two different antigens limits antigen loss and tumor escape. Hum Gene Ther. 2012;23:1054–1064. doi: 10.1089/hum.2012.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisgerault N, Kottke T, Pulido J, Thompson J, Diaz RM, Rommelfanger-Konkol D, et al. Functional cloning of recurrence-specific antigens identifies molecular targets to treat tumor relapse. Mol Ther. 2013;21:1507–1516. doi: 10.1038/mt.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roda JM, Wang Y, Sumner LA, Phillips GS, Marsh CB, Eubank TD. Stabilization of HIF-2a induces sVEGFR-1 production from tumor-associated macrophages and decreases tumor growth in a murine melanoma model. J Immunol. 2012;189:3168–3177. doi: 10.4049/jimmunol.1103817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakhova O, Zingg D, Schaefer SM, Hari L, Civenni G, Blunschi J, et al. Sox10 promotes the formation and maintenance of giant congenital naevi and melanoma. Nat Cell Biol. 2012;14:882–890. doi: 10.1038/ncb2535. [DOI] [PubMed] [Google Scholar]
- Zhuang D, Mannava S, Grachtchouk V, Tang WH, Patil S, Wawrzyniak JA, et al. C-MYC overexpression is required for continuous suppression of oncogene-induced senescence in melanoma cells. Oncogene. 2008;27:6623–6634. doi: 10.1038/onc.2008.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata K, Takeda K, Tomita Y, Tagami H, Shibahara S. Downstream region of the human tyrosinase-related protein gene enhances its promoter activity. Biochem Biophys Res Commun. 1992;184:568–575. doi: 10.1016/0006-291x(92)90627-w. [DOI] [PubMed] [Google Scholar]
- Linardakis E, Bateman A, Phan V, Ahmed A, Gough M, Olivier K, et al. Enhancing the efficacy of a weak allogeneic melanoma vaccine by viral fusogenic membrane glycoprotein-mediated tumor cell-tumor cell fusion. Cancer Res. 2002;62:5495–5504. [PubMed] [Google Scholar]
- Gingrich JR, Barrios RJ, Kattan MW, Nahm HS, Finegold MJ, Greenberg NM. Androgen-independent prostate cancer progression in the TRAMP model. Cancer Res. 1997;57:4687–4691. [PubMed] [Google Scholar]
- Overwijk WW, Theoret MR, Finkelstein SE, Surman DR, de Jong LA, Vyth-Dreese FA, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez M, Porosnicu M, Markovic D, Barber GN. Genetically engineered vesicular stomatitis virus in gene therapy: application for treatment of malignant disease. J Virol. 2002;76:895–904. doi: 10.1128/JVI.76.2.895-904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]