

Abstract
Mutations in fukutin-related protein (FKRP) gene cause a wide spectrum of disease phenotypes including the mild limb-girdle muscular dystrophy 2I (LGMD2I), the severe Walker-Warburg syndrome, and muscle-eye-brain disease. FKRP deficiency results in α-dystroglycan (α-DG) hypoglycosylation in the muscle and heart, which is a biochemical hallmark of dystroglycanopathies. To study gene replacement therapy, we generated and characterized a new mouse model of LGMD2I harboring the human mutation leucine 276 to isoleucine (L276I) in the mouse alleles. The homozygous knock-in mice (L276IKI) mimic the classic late onset phenotype of LGMD2I in both skeletal and cardiac muscles. Systemic delivery of human FKRP gene by AAV9 vector in the L276IKI mice, at either neonatal age or at the age of 9 months, rendered body wide FKRP expression and restored glycosylation of α-DG in both skeletal and cardiac muscles. FKRP gene therapy ameliorated dystrophic pathology and cardiomyopathy such as muscle degeneration, fibrosis, and myofiber membrane leakage, resulting in restoration of muscle and heart contractile functions. Thus, these results demonstrated that the treatment based on FKRP gene replacement was effective.
Introduction
Muscular dystrophies (MDs) are a class of hereditary, degenerative disorders of striated muscles. MDs are caused by defects in genes that encode a diverse group of proteins.1 Among them, a subset of MDs shares the same biochemical feature of reduced glycosylation of α-dystroglycan and diminished laminin binding activity, collectively termed dystroglycanopathies.2 They are a heterogeneous group of autosomal recessive diseases with a wide spectrum of clinical severities.3,4
Dystroglycan (DG) consists of a heavily glycosylated extracellular α subunit (α-DG) and a transmembrane β subunit (β-DG). α-DG and β-DG are encoded by a single gene and post-translationally cleaved to generate the two subunits.5 α-DG is a cell surface receptor for several extracellular matrix proteins such as laminin, agrin, and perlecan in muscle,6 and neurexin in brain.7 The trans-membrane β-DG anchors α-DG at the cell surface and binds intracellularly to dystrophin, which in turn binds to the actin cytoskeleton. Thus, DG functions as a molecular anchor, connecting the extracellular matrix with the cytoskeleton across the plasma membrane in skeletal muscle.8 Deficiencies of proteins and enzymes involved in the pathways of α-DG post-translational modification, e.g., glycosylation, are chiefly responsible for dystroglycanopathies. The dystroglycanopathies account for the second most common group of MDs, only less frequent than Duchenne muscular dystrophy (DMD).9 Currently, more than six proteins have been identified in DG glycosylation pathway.10,11,12,13,14,15 Specifically, protein O-mannosyltransferase 1 (POMT1), POMT2, and protein O-linked mannose N-acetylglucosaminyltransferase 1 (POMGnT1) are shown to initiate the first two steps of the O-mannosylation pathway, a unique type of O-linked glycosylation in α-DG.16 Fukutin-related protein (FKRP) and LARGE (like-acetylglucosaminyltransferase) are involved in phosphorylation of O-linked mannose in α-DG, which is required for laminin binding.5,17
Mutations in the FKRP gene cause a wide spectrum of disease severity in muscular dystrophies. There is a general genotype–phenotype correlation in patients, despite the fact that hypoglycosylation of α-DG can be variable in FKRP associated dystroglycanopathy.18 The milder, most common form is limb girdle muscular dystrophy 2I (LGMD2I), characterized by childhood or adult onset with a relatively benign disease course.4 Individuals with LGMD2I are mostly homozygous for the Leu276Ile (nucleotide C826A) mutation of FKRP and show a variable but apparent reduction in α-DG immunolabeling.18 LGMD patients with Duchenne-like severity typically have a moderate reduction in α-DG and contain compound heterozygous mutations, including the common C826A FKRP mutation and other missense or nonsense mutations.18,19 The more severe form is the congenital muscular dystrophy type 1C (MDC1C) characterized by early onset, inability to walk, and highly elevated serum creatine kinase (CK) levels.4 The MDC1C is a compound heterozygote between a nonsense and a missense mutation or two missense mutations.18 The most severe forms of FKRP deficiency containing missense mutations are clinically similar to Walker-Warburg syndrome (mutation in POMT1 gene) or muscle-eye-brain disease (mutation in POMGnT1 gene), which has striking structural brain and eye defects.20 Until recently, the lack of viable animal models has impeded research in pathogenesis and therapy. Deletion of the mouse FKRP gene or homozygous nonsense mutations are embryonic lethal.21 To circumvent embryonic lethality, we previously utilized gene delivery of RNA interference technology to knock down FKRP expression, via postnatal gene delivery, creating a FKRP deficient animal model.22 More recently, the FKRP P448L mutation knock-in animal model was generated, which resembles clinically severe MDC1C phenotypes with brain and eye involvement.21,23
In the current study, a mild LGMD2I animal model is generated by knocking in the common human L276I mutation, producing homozygous mice (L276IKI) that carry this mutation. The mouse model has been extensively characterized for their phenotypes, which are found to closely recapitulate resemble the clinical pathology and phenotypes of the LGMD2I patients. Using the L276IKI mice, we have tested adeno-associated virus (AAV)-mediated gene therapy for the treatment of LGMD2I. AAV vectors are highly efficient in transducing skeletal and cardiac muscle cells by in vivo gene delivery.24,25 The vectors have been broadly used to study other types of muscular dystrophies, such as DMD25,26,27 and laminin-α2 deficient congenital muscular dystrophy.28 Here, we have used AAV9 vector to systemically deliver the FKRP gene into the neonatal and adult L276IKI mice for long-term gene expression and therapeutic benefits. We found that overexpression of FKRP in both skeletal and cardiac muscle effectively restored the biochemical deficiency and normalized glycosylation of α-DG. Body wide FKRP gene expression showed no discernable toxicity, but prevented the development of dystrophic pathology in mice treated neonatally, and effectively ameliorated the pathology in mice treated at the adult age. More importantly, overexpression of FKRP resulted in contractile function recovery of skeletal muscles as well as cardiac muscles. Therefore, due to the lack of any effective treatment for FKRP deficient patients, the new mouse model and FKRP gene transfer study will facilitate our understanding of LGMD2I molecular pathogenesis as well as therapeutic developments.21,22
Results
FKRP L276IKI knock-in mice show both muscular dystrophy and cardiomyopathy
The LGMD2I mouse model was generated by knocking in the human mutation (nucleotide C826 to A) into the coding sequence of the mouse FKRP gene, resulting in amino acid replacement L276I.21,22 The founder mice were bred to produce homozygous FKRP knock-in mice with hybrid 129/C57BL6 background, termed hybrid L276IKI line. Additionally, the hybrid line was further back-crossed with the syngeneic C57/BL6 mice for more than 10 generations to produce a genetically homogeneous line, termed B6 L276IKI. The growth rate and life span of the homozygous mice were indistinguishable from their heterozygous littermates. Both hybrid and B6 L276IKI homozygous mice were bred normally.
Both lines of homozygous L276IKI mice displayed late onset of mild muscular dystrophic pathology. Muscle histology between heterozygous and homozygous mice was minimal at 3 months of age (Figure 1a). At 6 months of age, the centrally located nuclei (CN) in myofibers started to increase in the homozygous group (homo 9.4 ± 1.8% versus hetero 1.4 ± 0.6% in quadriceps, rectus femoris, muscle), ultimately reaching ~30% central nucleation in mice older than 9 months (Figure 1b). Compared to the severe phenotype of the FKRP P448LKI model, the muscle pathology, such as fibrosis and cellular infiltration, in FKRP L276IKI mice were often focal and not uniform in the same muscle groups. However, the degree of myofiber central nucleation and the overall interstitial fibrosis increased consistently with age.
Figure 1.

Characterization of homozygous FKRP L276IKImice. (a) Muscle pathology corresponding to age was displayed for both heterozygous and B6 L276IKI homozygous mice. Cryo-thin-sections of the quadriceps muscle from different ages were analyzed with H&E staining. Large amount of centrally nucleated myofibers were observed in the quadriceps of both the 6- and 9-month-old B6 L276IKI homozygous mice compared to the heterozygous mice. (b) Quantitation of centrally nucleated myofibers in homozygous versus heterozygous mice at different ages (n = 8; *P < 0.05; **P < 0.001). (c) Muscle leakage and fibrosis progression seen in 18 months old homozygous FKRP L276I mice. The diaphragm of homozygous FKRP L276IKI mice (18-month-old) displayed the most dramatic dystrophic pathology as revealed by the Evans blue dye leakage, mononuclear cell infiltration (H&E staining), and fibrosis infiltration (collagen staining). Sirius red was used to stain collagen (red), and Fast green was utilized for myofiber staining (green). (d) Quantification of collagen content in the diaphragm muscle shown in Figure 1c. The collagen content was expressed as a percentage of the collagen (Sirius red staining) versus the noncollagen protein (fast green staining). Statistics were calculated by an unpaired t-test with Welch's correction (*P < 0.05; n = 4). (e) The serum creatine kinase level was increased in homozygous mice. All homozygous B6 L276IKI mice had CK levels greater than 1,000 units/l, and revealed a trend that the serum CK levels increase with age (*P < 0.05 with one-way analysis of variance (ANOVA); n = 4–9). (f) The treadmill running experiment indicated homozygous mice ran shorter distance than heterozygous control (**P = 0.01 with one-way ANOVA; n = 5–9). (g) The in vitro eccentric contraction force measurement. There was no difference in force production after 10 cycles of eccentric contraction challenge in the TA muscles of the homozygous mice versus heterozygous mice (n = 6, P > 0.05).
The diaphragm of homozygous L276IKI mice was the muscle most severely affected, as shown by pronounced membrane leakage and fibrosis, especially in the older mice (Figure 1c). These results are similar to that of the mdx mice of DMD model.29 Evans blue dye was injected into 18-month-old heterozygous and homozygous mice. Evans blue is a widely used vital red fluorescent dye that is taken up by the cells with leaky cell membrane.25 We found that the diaphragm was the major muscle that took up the dye (Figure 1c). Other muscles including tibialis, gastrocnemius, quadriceps, and heart had limited dye uptake as shown by increased intensity of interstitial red fluorescence (Supplementary Figure S1; Figure 1c). Additionally, we observed significant increase in fibrosis of the diaphragm, which was revealed by Sirius red/fast green staining (Figure 1c). A Sirius red/fast green dye staining technique, which binds selectively to collagen and noncollagen protein, respectively, is a commonly used assay to display and quantify the amount of collagen deposition occurring in the tissues. Quantitative collagen content (an indicator of the extent of fibrosis) analysis showed an increase in the diaphragm of the homozygous versus heterozygous mice (2.4 ± 0.6% versus 1.3 ± 0.2%; n = 5, P < 0.05) (Figure 1d). Similarly, heart tissue fibrosis also increased in the homozygous B6 L276IKI mice (Figure 1c), signifying the presence of cardiomyopathy.
Another hallmark of dystrophic pathology in homozygous L276IKI mice is elevated serum CK levels, which indicate muscle damage.30 While the average serum CK levels in wild-type and heterozygous FKRP L276IKI mice were below 300 units/l, all homozygous B6 L276IKI mice (with C57/BL6 background) had CK levels greater than 1,000 units/l (Figure 1e). Moreover, a trend emerged as the serum CK levels increased with age (Figure 1e), which was consistent with the histopathology observations (Figure 1a).
The homozygous L276IKI mice had impaired muscle functions, such as shorter treadmill running distance and lower in vitro isometric force production. The running distance of the 9-month-old hybrid background heterozygous mice was 697 ± 97 meters, while the running distance of the age- and sex-matched homozygous L276IKI litter mates was only 353 ± 172 meters (n was 5 in the first group and 9 in the second group, P < 0.01) (Figure 1f). The in vitro contraction measurement of the TA muscles showed a decrease in the specific isometric tetanic force of the 13-month-old hybrid homozygous L276IKI mice compared to their heterozygous littermates (23.5 ± 2.5 versus 32.5 ± 4.5 N/cm2, P < 0.05, n = 5) (Supplementary Figure S2). Unlike the dystrophin-deficient mdx mice,31 the L276IKI mice did not show a significant difference from its control heterozygous group during the 10-cycle eccentric contraction assay (lengthening activation) (Figure 1g), again suggesting mild dystrophic phenotypes. The liver and kidney functions of the homozygous mice had no abnormalities as shown by blood urea nitrogen (BUN) and serum alanine transaminase (ALT) levels (Supplementary Figure S3).
Systemic FKRP gene transfer restored glycosylation of α-dystroglycan
Our next step was to investigate the therapeutic intervention by systemic gene transfer of FKRP. Mouse codon-optimized FKRP gene and the ubiquitous CB promoter (CMV enhancer/chicken β-actin promoter) were used for strong and sustained transgene expression.32 Myc tag was fused to the C-terminus of the FKRP coding sequence to facilitate detection. The optimized FKRP gene was packaged into AAV9 vector which would efficiently cross the blood vessel barrier and transduce whole-body muscle cells after systemic delivery.32
First, we delivered the AAV9-CB-FKRP vector into neonatal mice to see whether or not overexpression of FKRP can prevent muscular dystrophic pathology in early age. AAV9-CB-FKRP-myc (1 × 1011 vector genomes (vg)/mouse) was injected intraperitoneally into 3-day-old homozygous L276IKI mice (with hybrid background). At different time points (1 month and 9 months) after vector delivery, some of the mice (three from each group) were sacrificed for examination of transgene expression and evaluation of pathology improvement. Immunofluorescent (IF) staining against FKRP in cryo-thin-section of muscle tissue displayed strong transgene expression in all of the major muscle tissues examined (Figure 2a). FKRP expression appeared as punctate dots in the cytoplasmic compartment of the myofibers (Figure 2a), which was previously shown in the Golgi and endoplasmic reticulum (ER).23 Also, FKRP expression in skeletal muscles was confirmed by western blot (Figure 2b) and anti-myc staining (Supplementary Figure S4).
Figure 2.
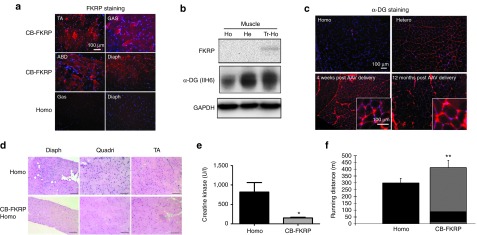
Improvement of muscle pathology and function after delivery of AAV9-CB-fukutin-related protein (FKRP)-myc vector in neonates. The AAV9-CB-FKRP-myc vector (1 × 1011 vg/pup) was delivered into 3-day-old homozygous pups. (a) Overexpression of FKRP on AAV-treated muscle. The mice (three per group) were sacrificed 1 month after adeno-associated virus (AAV) treatment, and immunofluorescent staining against FKRP was performed on the indicated tissues. ABD, abdominal muscle; Diaph, diaphragm muscle; GAS, gastrocnemius muscle; TA, tibialis muscle. (b) Western blot analysis of FKRP expression and glycosylated α-DG expression in quadriceps muscles from indicated mice. A total protein of 15 µg was loaded in each lane. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was the loading control here. (c) Hypoglycosylation of α-DG in quadriceps muscles from homozygous FKRP L276IKI mice and the restoration of α-DG glycosylation by AAV vector treatment. The insets were large magnification (×40) of the same area (rectus femoris quadriceps muscle) (d) Muscle pathology improvement revealed by H&E staining. The mice were sacrificed 9 months after treatment for pathology examination. (e) The serum creatine kinase levels were significantly decreased after AAV treatment. (*P < 0.05; n = 5 for homo group, and n = 9 for AAV9-CB-FKRP-myc treatment group). (f) Delivery of AAV9-CB-FKRP vector in neonatal mice (C57/BL6 background) also improved muscle function when evaluated by the treadmill running test (**P < 0.01, n = 4–8).
Similar to other dystroglycanopathy animal models,2,21 hypoglycosylation of α-DG is a feature of homozygous L276IKI mice. To examine if overexpression of FKRP in the skeletal muscle of homozygous L276IKI mice would restore the glycosylation of α-DG, we performed immunofluorescent staining and western blot with the widely used monoclonal antibody IIH6. IIH6 only recognizes the functional glycan epitopes of α-DG.2 The homozygous L276IKI skeletal muscle displayed weak or background levels of glycosylated α-DG by IF staining (Figure 2c). The weak glycosylation signal of α-DG was also revealed by western blot, indicating hypoglycosylation of α-DG in the skeletal muscle of the homozygous L276IKI mice (Figure 2b). In contrast, the AAV-CB-FKRP-myc treated homozygous muscle displayed strong immunofluorescent signal after IIH6 antibody staining. The glycosylation of α-DG was effectively restored even 12 months after vector delivery (Figure 2c), making it indistinguishable from the heterozygous controls. These results demonstrated that overexpression of FKRP in skeletal muscle via AAV vector delivery fully restored the glycosylation of the α-DG in the LGMD2I mouse model.
Systemic FKRP gene transfer effectively ameliorates muscle pathology
We found that overexpression of FKRP in the homozygous L276IKI mice (treated at the neonatal age) was able to normalize muscle pathology, as shown by H&E staining (Figure 2d). The AAV9-CB-FKRP-myc treatment of theL276IKI mice fully prevented the formation of centrally located nuclei in the myofibers and the mononuclear cell infiltration in the muscles (Figure 2d). As expected, the treated mice's serum CK levels returned to normal (151 ± 57 units/l) in comparison to their untreated counterparts (816 ± 546 units/l, P < 0.05, n = 5–9) (Figure 2e). Furthermore, the muscle function improved after gene therapy in the hybrid homozygous L276IKI mice. Treadmill tests showed that the treated homozygous mice ran much further distances (412.0 ± 53.7 meters) than their untreated counterparts (299.8 ± 33.7 meters, P < 0.001, n = 4–8).
Considering L276IKI mice manifested delayed disease onset, we also performed gene therapy in adult homozygous mice, in order to examine whether therapeutic effects could be observed after the dystrophic pathology already appeared. The AAV9-CB-FKRP-myc vectors were delivered into 9-month-old homozygous L276IKI mice (which corresponds to a 25-year-old human with similar symptoms) at a dosage of 6 × 1013 vg/kg via tail vein injection. Three months after vector delivery, three mice from each group were sacrificed and their tissues were utilized for pathology analysis. As anticipated, the treatment of these older mice was also able to efficiently restore the muscle morphology, shown by the H&E staining (Figure 3a). The centrally nucleated myofibers were significantly decreased in the treated homozygous mice (2.98 ± 0.82%) as compared to their untreated homozygous counterparts (13.89 ± 4.73%, P < 0.001, n = 3) (Figure 3c). The vector treatment ameliorated fibrosis infiltration as revealed by Masson's trichrome staining (Figure 3b). Quantitative data of collagen content failed to show statistical significance (P > 0.05, n = 5), but did indicate there was a decreasing fibrosis trend (Figure 3d). The treadmill running experiment demonstrated muscle function improvement by the treatment (Figure 3e). The treated homozygous mice ran much longer distance than the untreated littermates (697 ± 97 m versus 353 ± 172 m), and the running distance was comparable to the heterozygous controls (673 ± 108 m, P < 0.01, n = 5–9). These results indicated that delivering AAV9 vector encoding FKRP gene could be therapeutic even in older homozygous mice that already displayed dystrophic pathology.
Figure 3.

Delivery of AAV vector encoding fukutin-related protein (FKRP) in adult homozygous mice ameliorated dystrophic pathology and improved fibrosis infiltration. The AAV9-CB-FKRP vector was delivered into 9-month-old homozygous mice (hybrid background), and the mice were sacrificed 3 months after treatment. (a) H&E staining indicated that the skeletal muscle pathology was ameliorated after adeno-associated virus (AAV) treatment. Gas, gastrocnemius muscle; TA, tibialis muscle; Quadri, quadriceps (rectus femoris) muscle. (b) Masson's trichrome staining revealed a fibrosis infiltration reduction by the delivery of AAV vector encoding FKRP. (c) Quantification of centrally nucleated myofibers. All of the gastrocnemius muscle fibers (a minimum of 267 muscle fibers) were counted on each photo (objective ×10) of H&E staining, and at least three photos were included for each mouse. The percentile of centrally nucleated fibers was defined as the percentage of centrally nucleated fibers versus the total counted myofibers. The one-way analysis of variance (ANOVA) test was utilized for statistical analysis (**P = 0.0002; three animals were used for each group). (d) Quantification of collagen content in Gastrocnemius muscle. This analysis failed to show statistical significance (P = 0.08, n = 5), but did indicate there was a decreasing fibrosis trend. (e) Motor function recovery by AAV9-CB-FKRP treatment in older homozygous mice. The vector was delivered into 9-month-old male homozygous mice (hybrid background), and the treadmill test was performed 4 months after treatment (**P < 0.01 with one-way ANOVA; n = 5 for CB-FKRP homo and hetero groups, and n = 9 for homo group).
Prevention of cardiomyopathy and improvement of heart contractile capacity
It has been reported that LGMD2I patients often develop cardiomyopathy.33 In homozygous FKRP L276IKI mice, focal and scattered fibrosis was also observed in the cardiac muscle (Figure 1c). Therefore, we examined if overexpression of FKRP in the heart could improve cardiomyopathy. To achieve robust and persistent cardiac expression, we utilized an improved cardiac- and skeletal-muscle specific synthetic promoter,34,35 in addition to the ubiquitous CB promoter. The muscle-specific promoter contained a synthetic muscle promoter35 and an 100 bp fragment downstream of the +1 site in the chicken skeletal α-actin promoter, which was designated as syn100. The AAV9-syn100-FKRP and AAV9-CB-FKRP vectors were delivered into the neonatal homozygous L276IKI mice (3 days old). The untreated littermates were used as the control. Seven months after vector delivery, the mice were subjected to echocardiography analysis and subsequently FKRP gene expression analysis. AAV9-CB-FKRP and AAV9-syn100-FKRP vector rendered strong FKRP expression in both cardiac and skeletal muscle as evidence by the IF staining (Figure 4a) and the western blot (Figure 4b). Similar to the expression pattern seen in skeletal muscle, the expression of FKRP in the heart muscle cytoplasm appeared as punctate dots, and the wild-type FKRP in heterozygous and homozygous mice could not be detected by the anti-FKRP antibody (Figure 4a). As expected, overexpression of FKRP by AAV9-syn100-FKRP and AAV9-CB-FKRP in homozygous mice heart restored the glycosylation of α-DG.
Figure 4.
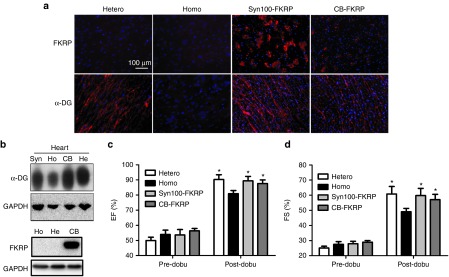
Restoration of α-DG glycosylation and improvement of cardiac contractile capacity by delivery of adeno-associated virus (AAV)9 encoding fukutin-related protein (FKRP) gene. The AAV9-syn100-FKRP and AAV9-CB-FKRP vectors (3 × 1011 vg/pup) were delivered into the neonatal mice, and age matched heterozygous and homozygous mice were used as controls. Gene expression and heart function examinations were performed 7 months after vector delivery. (a) Overexpression of FKRP via AAV vector, driven by muscle-specific syn100 promoter or ubiquitous CB promoter, all restored the glycosylation of α-DG in the homozygous FKRP L276IKI mice shown by the IF staining. (b) The western blot further indicated that overexpression of FKRP in homozygous mice completely restored the glycosylation of α-DG. For the western blot of FKRP expression, homozygous mice were treated by AAV9-CB-FKRP vector. (c) The dobutamine stress Echo indicated improvement in ejection fraction (EF) for both syn100-FKRP and CB-FKRP treated groups after dobutamine stimulation. (d) The dobutamine stress Echo displayed enhanced cardiac contractile function in fractional shortening (FS) for both syn100-FKRP- and CB-FKRP-treated groups after dobutamine stimulation. (*P < 0.05, n = 3 for treated group, and n = 8 for hetero and homo groups for both c and d).
Finally, we investigated if gene therapy could improve cardiac function. Similar to the mdx mice, the FKRP L276IKI mice displayed a mild cardiac function deficit, which is not readily detectable by regular echocardiography.36 Therefore, we opted to evaluate the cardiac function by echocardiography (Echo) with a dobutamine challenge. Dobutamine is a sympathomimetic drug that stimulates ß1-adrenergic receptors in the heart, resulting in an increase in heart rate, contractility and demand for myocardial oxygen.37 Clinically, the dobutamine stress test provides a more revealing diagnostic and prognostic examination for cardiomyopathy patients.37 Echocardiography was performed before and 2 minutes after dobutamine administration on the 7-month-old homozygous, heterozygous, AAV9-syn100-FKRP-treated, and AAV9-CB-FKRP-treated homozygous mice.38 Before dobutamine stimulation, no significant difference of cardiac systolic function was found among the four groups of mice as determined by ejection fraction (EF) and fractional shortening (FS) (Figure 4c,d). EF and FS are the most commonly used indexes of left ventricle contractile function. EF represents the volumetric fraction of blood pumped out of the left and right ventricle with each heartbeat, while FS measures the change in the diameter of the left ventricle between the contracted and relaxed cycles. After dobutamine stimulation, however, the EF (89.4 ± 5.1% for syn100 promoter, 87.6 ± 5.3% for CB promoter) and FS (59.75 ± 6.0% for syn100 promoter, 57.09 ± 7% for CB promoter) of the AAV-treated homozygous mice were significantly higher than the untreated homozygous mice (80.9 ± 5.1% for EF, 49.0 ± 4.9% for FS, n = 3–8, P < 0.05), and similar to the heterozygous control littermates (90.2 ± 5.5% for EF, 60.8 ± 8.2%) (Figure 4c,d). These results demonstrated that the heart of the homozygous FKRP L276IKI mice indeed have compromised contractile capacity, and that overexpression of FKRP can completely restore original heart function.
Discussion
Dystroglycanopathies have recently been recognized as the most common form of muscular dystrophies second to Duchenne muscular dystrophy. FKRP-related dystroglycanopathies are associated with a wide range of muscular dystrophies from the most common, mild form of LGMD2I with adult onset to the rare but severe Walker-Warburg syndrome or muscle eye-brain disease with brain and eye involvement.3,18,20 Chan et al. 21 created a human FKRP mutation P448L knock-in mouse model P448LKI, which represents the severe congenital muscular dystrophies with early onset and with brain and eye involvement. Here, we described the FKRP L276IKI model with late onset and mild muscular dystrophic pathology reminiscent of the LGMD2I patients. The FKRP L276IKI model mimicked the LGMD2I pathology in nearly all aspects, including the major histopathology as well as the biochemical deficiencies. Systemic gene delivery of FKRP in either neonatal mice or adult mice was able to achieve body wide FKRP gene expression and restoration of the α-dystroglycan glycosylation. Biochemical correction also resulted in histopathological correction for the homozygous FKRP L276IKI mice. Furthermore, both skeletal muscle and cardiac muscle functions were recovered as shown by the treadmill running distance and the echocardiography results. Overexpression of FKRP did not reveal any discernable toxicity, suggesting gene therapy with FKRP gene replacement is a safe approach. Therefore, FKRP L276IKI represents a useful model that can be used to study the mechanisms of pathogenesis as well as new therapies in the most common form of LGMD.
The disease phenotype of the homozygous B6 FKRP L276IKI only slightly differed from that of the homozygous hybrid FKRP L276IKI. Only the serum CK level of homozygous B6 FKRP L276IKI was higher (Figure 1e) than the hybrid FKRP L276IKI (Figure 2e); therefore, minimal strain variations was indicated in this study. Furthermore, the mild FKRP L276IKI model of LGMD2I can also be converted into a more severe animal model by creating a compound heterozygous mutation line through breeding with a severe FKRP mouse line, which carries other missense mutations or a stop codon mutation. In human patients, some correlation between FKRP mutation and disease phenotype has been found.18 For example, the L276I (C826A) mutation causes the mild phenotype LGMD2I, but the P448L mutation causes severe congenital muscular dystrophy. The homozygous P448LKI mouse model recreates the severe clinical phenotype.21 The compound heterozygous mutation between the common L276I and either other missense mutations or a nonsense mutation causes the intermediate Duchenne-like severity; the compound heterozygous FKRP mutation between a nonsense mutation and a missense mutation or one that carries two missense mutations triggers the onset of the severe phenotype, with or without eye and brain involvement.18
In addition to the muscle pathology, cardiomyopathy is often found in LGMD2I patients.39,40 We also observed cardiac fibrosis and cell infiltration in the FKRP L276IKI model. However, echocardiography did not detect any significant difference between the homozygotes and their heterozygous carrier control mice. No differences were found in the gene therapy treated mice either, despite overexpression of the FKRP gene in the heart. This phenomenon brought to mind previous findings in the mdx mice, which also displayed cardiac fibrosis and cardiomyopathy, but echocardiography variations were insignificant until mice reached an older age.36 Nonetheless, the contractile function deficit of the mdx heart can be exacerbated becoming readily detectable when the cardiac stress test was induced by dobutamine treatment.36 Similarly, the FKRP L276IKI mice also displayed significant cardiac function deficits by dobutamine-induced cardiac stress, as shown by the echocardiography (Figure 4). Gene therapy treated mice showed functional improvement compared to the heterozygous controls, suggesting efficient function recovery by FKRP overexpression without evidence of cardiac toxicity.
We did not see systemic toxicity associated with either AAV9-CB-FKRP vector that contains a ubiquitous promoter or the AAV9-Syn100-FKRP vector that contains a striated muscle-specific promoter. Furthermore, no additional functional improvements were observed between the syn100 promoter and the CB promoter. Although the FKRP gene is expressed in many nonmuscle tissues,9 the liver and kidney functions were found to be normal in the mutant mice, which was verified by their normal serum ALT and BUN levels (Supplementary Figure S3). Understandably, a striated muscle-specific promoter is preferable for the purpose of systemic gene therapy. However, nonspecific expression of FKRP in the liver and the kidney was not found to cause any harm and could even provide beneficial effect to those tissues. Previously, overexpression of the LARGE gene induced hyperglycosylation of α-DG in skeletal and cardiac muscle causing toxicity.9 LARGE has a similar function to FKRP, since it phosphorylates the O-linked mannose on α-DG. Nevertheless, we did not observe similar hyperglycosylation effects associated with the overexpression of FKRP in our L276IKI model, suggesting FKRP gene therapy has a large safety margin and is an excellent approach for treating FKRP deficiency and other related dystroglycanopathties.
Materials and Methods
Generation of the FKRP animal model and genotyping. The method used to create the knock-in mice was carefully described previously.21 Briefly, the FKRP KI mice were generated by the UNC core facility. The targeting vector was engineered with a c.826 C>A point mutation resulting in an amino acid change from leucine to isoleucine at position 276. The neomycin resistant (Neor) cassette was placed into intron 2 about 140 bp upstream from the coding exon 3. The PCR primers for genotyping the heterozygous and homozygous mice were listed as follows: FKRP-WT-F: TTC TTG CCT CGG ATT GTG TAT G; FKRP-WT-R: TCA CTC TCC AAG GGC CTA CAG C; Neo-F-FKRP: AAC AAG ATG GAT TGC ACG CAG G; Neo-R-FKRP: TGG TCG AAT GGG CAG GTA G.
Plasmid construction and AAV vector production. The codon optimized human FKRP cDNA with myc tag fragments was synthesized from a commercial company (GeneArt, Grand Island, NY). By digesting with restriction enzymes, NotI and SalI, the cDNA fragments were cohesively ligated to the AAV backbone vector pXX-UF1, containing a CB promoter.41,42 The final construct was named pXX-UF1-CB-FKRP-myc. Syn100 was based upon a synthetic muscle promoter (SP c5-12), which was screened from randomly assembled myogenic element libraries.35 To increase tissue specificity and promoter strength, the downstream +100 bp sequence (cga gct acc cgg agg agc ggg agg cgt ctc tgc cag cgg tcc gac gcg cag tca gca cca ggt agg tgg gca ccg cgc cgt gcc gtg ccg tgc cgt gccg) of chicken skeletal α-actin promoter was added to the end of SP c5-12, and the new promoter was designated as syn100. To construct AAV vector plasmid containing syn100 promoter (pXX-UF1-syn100-FKRP), the PCR product of syn100 promoter (flanked by KpnI and HpaI) was cloned into the KpnI and HpaI sites of pXX-UF1-CB-FKRP in order to replace the CB promoter.
AAV vectors were produced by triple transfection method43 and the virus was purified by polyethylene glycol (PEG) precipitation followed by CsCl centrifugation.44 The virus titer was determined by both dot-blot and real-time PCR methods.45,46 The concentration of the viral vectors was in the range of 2 × 1012 to 5 × 1012 vgs per milliliter (vg/ml).
Collagen staining and quantification. Two methods, Masson's Trichrome staining and Sirius red/Fast green staining, were used for collagen display. The Masson trichrome stain kit was commercially available (IMEB, San Marcos, CA; cat# K7228), and the instructor's protocol was strictly followed. For Sirius red/Fast green staining, first, 10-μm cryo-section tissues were fixed with ice-cold acetone for 5 minutes. Following three repeated washings with tap water, the slides were then stained in 0.1% fast green for 15–20 minutes, and next in 0.1% Sirius red for 4 minutes. The stained slides were subjected to either dehydration and mounting for examination and pictures or elution for collagen quantification.
The collagen and noncollagen proteins were eluted from the stained slides using a dye extraction solution (a 1:1 mixture of 0.1 N NaOH and methanol) with gentle pipetting. Then, samples were quantified by protein measurement at wavelengths 540 and 605 nm. Formulas to calculate the collagen and total noncollagen protein are as follows: collagen (µg/section) = (A540 nm−(A605 nm × 0.291))/37.8 × 1,000, where A540 nm and A605 nm are the absorbance at 540 and 605 nm, respectively; noncollagen protein (µg/section) = A605 nm/2.04 × 1,000. The ratio of collagen to noncollagen protein was presented in the Figures 1d and 3d.
Mice and vector administration. All protocols involving animal experiments were approved by the University of North Carolina Animal Care and Use Committee. C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). The L276I FKRP knock-in mice were generated and adapted from the UNC core facility. For systemic studies, the AAV serotype 9 FKRP vectors were delivered into the neonates (3–5 days old, 10 mice/group) of B6 background L276I FKRP KI mice by intraperitoneal injection (total 1 × 1011 vg/mouse).47 For adult injection, the vectors (6 × 1013 vg/kg) were delivered into the 9-month-old male mice through tail vein injection.
Immunofluorescent staining, western blot, and histology examination. The immunofluorescent staining was performed according to the previously published protocol.31,48 Since the anti-α-DG antibody (kindly provided by Dr Kevin P Campbell) was a monoclonal antibody (1:50 dilution), the mouse on mouse blocking reagent (VECTOR MKB-2213) was applied to the 8-µm cryo-section tissues right after the regular blocking procedure (with 10% horse serum) to prevent high background staining. For FKRP staining, the cryo-thin section tissues were fixed in 4% paraformaldehyde for 15 minutes. The rabbit polyclonal anti-FKRP antibody (raised against the last 15 residues of mouse FKRP sequence, NPE YPN PAL LSL TGG, kindly provide by Dr Lu's laboratory) was used at 1:400 dilution. The rabbit polyclonal anti-myc antibody was from Abcam (Cambridge, MA; Catalog # ab9106). It was applied at 1:200 dilution. Photographs were taken with a Nikon Eclipse TE300 microscope, using a SPOT RT SLIDER digital camera (Diagnostic Instrument, Life Technologies, Grand Island, NY).
The protocol of western blot against α-DG has been described previously.21 Briefly, the total proteins were dissolved in TX-100 buffer (1% Triton X-100, 50 mmol/l Tris, PH 8.0, 150 mmol/l NaCl, 0.1% sodium dodecyl sulfate (SDS)) supplemented with protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). For the western blot against FKRP, total proteins were extracted from tissues of interest using lysis buffer containing 4% SDS, 125 mmol/l Tris PH 8.8, 4% glycerol, 100 mmol/l DTT (dithiothreitol), 0.01% BPB (bromophenol blue), and protease inhibitor cocktail. Protein concentration was determined by BCA protein assay kit (Pierce, Rockford, IL; Cat # 23227). Proteins (15 µg for each sample) were separated by 8–10% SDS–polyacrylamide gel electrophoresis (PAGE) and transferred to polyvinylidene fluoride membranes and probed with α-DG antibody (1:1,000), or FKRP antibody (1:4,000), and developed with horseradish peroxidase–enhanced chemiluminescence.
H&E staining was performed for histology examination. In order to calculate the centrally located nuclei, three images (objective lens ×10) were taken from each muscle. All the fibers (roughly 1,000 fibers from each muscle) were counted from printed images.
Serum creatine kinase enzyme assay. Serum CK enzyme activities were measured with a creatine kinase reagent kit (Pointe Scientific, Canton, MI; catalog # C7512-120). The procedure was followed exactly according to the manufacturer's instruction.
In vivo myofiber plasma membrane integrity test. Evans blue dye (10 mg/ml phosphate-buffered saline) was injected into the tail vein of heterozygous and homozygous FKRP L276I mice at the dose of 0.1 mg/g of body weight.25 After dye injection, mice were subjected to treadmill running (at a 15° downward angle at a rate of 15 m/minute) for 20 minutes. At 16 hours after Evans blue injection, the mice were sacrificed and muscles were collected and cryo-sectioned. Evans blue dye-positive myofibers were observed under the fluorescent microscope with rhodamine filters.
Muscle function analysis. The fore limb grip force was performed using the digital force gauge (Chatillon Ametek, Largo, FL) as previously described.49 The treadmill tests were described in our previous study.28 Briefly, both treated and nontreated mice were placed on a motorized treadmill (Harvard Apparatus, Holliston, MA; model 7054) to run at a 15° downward angle at a rate of 15 m/minute. The test was stopped after the animal spent 5 seconds sitting on the shock grid without attempting to re-enter the treadmill.
The in vitro contraction functional measurement was performed according to the previously published protocol, except for using a different model of transducer (Model 300B, dual mode; Aurora Scientific, Aurora, Ontario, Canada).48 Briefly, the TA muscle was isolated by removing the overlying bicep femoris and gently opening the fascia of the anterior compartment. The distal portion of the TA tendon was secured with a 4-0 silk suture, and the entire TA was removed with its tibial origin intact. The TA tibial origin was fixed by securing the head of the tibia with a vascular clamp mounted in series to a micropositioner near the base of the tissue chamber. The tissue chamber was constantly perfused with mammalian Ringer's solution aerated with 95% O2–5% CO2 and maintained at 25 °C. Care was taken to position the tibia in a vertical orientation so that the TA fibers were in vertical alignment. The muscle was stimulated by using the single pulse program (Stimulator model No 701A and A/D interface). Then, the muscle length was adjusted, and the muscle was restimulated with the single pulse program in order to find the optimal wave length and maximal twitch force. Two minutes of resting time were given between stimulations. Tetanic force measurement and eccentric contraction testing were performed using fixed protocols specifically for tetanic stimulation and stretch stimulation. For tetanic force measurement, different stimulating frequencies (50, 75, 100, and 125 Hz) were utilized, and the maximal tetanic force (Po) was obtained. For eccentric contraction test (10 cycles), the specific value of individual muscle length was entered into the program to guarantee an accurate lengthening stretch. A 2-minute resting period was given between stimulation cycles. Following these measurements, the stimulated muscle was weighed on an analytic balance (Mettler Toledo, Columbia, MD; D521512), after the tendon and bone attachments were removed and the muscle was blotted dry. The specific tetanic forces (N/cm2) were calculated using the following formula:
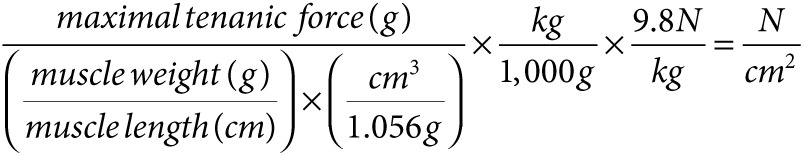 |
Echocardiography. The echocardiography (Echo) analysis was described previously.38 Dobutamine was purchased from Sigma-Aldrich (catalog # D0676 -10MG). Experimental mice were sedated with 2.5% avertin (150 ìl/10 g body weight). The freshly diluted dobutamine (2.25 µg/g body weight) was immediately injected into the mice via intraperitoneal delivery. The heart rate was increased by roughly 100 beats per minute after dobutamine stimulation. The second Echo was performed 2 minutes after dobutamine stimulation once the heart rate remained stable. The three M-mode graphs were saved for analysis from each Echo. There were three to eight mice involved in each group.
Statistical analysis. Values are expressed as means ± SD. Welch's t-test was applied when comparing two groups. When comparing three groups, one-way analysis of variance plus Dunnett post-test was applied using Graph Pad Prism software (GraphPad Software, La Jolla, CA). A P < 0.05 was considered statistically significant.
SUPPLEMENTARY MATERIAL Figure S1. The muscle pathology of homozygous FKRP L276IKI mice was focal. Figure S2. The muscle forces generated by homozygous FKRP L276IKI mice were weaker. Figure S3. Serum ALT and BUN levels. Figure S4. Expression of FKRP confirmed by anti-myc staining.
Acknowledgments
We sincerely thank Kevin P Campbell (University of Iowa) for providing the IIH6 antibody. This project was supported by NIH grant R01 NS082536 to X.X. and the McColl-Lockwood Foundation Endowment to Q.L. No conflict of interest to disclose.
Supplementary Material
The muscle pathology of homozygous FKRP L276IKI mice was focal.
The muscle forces generated by homozygous FKRP L276IKI mice were weaker.
Serum ALT and BUN levels.
Expression of FKRP confirmed by anti-myc staining.
References
- Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- Michele DE, Barresi R, Kanagawa M, Saito F, Cohn RD, Satz JS, et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- Godfrey C, Clement E, Mein R, Brockington M, Smith J, Talim B, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130 Pt 10:2725–2735. doi: 10.1093/brain/awm212. [DOI] [PubMed] [Google Scholar]
- Mercuri E, Brockington M, Straub V, Quijano-Roy S, Yuva Y, Herrmann R, et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann Neurol. 2003;53:537–542. doi: 10.1002/ana.10559. [DOI] [PubMed] [Google Scholar]
- Kuga A, Kanagawa M, Sudo A, Chan YM, Tajiri M, Manya H, et al. Absence of post-phosphoryl modification in dystroglycanopathy mouse models and wild-type tissues expressing non-laminin binding form of α-dystroglycan. J Biol Chem. 2012;287:9560–9567. doi: 10.1074/jbc.M111.271767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowe MA, Deyst KA, Leszyk JD, Fallon JR. Identification and purification of an agrin receptor from Torpedo postsynaptic membranes: a heteromeric complex related to the dystroglycans. Neuron. 1994;12:1173–1180. doi: 10.1016/0896-6273(94)90324-7. [DOI] [PubMed] [Google Scholar]
- Sugita S, Saito F, Tang J, Satz J, Campbell K, Südhof TC. A stoichiometric complex of neurexins and dystroglycan in brain. J Cell Biol. 2001;154:435–445. doi: 10.1083/jcb.200105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barresi R, Campbell KP. Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci. 2006;119 Pt 2:199–207. doi: 10.1242/jcs.02814. [DOI] [PubMed] [Google Scholar]
- Brockington M, Torelli S, Sharp PS, Liu K, Cirak S, Brown SC, et al. Transgenic overexpression of LARGE induces α-dystroglycan hyperglycosylation in skeletal and cardiac muscle. PLoS One. 2010;5:e14434. doi: 10.1371/journal.pone.0014434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longman C, Brockington M, Torelli S, Jimenez-Mallebrera C, Kennedy C, Khalil N, et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet. 2003;12:2853–2861. doi: 10.1093/hmg/ddg307. [DOI] [PubMed] [Google Scholar]
- van Reeuwijk J, Janssen M, van den Elzen C, Beltran-Valero de Bernabé D, Sabatelli P, Merlini L, et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J Med Genet. 2005;42:907–912. doi: 10.1136/jmg.2005.031963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran-Valero de Bernabe D, Currier S, Brunner HG. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Human Genet. 2002;71:1033–1043. doi: 10.1086/342975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida A, Kobayashi K, Manya H, Taniguchi K, Kano H, Mizuno M, et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell. 2001;1:717–724. doi: 10.1016/s1534-5807(01)00070-3. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388–392. doi: 10.1038/28653. [DOI] [PubMed] [Google Scholar]
- Brockington M, Yuva Y, Prandini P, Brown SC, Torelli S, Benson MA, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet. 2001;10:2851–2859. doi: 10.1093/hmg/10.25.2851. [DOI] [PubMed] [Google Scholar]
- Manya H, Chiba A, Yoshida A, Wang X, Chiba Y, Jigami Y, et al. Demonstration of mammalian protein O-mannosyltransferase activity: coexpression of POMT1 and POMT2 required for enzymatic activity. Proc Natl Acad Sci USA. 2004;101:500–505. doi: 10.1073/pnas.0307228101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida-Moriguchi T, Yu L, Stalnaker SH, Davis S, Kunz S, Madson M, et al. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science. 2010;327:88–92. doi: 10.1126/science.1180512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SC, Torelli S, Brockington M, Yuva Y, Jimenez C, Feng L, et al. Abnormalities in alpha-dystroglycan expression in MDC1C and LGMD2I muscular dystrophies. Am J Pathol. 2004;164:727–737. doi: 10.1016/s0002-9440(10)63160-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D, Zhang W, Wang W, Wang Z, Yuan Y. Asian patients with limb girdle muscular dystrophy 2I (LGMD2I) J Clin Neurosci. 2011;18:494–499. doi: 10.1016/j.jocn.2010.08.010. [DOI] [PubMed] [Google Scholar]
- Beltran-Valero de Bernabe D, Voit T, Longman C, Steinbrecher A, Straub V, Yuva Y, et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet. 2004;41:e61. doi: 10.1136/jmg.2003.013870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YM, Keramaris-Vrantsis E, Lidov HG, Norton JH, Zinchenko N, Gruber HE, et al. Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum Mol Genet. 2010;19:3995–4006. doi: 10.1093/hmg/ddq314. [DOI] [PubMed] [Google Scholar]
- Wang CH, Chan YM, Tang RH, Xiao B, Lu P, Keramaris-Vrantsis E, et al. Post-Natal knockdown of fukutin-related protein expression in muscle by long-termRNA interference induces dystrophic pathology [corrected] Am J Pathol. 2011;178:261–272. doi: 10.1016/j.ajpath.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Lu PJ, Wang CH, Keramaris E, Qiao C, Xiao B, et al. Adeno-associated virus 9 mediated FKRP gene therapy restores functional glycosylation of α-dystroglycan and improves muscle functions. Mol Ther. 2013;21:1832–1840. doi: 10.1038/mt.2013.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Li J, Samulski RJ. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J Virol. 1996;70:8098–8108. doi: 10.1128/jvi.70.11.8098-8108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Li J, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci USA. 2000;97:13714–13719. doi: 10.1073/pnas.240335297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- Kornegay JN, Li J, Bogan JR, Bogan DJ, Chen C, Zheng H, et al. Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs. Mol Ther. 2010;18:1501–1508. doi: 10.1038/mt.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao C, Li J, Zhu T, Draviam R, Watkins S, Ye X, et al. Amelioration of laminin-alpha2-deficient congenital muscular dystrophy by somatic gene transfer of miniagrin. Proc Natl Acad Sci USA. 2005;102:11999–12004. doi: 10.1073/pnas.0502137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, et al. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- Mokuno K, Riku S, Sugimura K, Takahashi A, Kato K, Osugi S. Serum creatine kinase isoenzymes in Duchenne muscular dystrophy determined by sensitive enzyme immunoassay methods. Muscle Nerve. 1987;10:459–463. doi: 10.1002/mus.880100513. [DOI] [PubMed] [Google Scholar]
- Watchko J, O'Day T, Wang B, Zhou L, Tang Y, Li J, et al. Adeno-associated virus vector-mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum Gene Ther. 2002;13:1451–1460. doi: 10.1089/10430340260185085. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, et al. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. 2005;23:321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- Sveen ML, Thune JJ, Køber L, Vissing J. Cardiac involvement in patients with limb-girdle muscular dystrophy type 2 and Becker muscular dystrophy. Arch Neurol. 2008;65:1196–1201. doi: 10.1001/archneur.65.9.1196. [DOI] [PubMed] [Google Scholar]
- Wang B, Li J, Fu FH, Chen C, Zhu X, Zhou L, et al. Construction and analysis of compact muscle-specific promoters for AAV vectors. Gene Ther. 2008;15:1489–1499. doi: 10.1038/gt.2008.104. [DOI] [PubMed] [Google Scholar]
- Li X, Eastman EM, Schwartz RJ, Draghia-Akli R. Synthetic muscle promoters: activities exceeding naturally occurring regulatory sequences. Nat Biotechnol. 1999;17:241–245. doi: 10.1038/6981. [DOI] [PubMed] [Google Scholar]
- Wu B, Moulton HM, Iversen PL, Jiang J, Li J, Li J, et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc Natl Acad Sci USA. 2008;105:14814–14819. doi: 10.1073/pnas.0805676105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertes H, Sawada SG, Ryan T, Segar DS, Kovacs R, Foltz J, et al. Symptoms, adverse effects, and complications associated with dobutamine stress echocardiography. Experience in 1118 patients. Circulation. 1993;88:15–19. doi: 10.1161/01.cir.88.1.15. [DOI] [PubMed] [Google Scholar]
- He B, Tang RH, Weisleder N, Xiao B, Yuan Z, Cai C, et al. Enhancing muscle membrane repair by gene delivery of MG53 ameliorates muscular dystrophy and heart failure in δ-Sarcoglycan-deficient hamsters. Mol Ther. 2012;20:727–735. doi: 10.1038/mt.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth KG, Willis TA, Bates MG, Dixon BJ, Lochmüller H, Bushby K, et al. Subepicardial dysfunction leads to global left ventricular systolic impairment in patients with limb girdle muscular dystrophy 2I. Eur J Heart Fail. 2013;15:986–994. doi: 10.1093/eurjhf/hft057. [DOI] [PubMed] [Google Scholar]
- Wahbi K, Meune C, Hamouda el H, Stojkovic T, Laforêt P, Bécane HM, et al. Cardiac assessment of limb-girdle muscular dystrophy 2I patients: an echography, Holter ECG and magnetic resonance imaging study. Neuromuscul Disord. 2008;18:650–655. doi: 10.1016/j.nmd.2008.06.365. [DOI] [PubMed] [Google Scholar]
- Li J, Dressman D, Tsao YP, Sakamoto A, Hoffman EP, Xiao X. rAAV vector-mediated sarcogylcan gene transfer in a hamster model for limb girdle muscular dystrophy. Gene Ther. 1999;6:74–82. doi: 10.1038/sj.gt.3300830. [DOI] [PubMed] [Google Scholar]
- Qiao C, Yuan Z, Li J, He B, Zheng H, Mayer C, et al. Liver-specific microRNA-122 target sequences incorporated in AAV vectors efficiently inhibits transgene expression in the liver. Gene Ther. 2011;18:403–410. doi: 10.1038/gt.2010.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuso E, Mingozzi F, Montane J, Leon X, Anguela XM, Haurigot V, et al. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 2010;17:503–510. doi: 10.1038/gt.2009.157. [DOI] [PubMed] [Google Scholar]
- Rohr UP, Heyd F, Neukirchen J, Wulf MA, Queitsch I, Kroener-Lux G, et al. Quantitative real-time PCR for titration of infectious recombinant AAV-2 particles. J Virol Methods. 2005;127:40–45. doi: 10.1016/j.jviromet.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Veldwijk MR, Topaly J, Laufs S, Hengge UR, Wenz F, Zeller WJ, et al. Development and optimization of a real-time quantitative PCR-based method for the titration of AAV-2 vector stocks. Mol Ther. 2002;6:272–278. doi: 10.1006/mthe.2002.0659. [DOI] [PubMed] [Google Scholar]
- Qiao C, Li J, Jiang J, Zhu X, Wang B, Li J, et al. Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum Gene Ther. 2008;19:241–254. doi: 10.1089/hum.2007.159. [DOI] [PubMed] [Google Scholar]
- Xiao X, Li J, Tsao YP, Dressman D, Hoffman EP, Watchko JF. Full functional rescue of a complete muscle (TA) in dystrophic hamsters by adeno-associated virus vector-directed gene therapy. J Virol. 2000;74:1436–1442. doi: 10.1128/jvi.74.3.1436-1442.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojak A, Hammer D, Wolf H, Wagner R. Muscle specific versus ubiquitous expression of Gag based HIV-1 DNA vaccines: a comparative analysis. Vaccine. 2002;20:1975–1979. doi: 10.1016/s0264-410x(02)00081-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The muscle pathology of homozygous FKRP L276IKI mice was focal.
The muscle forces generated by homozygous FKRP L276IKI mice were weaker.
Serum ALT and BUN levels.
Expression of FKRP confirmed by anti-myc staining.