

Abstract
We hypothesized that the combination of oncolytic virotherapy with immune checkpoint modulators would reduce tumor burden by direct cell lysis and stimulate antitumor immunity. In this study, we have generated attenuated Measles virus (MV) vectors encoding antibodies against CTLA-4 and PD-L1 (MV-aCTLA-4 and MV-aPD-L1). We characterized the vectors in terms of growth kinetics, antibody expression, and cytotoxicity in vitro. Immunotherapeutic effects were assessed in a newly established, fully immunocompetent murine model of malignant melanoma, B16-CD20. Analyses of tumor-infiltrating lymphocytes and restimulation experiments indicated a favorable immune profile after MV-mediated checkpoint modulation. Therapeutic benefits in terms of delayed tumor progression and prolonged median overall survival were observed for animals treated with vectors encoding anti-CTLA-4 and anti-PD-L1, respectively. Combining systemic administration of antibodies with MV treatment also improved therapeutic outcome. In vivo oncolytic efficacy against human tumors was studied in melanoma xenografts. MV-aCTLA-4 and MV-aPD-L1 were equally efficient as parental MV in this model, with high rates of complete tumor remission (> 80%). Furthermore, we could demonstrate lysis of tumor cells and transgene expression in primary tissue from melanoma patients. The current results suggest rapid translation of combining immune checkpoint modulation with oncolytic viruses into clinical application.
Introduction
Oncolytic viruses (OV) which replicate selectively in tumor cells are an emerging modality of cancer treatment with promising results in both preclinical studies and clinical trials. The Measles virus vaccine strain (MV) has been developed as a vector platform to target multiple tumor entities and trials recruiting patients with ovarian cancer, glioma, myeloma, and mesothelioma are ongoing.1 Aside from direct cytopathic effects and lysis of tumor cells, interactions of OV with the immune system can trigger systemic antitumor immunity. OV can evoke innate as well as adaptive immune effectors: Pathogen-associated molecular patterns provided by the viral vector induce cytokine production. Oncolysis leads to the release of tumor-associated antigens in an inflammatory milieu which may facilitate induction of cellular immunity.2,3 Recently, we could demonstrate the potential of oncolytic MV to support the induction of a specific antitumor immune response in terms of a tumor vaccination effect.4 OV have been modified to express immunomodulatory transgenes to further enhance these effects.2 Arming of MV with interferon-β5 and granulocyte macrophage colony-stimulating factor4,6 led to improved outcome in preclinical studies. The Vaccinia virus JX-594 and the Herpes virus talimogene laherparepvec (T-VEC), both harboring granulocyte macrophage colony-stimulating factor, have shown promising results in clinical phase 2 and 3 trials.7,8
Antibodies targeting the T cell inhibitory factors cytotoxic T lymphocyte antigen 4 (CTLA-4), programmed death-1 (PD-1) and its ligand programmed death-1 ligand 1 (PD-L1) have been celebrated as a recent breakthrough in cancer immunotherapy.9 The anti-CTLA-4 antibody Ipilimumab improved survival in patients with metastatic melanoma,10 leading to its approval by the US Food and Drug Administration. Antibodies against PD-111 and PD-L112 have shown efficacy against a broad range of advanced tumors. However, not all patients respond and severe immune-related adverse events are frequent in systemic immunotherapy, with Grade 3–4 immune-related adverse events occurring in around 25–30% of patients treated with Ipilimumab.9
We reasoned that tumor-restricted expression of immune checkpoint modulators may limit adverse events. Moreover, combining OV-mediated tumor vaccination with inhibition of the CTLA-4 and PD-1/PD-L1 pathways may prove highly beneficial.
In this study, we have generated MV vectors encoding antibodies against the T cell inhibitory factors CTLA-4 and PD-L1. We have established a new, fully immunocompetent MV-susceptible mouse model of malignant melanoma. In this model, we observe a favorable profile of immune effectors and therapeutic benefits after treatment with MV-aCTLA-4 and MV-aPD-L1. Combining MV with systemic antibody administration also improves therapeutic outcome. We demonstrate oncolytic efficacy of MV-aCTLA-4 and MV-aPD-L1 in a human xenograft model with high rates of complete remissions. Furthermore, we demonstrate viral replication and transgene expression in primary tissue samples from melanoma patients. This study is proof-of-concept that immunovirotherapy of cancer can be enhanced by both vector-mediated and systemic immune checkpoint modulation. The concept can be broadly applied to a range of OV platforms. This work leads the way for future clinical trials combining immune checkpoint modulation with oncolytic viruses.
Results
Cloning and characterization of targeted MV encoding anti-CTLA-4 and anti-PD-L1
Recombinant MVs encoding antibodies against the T cell inhibitory factors CTLA-4 and PD-L1 (MV-aCTLA-4 and MV-aPD-L1) were generated by inserting respective antibody-coding sequences into an additional transcription unit downstream of the hemagglutinin gene (Figure 1a). Control viruses encoding the antibody constant region only (MV-IgG Fc) were cloned analogously. Transgene expression was assessed by immunoblot and ELISA of cell culture supernatants after infection (Figure 1b,c). After infection of human melanoma cell lines Sk-Mel-28 and Mel888, characteristic MV-mediated syncytia formation was observed (Figure 1d).
Figure 1.

Cloning and characterization of recombinant Measles virus (MV) vectors. (a) Schematic representation of recombinant MV genomes. X: empty or EGFP, Y: aCTLA-4, aPD-L1, IgG Fc, or EGFP. H: Measles attachment protein hemagglutinin with native tropism; HαCD20: H retargeted to CD20. (b) Transgene expression. Thirty-six hours after infection at MOI 3, cells culture supernatants were collected. After immunoprecipitation with Protein A Sepharose immunoblot analysis was performed with anti-HA antibody. (c) Binding of MV-encoded aCTLA-4 and aPD-L1 to their cognate antigens was assessed by enzyme-linked immunosorbent assay (ELISA). (d) Syncytia formation and lysis of human melanoma cell lines. Sk-Mel-28 and Mel888 cells were infected with recombinant MV encoding EGFP and aCTLA-4 or aPD-L1 at MOI 1 and images were taken 36 hours p.i. (e) Targeted infection. Parental Vero and B16 as well as Vero-CD20 and B16-CD20 which stably express the CD20 surface antigen were infected with MV-EGFP with unmodified tropism and retargeted MV-EGFP HαCD20 at an MOI of 1. Images of cells 24 hours after infection (Vero, Vero-CD20) and 48 hours after infection (B16, B16-CD20) are shown. Scale bars: 100 µm. Replication and cytotoxicity in (f) human and (g) murine cells. Top panels: Viral growth kinetics. One-step growth curves of parental MV ld-EGFP, MV H-EGFP, MV-IgG Fc, MV-aCTLA-4, and MV-aPD-L1. Cells were harvested at designated time points and progeny viral particles were quantified by titration assays. Bottom panels: Cytotoxic effects. Cell viability was assessed by XTT assay. Mean values from triplicate infections are shown. Error bars indicate standard deviations. Viability of mock-treated cells was set to 100%. XTT, 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide.
While oncolytic efficacy can be addressed in xenografts, the investigation of immunotherapeutic effects requires an immunocompetent model. We have recently established a new, fully immunocompetent MV-susceptible mouse model of malignant melanoma, B16-CD20. Since murine cells lack MV receptors, B16 cells were transduced to stably express the CD20 surface antigen. Recombinant MV which do not bind the natural receptors CD46 and SLAM but instead are retargeted to CD20 via a single-chain antibody fused to the hemagglutinin protein (HαCD20) were generated. Specificity of infection was assessed in cell lines with and without CD20 surface expression (Figure 1e and Supplementary Figure S1). While MV with unmodified tropism infects only primate cells, CD20-targeted MV leads to syncytia formation on primate and murine CD20-positive cells. B16-CD20 cells are syngeneic to C57BL/6 mice and express PD-L1 (Supplementary Figure S2).
Growth kinetics of the new recombinant MV compared to control viruses encoding EGFP in additional transcription units upstream of the nucleocapsid gene (MV ld-EGFP) or downstream of the hemagglutinin gene (MV H-EGFP) revealed a slight attenuation of antibody-encoding viruses compared to MV H-EGFP, while growth kinetics of antibody-encoding viruses were comparable to MV encoding EGFP in the leader position of the genome (Supplementary Figure S3).
MV replication and oncolysis in melanoma cell lines
Replication of MV encoding anti-CTLA-4 and anti-PD-L1 compared to parental MV was characterized by determining progeny particles after infection of melanoma cell lines. Kinetics of viral replication differed between Sk-Mel-28 and Mel888 (Figure 1f, top panels): Rapid replication was observed in Mel888 cells with maximum titers 24 hours after infection. Release of progeny particles was delayed in Sk-Mel-28 cells, but higher maximum titers were reached which were comparable to maximum titers observed in Vero producer cells (Supplementary Figure S3a). Cytopathic effects of oncolytic MV on human and murine melanoma cell lines were quantified by cell viability assays (Figure 1f,g, bottom panels). Again kinetics differed between cell types, with rapid cell killing in MV-infected Mel888. In both cell lines, cell viability was reduced to below 10% compared to mock-treated cells 96 hours after infection. Viral replication was limited in B16-CD20 melanoma cells (Figure 1g, top panel). MV infection only reduced cell viability to 20–50% compared to mock-treated controls in murine cells (Figure 1g, bottom panel).
Immunovirotherapy in a syngeneic melanoma model
With oncolysis being limited in this nonpermissive host, immunocompetent murine models are suitable to address immunotherapeutic effects of oncolytic MV in vivo. Therapeutic efficacy of immunovirotherapy was assessed in C57BL/6 mice with subcutaneous B16-CD20 tumors. Mice were treated by five intratumoral injections of carrier fluid (mock) or CD20-targeted MV encoding anti-CTLA-4 or anti-PD-L1 or the antibody constant region (IgG Fc) as a control. Expression of anti-CTLA-4, anti-PD-L1 and IgG Fc in vivo were confirmed by RT-PCR and immunohistochemistry (Figure 2a,b). Treatment with MV led to a delay in tumor progression (Figure 2c). Tumor volumes on day 15 after implantation revealed a significantly lower tumor volume in mice treated with MV-aCTLA-4 compared to mock and MV-IgG Fc controls (P < 0.001 and P < 0.05 by analysis of variance (ANOVA) and Tukey's multiple comparison test, respectively) (Figure 2d). In case of mice treated with MV-aPD-L1, a subgroup of mice experienced partial tumor remission. While reduced tumor volumes at early time points did not prolong overall survival in mice treated with MV-aCTLA-4, mice responding to MV-aPD-L1 also survived longer compared to mock and MV-IgG Fc controls (P = 0.0016 and P = 0.031 in log rank test, respectively) (Figure 2e).
Figure 2.
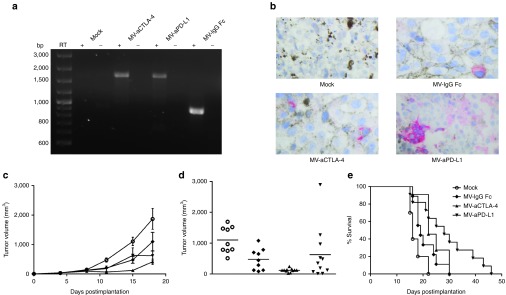
Immunovirotherapy in immunocompetent melanoma model. 1 × 106 B16-CD20 cells were implanted subcutaneously into the flank of C57BL/6 mice. When tumors reached an average volume of 40 mm3, animals were subject to mock treatment (carrier fluid; n = 10) or intratumoral injection of 2 × 106 viral particles of MV-IgG Fc (n = 9), MV-aCTLA-4 (n = 11), or MV-aPD-L1 (n = 11) on 5 consecutive days. (a) Tumors were explanted 24 hours after the last treatment. RNA was extracted and RT-PCR was performed with primers specific for aCTLA-4, aPD-L1, and IgG Fc, respectively. RT- indicates controls without reverse transcriptase. (b) Immunohistochemistry of explanted tumors with an antibody specific for the IgG constant region. Magnification: 500-fold. (c) Tumor volumes were determined every third day. Mean tumor volumes of mock-treated mice and mice treated with the indicated viruses are shown. Error bars represent standard error of the mean. (d) Distribution of tumor volumes for each group on day 15 postimplantation. Dots represent tumor volumes of individual mice. (e) Kaplan–Meier survival analysis.
To investigate possible mechanisms mediating therapeutic benefits of MV-mediated checkpoint blockade, tumor-infiltrating lymphocytes were characterized by flow cytometry and splenocytes from treated mice were restimulated with B16-CD20 tumor cells (Figure 3). Treatment with MV-aCTLA-4 and MV-aPD-L1 led to a significant increase in CD3+ T cells in the tumor (P < 0.05 and P < 0.01 by ANOVA and Tukey's multiple comparison test compared to mock controls, respectively) and a decrease in FoxP3+ regulatory T cells (Figure 3a,b). Treatment with MV-aPD-L1 was also associated with increased levels of CD8+ cytotoxic T cells (Figure 3c, P < 0.01 compared to mock controls) and activated, interferon-γ (IFNγ) expressing CD8+ cells (Figure 3d, P < 0.05 compared to mock controls). CD8+/Treg ratios (Figure 3e) were significantly increased after treatment with MV-aPD-L1 (P < 0.05 compared to mock controls).
Figure 3.

Tumor-infiltrating lymphocytes and IFNγ response after immunovirotherapy. 1 × 106 B16-CD20 cells were implanted subcutaneously into the flank of C57BL/6 mice and treated with five intratumoral injections of mock treatment, MV-IgG Fc, MV-aCTLA-4, or MV-aPD-L1 (n = 6 each). Tumors were explanted and splenocytes were isolated 24 hours after the last treatment. Single cell suspensions were prepared and tumor-infiltrating lymphocytes were analyzed by flow cytometry. Mean values are shown; error bars indicate standard error of the mean. (a) Percentage of CD3+ T cells among all cells. (b) Percentage of CD25+ FoxP3+ regulatory T cells among all CD4+ cells. (c) Percentage of CD8+ among all CD3+ T cells. (d) Percentage of IFNγ+ within total CD8+ T cells. (e) Ratios of CD8+/FoxP3+ cells. (f) Splenocytes harvested 24 hours after the last treatment (“early”) were cocultivated with B16-CD20 tumor cells. Cell culture supernatants were collected after 24 hours and IFNγ concentration was determined by enzyme-linked immunosorbent assay (ELISA). Mean values (n = 4 per group) are shown. Error bars represent standard error of the mean. (g) Splenocytes were isolated when tumors reached a volume exceeding 1,500 mm3 (“late”) and cocultivated with B16-CD20 tumor cells. After 24 hours, IFNγ release was determined by ELISA. Mean values (n = 4 for mock and MV-IgG Fc; n = 5 for MV-aCTLA-4 and MV-aPD-L1) and standard error of the mean are depicted.
Splenocytes from treated mice were isolated 24 hours after the last virus treatment (“early”) and when tumor volumes reached 1,500 mm3 (“late”) and cocultivated with B16-CD20 tumor cells (Figure 3f,g). At the early time point, splenocytes from mice treated with MV-aCTLA-4 released increased levels of IFNγ (Figure 3f). At late time points, splenocytes from mice treated with MV-aPD-L1 showed increased IFNγ release (Figure 3g).
MV-mediated compared to systemic checkpoint blockade
MV-mediated checkpoint blockade was compared to systemic administration of anti-CTLA-4 and anti-PD-L1 antibodies in the B16-CD20 model (Figure 4). Mice were treated by four intratumoral injections of carrier fluid (mock), MV encoding anti-CTLA-4 or anti-PD-L1 antibodies, intraperitoneal administration of anti-CTLA-4 or anti-PD-L1 antibodies on days 6, 9, 12, and 15 after implantation, or the combination of intraperitoneal antibodies and control MV (Figure 4a).
Figure 4.

Measles virus (MV)-mediated compared to systemic checkpoint blockade. 1 × 106 B16-CD20 cells were implanted subcutaneously into the flank of C57BL/6 mice. Mice (n = 10 per group) received four intratumoral (i.t.) injections and four intraperitoneal (i.p.) injections of carrier fluid and saline, respectively (mock), carrier fluid and checkpoint modulating antibody (aCTLA-4, aPD-L1), MV encoding aCTLA-4 or aPD-L1 and saline (MV-aCTLA-4, MV-aPD-L1) or control MV and checkpoint modulating antibody (MV + aCTLA-4, MV + aPD-L1). (a) Schematic depicting the treatment schedule. (b) Distribution of tumor volumes on day 14 after implantation and (c) Kaplan–Meier survival analysis for CTLA-4 blockade. (d) Distribution of tumor volumes on day 14 after implantation and (e) Kaplan–Meier survival analysis for PD-L1 blockade.
On day 14 after implantation, mice treated with MV encoding anti-CTLA-4 as well as mice receiving intratumoral MV and intraperitoneal anti-CTLA-4 had significantly lower tumor volumes compared to mock controls (Figure 4b; P < 0.005 by ANOVA and Tukey's test, respectively). Mice treated with MV-aCTLA-4 survived longer than mock controls (Figure 4c; P = 0.0088 in log rank test), but there were no significant differences in survival compared to systemic anti-CTLA-4 treatment (P = 0.18). Treatment with intratumoral control MV and intraperitoneal anti-CTLA-4 prolonged survival significantly compared to mock treatment (P < 0.0001), treatment with anti-CTLA-4 only (P = 0.0016) or MV-aCTLA-4 (P = 0.0255) (Figure 4c). Treatment with MV-aPD-L1 or control MV with systemic administration of anti-PD-L1 led to significantly reduced tumor volumes on day 14 (P < 0.005, respectively) compared to mock controls (Figure 4d). Treatment with MV-aPD-L1 prolonged survival significantly compared to mock treatment (P = 0.0064) and systemic treatment with anti-PD-L1 (P = 0.046) (Figure 4e). Treatment with control MV and anti-PD-L1 prolonged survival significantly compared to mock treatment (P = 0.007) and anti-PD-L1 only (P = 0.008). There were no statistically significant differences in terms of survival between MV-aPD-L1 and combination treatment of control MV with anti-PD-L1 (P = 0.21).
Oncolytic efficacy in human melanoma xenografts
Oncolytic efficacy of MV-aCTLA-4 and MV-aPD-L1 was assessed in a xenograft model of human melanoma. NOD/SCID mice with subcutaneous Mel888 tumors were treated by intratumoral injection of carrier fluid (mock treatment), parental MV-EGFP, MV-aCTLA-4 or MV-aPD-L1 on 5 consecutive days. MV treatment led to a significant delay in tumor progression (Figure 5a,b). On day 16 after implantation (4 days after the last treatment), mock-treated mice had a mean tumor volume of 120 mm3, while the mean tumor volume in mice treated with MV was 13 mm3 (MV-EGFP), 30 mm3 (MV-aCTLA-4), and 31 mm3 (MV-aPD-L1), respectively (P < 0.01 for MV-EGFP and P < 0.05 for MV-aCTLA-4 and MV-aPD-L1, respectively by ANOVA and Tukey's multiple comparison test compared to mock controls). MV treatment led to a significant survival benefit (Figure 5c). Median overall survival was 24 days for mock controls, whereas all but one of the MV-treated mice survived over 50 days after tumor implantation (P = 0.013 for MV-EGFP, P = 0.028 for MV-aCTLA-4, and P = 0.0013 for MV-aPD-L1 in log rank test). On day 50 after implantation, four MV-treated mice had detectable tumors, while six of seven (MV-EGFP), five of six (MV-aCTLA-4), and five of seven (MV-aPD-L1) mice showed complete tumor remission (Figure 5d).
Figure 5.
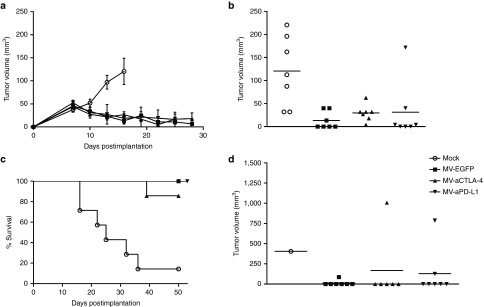
Oncolytic efficacy in human melanoma xenografts. 5 × 106 Mel888 cells were implanted subcutaneously into the flank of NOD/SCID mice. When tumors reached an average volume of 40 mm3, animals were subject to mock treatment (carrier fluid; n = 7) or intratumoral injection of 2 × 106 viral particles of MV-EGFP, MV-aCTLA-4, or MV-aPD-L1 (n = 7 per group) on 5 consecutive days. (a) Tumor volumes were determined every third day. Mean tumor volumes of mock-treated mice and mice treated with the indicated viruses are shown. Error bars represent standard error of the mean. (b) Distribution of tumor volumes for each group on day 16 postimplantation. Dots represent tumor volumes of individual mice. (c) Kaplan–Meier survival analysis. (d) Distribution of tumor volumes for each group on day 50 postimplantation. Dots represent tumor volumes of individual mice.
Viral replication in primary melanoma explants
To test whether primary human cancer cells can be transduced with MV-aCTLA-4 and MV-aPD-L1, short-term cultures were established from three individual melanoma biopsies. Cells were inoculated with MV-aCTLA-4 and MV-aPD-L1 additionally encoding EGFP as a reporter gene. At 48 hours p.i., EGFP expression and syncytia formation were observed in all three melanoma samples (Figure 6a). Viral replication and oncolysis were quantified by titration of progeny particles and cell viability assays (Figure 6b). To mimic virotherapy in the tumor environment, tissue slices from biopsy material were inoculated with MV (Supplementary Figure S4), demonstrating viral spread within primary tumors.
Figure 6.
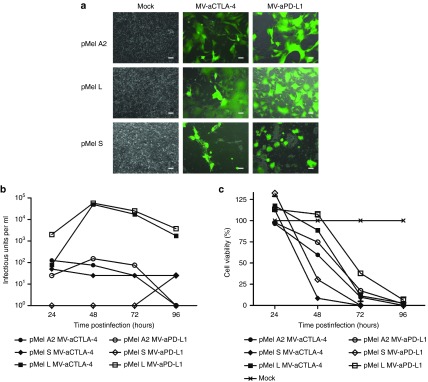
Infection of primary melanoma. Short-term cultures were prepared from melanoma biopsies and inoculated with recombinant Measles virus (MV) encoding aCTLA-4 or aPD-L1 and EGFP as a reporter gene. (a) Images were taken 48 hours p.i. (MOI 1). Scale bars: 100 µm. Low-passage cells from primary melanoma specimens were infected with MV-aCTLA-4 and MV-aPD-L1. (b) After infection at MOI 3, progeny particles were determined at designated time points. (c) At designated time points after infection at MOI 1, cell viability was determined by XTT assay. Viability of mock-treated cells was defined as 100%. XTT, 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide.
Discussion
This study demonstrates that MV immunovirotherapy can be enhanced by immune checkpoint modulation. MV vectors have evolved toward a versatile oncolytic platform which allows for efficient targeting, arming, and shielding. MV have been engineered for expression of reporter genes, prodrug convertases and cytokines.13 To our knowledge, vector-mediated delivery of antibodies by recombinant negative-strand RNA viruses has not been attempted previously. We show that MV vectors can be engineered for local expression of therapeutic antibodies without compromising viral replication, substantially expanding the possibilities of MV oncolytic vector engineering.
In vitro data demonstrate transgene expression and function, efficient replication, and lysis of human melanoma cell lines by MV-aCTLA-4 and MV-aPD-L1. Interestingly, kinetics of replication and cell killing differed between cell lines, possibly reflecting differences in the biology of individual tumor cell clones.
Oncolytic efficacy of MV-aCTLA-4 and MV-aPD-L1 was assessed in human melanoma xenografts in vivo. The novel, antibody-encoding MV vectors proved to be as efficient as the parental MV-EGFP control virus: In all MV-treated mice, tumor regression was observed without significant differences between the treatment groups. Importantly, complete remission was achieved in 80% of treated mice. However, not all animals remained tumor-free and even in those without detectable tumors, residual tumor cells may remain. In these cases, systemic antitumor immunity could prevent relapse and also destroy micrometastases at distant sites. In the context of oncolysis, the release of tumor-associated antigens coupled with viral pathogen-associated molecular patterns can promote antitumor immunity. OV vectors have been engineered to express various cytokines, e.g., IL-2, interferon, and granulocyte macrophage colony-stimulating factor, to further enhance these effects. While arming of OV with immunostimulatory molecules has been studied extensively, modulation of immune checkpoints in this context has not been broadly applied to date. A recent study reported oncolytic efficacy of an adenovirus encoding anti-CTLA-4 in prostate and lung cancer xenografts.14 However, the physiology and pathophysiology of these molecules designate them as attractive targets in immunovirotherapy trials: CTLA-4 is expressed exclusively on T cells and exerts its inhibitory function mainly in the priming phase of T cell activation. Though the precise mechanisms remain to be resolved, CTLA-4 appears to compete with the costimulatory molecule CD28 for binding to CD80 and CD86. In addition, CTLA-4 negatively influences TCR signaling and sequesters costimulatory receptors.15 The physiological role of CTLA-4 is the maintenance of self-tolerance, as illustrated by the lethal immune hyperactivation phenotype of CTLA-4 knockout mice.16 CTLA-4 blockade enhances T helper cell function and inhibits regulatory T cells.17 The PD-1/PD-L1 pathway limits T cell activation during inflammation and contributes to peripheral tissue tolerance. PD-1 expression is induced upon T cell activation and high levels of PD-1 are observed on tumor-infiltrating lymphocytes (TILs). PD-L1 is expressed in various tissues and is often upregulated in malignant tumors. PD-1 ligation dampens T cell activation especially during the effector phase, leads to T cell exhaustion or anergy during chronic antigen exposure and also inhibits NK cells and B cells.18 Antagonists of PD-1 or its ligands can at least in part restore immune effector function.19
The clinical success of targeting the CTLA-4 and PD-1/PD-L1 pathways most prominently in melanoma, but also in other tumor entities has been overwhelming.9 These therapeutics are now extensively tested in combination with other treatment modalities. For instance, Ipilimumab has been combined with tumor peptide vaccines,10 chemotherapy,20 and small molecules.21 With oncolytic viruses progressing toward broader clinical application, the combination of virotherapy with CTLA-4 and PD-1/PD-L1 blockade is highly warranted.
To study immune effects in the context of OV therapy, immunocompetent models are mandatory. Here, we introduce the B16-CD20 melanoma model which is syngeneic to fully immunocompetent C57BL/6 mice. Specific targeting of B16-CD20 with retargeted MV was demonstrated in vitro. MV replication is restricted in nonprimate cells, leading to a limited number of progeny particles and incomplete lysis of tumor cells in vitro. Thus, tumor reduction by MV in murine models may be limited, especially in an aggressive model such as B16. Nevertheless, this model is suited to study immune-mediated mechanisms during MV oncolysis, providing a second tumor model aside from MC38cea22 to study MV immunovirotherapy. MV-aCTLA-4 and MV-aPD-L1 showed therapeutic benefits in the immunocompetent B16-CD20 model. Tumor progression was delayed significantly in mice treated with MV-aCTLA-4 and in a subgroup of mice treated with MV-aPD-L1. This led to a significantly prolonged median overall survival of mice treated with MV-aPD-L1. No signs of immune-mediated toxicity were observed. Analysis of tumor-infiltrating lymphocytes indicated favorable immune profiles with an increase of total T cells accompanied by a reduction in regulatory T cells after treatment with MV-aCTLA-4 and MV-aPD-L1. Treatment with MV-aPD-L1 was also associated with increased levels of activated cytotoxic T cells and an increased CD8/Treg ratio.
As indicated by restimulation experiments, MV-aCTLA-4 may enhance antitumor immunity at early time points after treatment, whereas the effect of MV-aPD-L1 becomes apparent at later time points. This is consistent with the current model of CTLA-4 acting mainly in the early phase of an immune response in lymphoid organs, while PD-L1 signaling occurs at later phases of T cell activation in the periphery.17 In line with this, systemic administration of anti-CTLA-4 prolonged survival compared to local, MV-mediated expression. Though MV-aCTLA-4 may lead to transient antibody expression in draining lymph nodes, systemic therapy may be required for an optimal response. In case of anti-PD-L1, efficacy of local expression was comparable to systemic administration, which is consistent with high levels of PD-1/PD-L1 signaling within the tumor microenvironment. A more detailed analysis of immune phenomena over time during immunovirotherapy might further elucidate the mechanisms underlying the effects observed in the B16-CD20 model. However, murine models cannot fully represent clinical reality. Ultimately, oncolytic viruses will have to prove effective in clinical settings. In an ex vivo approach we could demonstrate viral replication and transgene expression in primary tissue samples from melanoma patients.
In summary, we have demonstrated that combining oncolytic MV with anti-CTLA-4 and anti-PD-L1 leads to therapeutic benefits in vivo. The present study supports combining oncolytic viruses and checkpoint modulation in future clinical trials.
Materials and Methods
Cloning of recombinant MV vectors. The anti-CTLA-4 p4F10-γ1 cassette encoding the murine Ig kappa-chain V-J2-C signal peptide, the single chain variable domain (scFv) from the hybridoma antibody UC10-F10-11 against murine CTLA-4, a (Gly4Ser)3 linker and the hinge CH2-CH3 region of human IgG1 with an N-terminal HA tag23 was a kind gift of A Lieber. The anti-CTLA-4 cassette was amplified by polymerase chain reaction (PCR) using primers 5′ - CCCTTTACGCGTGCCACCATGGAGACAGACACACTCCTGCTA - 3′ and 5′ - CCCTTTGGCGCGCCTAATTCAGATCTTCTTCTGAGATGAG - 3′ with MluI and AscI restriction sites, respectively, generating a 1,590 bp fragment fulfilling the rule of six. After restriction digest, the PCR product was inserted into the MauBI restriction site of an MV full length genome with an additional transcription unit downstream of the hemagglutinin open reading frame (MV H-ATU).
To generate the anti-PD-L1 cassette, the cloning vector pEX-A anti-PD-L1 scFv encoding murine Ig κ-chain V-J2-C signal peptide and the single chain variable region of the YW234.55.S70 antibody against murine PD-L1 with codon optimization for expression in murine cells was obtained by gene synthesis (Eurofins MWG Operon, Ebersberg, Germany). The complete anti-PD-L1 cassette was generated by PCR with pEX-A anti-PD-L1 scFv as template and with primers 5′ - CCCTTTACGCGTGCCACCATGGAGACAGACACACTCCTGCTA - 3′ and 5′ - TTGTCACAAGATTTGGCCTCGTCGAC - 3′ and amplification of the constant region of the anti-CTLA-4 p4F10-γ1 cassette with primers 5′ - GTCGACGAGGCCAAATCTTGTGACAA - 3′ and 5′ - CCTTTGGCGCGCCTATCAATTCAGATCTTCTTCTGAGATGAG - 3′. The complete anti-PD-L1 cassette was generated by overlap-PCR with the two PCR products as template and with the flanking primers 5′ - CCCTTTACGCGTGCCACCATGGAGACAGACACACTCCTGCTA - 3′ and 5′ - CCCTTTGGCGCGCCTATCAATTCAGATCTTCTTCTGAGATGAG - 3′. After restriction digest with MluI and AscI, the complete 1,594 bp cassette was inserted into the MauBI site of MV H-ATU.
To generate MV IgG Fc, two PCR reactions with anti-CTLA-4 p4F10-γ1 as template were carried out using primers 5′ - CCCTTTACGCGTGCCACCATGGAGACAGACACACTCCTGCTA - 3′ and 5′ - GGGCCCAGCCGGCCGTCGACGAGGCCAAA - 3′; 5′ - GGGGCCCAGCCGGCCGTCGACGAGGCCAAA - 3′ and 5′ - CCTTTGGCGCGCCTATCAATTCAGATCTTCTTCTGAGATGAG - 3′. Subsequently, overlap PCR with both PCR products and the outer primers 5′ - CCCTTTACGCGTGCCACCATGGAGACAGACACACTCCTGCTA - 3′ and 5′ - CCCTTTGGCGCGCCTATCAATTCAGATCTTCTTCTGAGATGAG - 3′ was performed to obtain the complete IgG Fc cassette. After restriction digest with MluI and AscI, the complete 864 bp cassette was inserted into the MauBI site of MV H-ATU. The control virus MV ld -EGFP has been described previously.24 To obtain MV H-EGFP, the EGFP ORF was inserted as a MluI-PauI fragment (732 bp) into the MauBI site of MV H-ATU.
MV-NIS was generated by insertion of the human NIS ORF as a 1,944 bp MluI-PauI fragment into MV H-ATU.
To generate CD20-targeted MV, the PacI-SpeI fragment of the respective vectors was replaced by the PacI-SpeI fragment excised from pCG-HáCD20 which encodes a CD46/SLAM-blinded hemagglutinin fused to a single-chain variable fragment (scFv) specific for human CD20 and a six histidine tag.24
Primers were obtained from Eurofins MWG Operon. All constructs were verified by restriction digest and sequencing.
Cell culture. Vero African green monkey kidney cells were obtained from ATCC (Manassas, Virginia) and cultured for less than 6 months after receipt. Vero-αHis cells which stably express an antibody against the six histidine tag as a pseudoreceptor25 were a kind gift of SJ Russell. Mel888, Sk-Mel-28, and B16 cells have been described previously.26 Vero-CD20 and B16-CD20 cells were generated by transduction of Vero and B16 cells with a SIN lentiviral vector harboring human CD20. Vero cell lines and Sk-Mel-28 were cultured in Dulbecco's modified Eagle's medium (Invitrogen, Darmstadt, Germany) supplemented with 10% (vol/vol) fetal bovine serum (BioSera, Boussens, France). Mel888 and B16 cell lines were cultured in Roswell Park Memorial Institute medium (Invitrogen) with 10% fetal bovine serum. Melanocytic origin of Mel888 and Sk-Mel-28 was verified by RT-PCR with primers specific for tyrosinase and melan-A. All cell cultures were maintained at 37 °C in a humidified atmosphere with 5% CO2 and routinely tested for mycoplasma contamination.
Virus propagation and titration. Recombinant MV particles were generated from cDNA constructs according to Radecke et al.27 MV with natural tropism were propagated on Vero cells, fully retargeted viruses were propagated on Vero-αHis cells. For propagation, cells were inoculated with MV at a multiplicity of infection (MOI) of 0.03 and cultured at 32 °C. When syncytia had formed across the entire cell layer (ca. 68 hours after infection), cells were scraped and viral particles were released by one freeze-thaw cycle. Cell debris was removed by centrifugation (5 minutes, 5,000 × g, 4 °C). Titers of virus stocks were determined by performing serial 1:10 dilutions in 96-well plates with 1 × 104 Vero (for MV with unmodified tropism) or Vero-αHis cells (for retargeted MV) per well in a total volume of 200 mm3. After 48 hours, individual syncytia were counted and titers were calculated as cell infectious units per ml (ciu/ml).
Infection experiments. All infection experiments were performed with virus stocks from the fourth passage. Cells were incubated at 37 °C with virus suspended in OptiMEM (Invitrogen) with gentle shaking of the plate every 15 minutes. After 2 hours, the inoculate was removed and replaced by culture media. To assess infection of different cell lines, 1 × 105 cells seeded in 12-well plates were infected at an MOI of 1. Cells were monitored for syncytia formation and images were taken using a Zeiss Axiovert 200 fluorescence microscope (Carl Zeiss, Jena, Germany) and Axiovision 4.7 software (Carl Zeiss).
Western blot analysis. 2 × 105 cells were infected at an MOI of 3 in six-well plates. Thirty-six hours after infection, cells were lysed in 200 µl radio immunoprecipitation assay buffer (RIPA). Cell debris was spun down (10 minutes, 10,000 × g, 4 °C) and the supernatant was resolved in 1× Laemmli buffer (Carl Roth, Karlsruhe, Germany). Samples were denatured at 95 °C for 5 minutes and subsequently subject to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) on a 12% polyacrylamide gel (Mini-PROTEAN TGX; BioRad, Munich, Germany) at 200 V for 40 minutes. After SDS-PAGE, separated proteins were transferred onto a polyvinylidene fluoride membrane (Immobilon; Merck Millipore, Schwalbach) activated with methanol (Carl Roth) using a wet blot chamber (Mini-PROTEAN Tetra Cell; BioRad), filter paper (Whatman, Dassel, Germany) and Novex TrisGlycine Transfer Buffer (Invitrogen). After blocking of nonspecific binding sites with 5% skim milk powder (Carl Roth) in TBS-T (Carl Roth) for 30 minutes at room temperature, membranes were probed with mouse anti-HA (Sigma-Aldrich, Taufkirchen, Germany) diluted 1:10,000 in 5% skim milk powder in TBS-T over night at 4 °C. After washing three times for 5 minutes in TBS-T, secondary anti-mouse HRP (Bethyl, Montgomery, TX) was added at a 1:10,000 dilution in 5% skim milk powder in TBS-T and incubated for 2 hours at room temperature. After three washing steps, SuperSignal West PICO Chemiluminescent Substrate (Fisher Scientific, Schwerte, Germany) was added for detection of bound antibodies and signals were recorded using a ChemiDOC XRS Imaging System (BioRad).
ELISA. Ninety-six well plates (Nunc Maxisorp, Thermo Fisher Scientific, Schwerte, Germany) were coated with 100 ng of recombinant CTLA-4 or PD-L1 (Life Technologies, Darmstadt, Germany). Wells were blocked and serial dilutions of tissue culture supernatants were added. After 2-h incubation and washing, the antibodies were detected with anti-HA Biotin (clone 3F10; Roche, Mannheim, Germany), Peroxidase-conjugated Streptavidin (Dianova, Hamburg, Germany) and 1-Step Ultra TMB-ELISA (Thermo Scientific). Absorbance measurements were performed using an Infinite M200 Pro microplate reader and i-control software (Tecan, Männedorf, Switzerland).
One-step growth curves. 1 × 105 cells in 12-well plates were infected as described above. At designated time points, cells were scraped in their culture medium. After one freeze-thaw cycle, titers were determined in quadruplicates.
Cell viability assays. After infection of cells in 12-well plates, cell viability was assessed using the Cell Proliferation Kit III (XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide); PromoKine, Heidelberg, Germany) according to the manufacturer's instructions. Absorbance measurements were performed using an Infinite microplate reader and XFluor software (Tecan).
In vivo experiments. All animal experimental procedures were approved by the Animal Protection Officer at the German Cancer Research Center and by the regional council according to the German Animal Protection Law.
Immunocompetent model. 1 × 106 B16-CD20 cells were implanted into the right flank of 6–8 weeks old female C57BL/6 mice (Harlan, Rossdorf, Germany). When tumors reached an average volume of 40 mm3, mice received 100 µl carrier fluid (mock, n = 10) or 2 × 106 viral particles in 100 µl (MV HαCD20-IgG Fc, n = 9; MV HαCD20-aCTLA-4, n = 11 or MV HαCD20-aPD-L1, n = 11) by intratumoral injection on 5 consecutive days (1 × 107 viral particles in total). Tumor diameters were measured every third day using a caliper and tumor volumes were calculated as : V (mm3) = (largest diameter (mm)) × (smallest diameter (mm))2/2. Mice were sacrificed when tumor volumes reached 1,500 mm3, when tumor ulceration occurred or when mice showed signs of severe illness such as apathy, respiratory distress, or weight loss over 20%.
For comparison with systemic administration of anti-CTLA-4 and anti-PD-L1 antibodies, mice received intratumoral (i.t.) injections of either carrier fluid or 2 × 106 viral particles on 4 consecutive days. Antibodies or saline were administered intraperitoneally (i.p.) on days 6, 9, 12, and 15 after tumor implantation. Mock controls (n = 10) received carrier fluid i.t. and saline i.p. Mice receiving i.t. MV HαCD20-aCTLA-4 or MV HαCD20-aPD-L1 (n = 10 per group) received saline i.p. Mice receiving i.p. doses of 100 µg anti-CTLA-4 (clone 9H10; eBioscience, Frankfurt am Main, Germany) or anti-PD-L1 (clone MIH5; eBioscience) received i.t. injections with either carrier fluid or control MV (MV HαCD20-NIS, n = 10 per group). Monitoring of tumor volumes was performed as described above. Mice were sacrificed according to the criteria stated above.
Xenograft model. 5 × 106 Mel888 cells were implanted into the flank of 8 weeks old female NOD/SCID mice (Charles River, Sulzbach, France). When tumors had reached an average volume of 40 mm3, mice received 100 µl carrier fluid (mock, n = 7) or 2 × 106 viral particles in 100 µl (n= 7 per group) by intratumoral injection on 5 consecutive days (1 × 107 viral particles in total). Mice were sacrificed when tumor volumes reached 1,500 mm3 or when tumor ulceration occurred.
RT–PCR. RNA was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and first-strand complementary DNA (cDNA) was synthesized using the SuperScriptIII System (Invitrogen) according to the manufacturers' instructions.
Immunohistochemistry. Slides were freshly cut to 3 µm from formalin fixed paraffin embedded material. Immunohistochemistry of tumor sections was performed by incubating the slides in Target Retrieval Solution S1699 (Dako, Hamburg, Germany) for 30 minutes in a pressure cooker, subsequently Avidin/Biotin Blocking Kit (Vector Laboratories, Burlingame, CA) was used for 10 minutes. Slides were then incubated with Biotin-labeled polyclonal rabbit anti-human Fc antibody (dilution 1:500, Dianova, Hamburg, Germany), followed by an incubation with Streptavidin AP /SS HK331-9K (BioGenex, Fremont, CA) for 25 minutes. Color development was done with Permanent AP Red Chromogen (Zytomed, Berlin, Germany). Slides were counterstained with hematoxilin and reviewed by a board cerified pathologist (W.W.).
Flow cytometry. For characterization of tumor-infiltrating lymphocytes, single cell suspensions were generated from explanted tumors and stained with antibodies CD45.2-PerCP-Cy5.5 (clone 104), CD3-AlexaFluor700 (clone 17A2), CD4-APC-Cy7 (clone GK1.5), CD8-APC (clone 53–6.7), CD25-PE-Cy7 (clone clone PC61) (BD Biosciences, Heidelberg). Intracellular staining with FoxP3-FITC (clone FJK-16s, eBioscience, San Diego California) and IFNγ-PE (clone XMG1.2, BD Biosciences) was performed using the FoxP3/Transcription Factor Staining Buffer Set (eBioscience) according to the manufacturer's instructions. Flow cytometry was performed with AriaII and FACS DIVA Software (BD Biosciences). Only samples with over 5,000 events were taken into account.
Restimulation experiments. Mice were sacrificed 24 hours after the last of five treatments (“early”) and when tumor volumes exceeded 1,500 mm3 (“late”, between 18 and 42 days after implantation). Spleens were isolated aseptically and single-cell suspensions were prepared. Splenocytes and B16-CD20 tumor cells were cocultured at a ratio of 10:1 (“early”) or 3:1 (“late”), respectively. After 24 hours of coculture, IFNγ concentration in culture supernatants was determined by ELISA (BD Opteia, Heidelberg, Germany) according to the manufacturer's protocol.
Primary melanoma. Melanoma biopsies were obtained from patients at the Department of Dermatology, University Hospital Heidelberg according to the Declaration of Helsinki Principles and to the local Ethics Committee's guidelines with patients' informed consent. Low-passage cultures were derived from biopsies as in ref. [26] and were cultivated in Roswell Park Memorial Institute supplemented with 10% fetal bovine serum, 1% antibiotic/antimycotic solution (Invitrogen), 1 mmol/l Hepes (Invitrogen) and 100 µg/ml gentamicin (Invitrogen). Melanocytic origin of cultures was verified by RT-PCR with primers specific for tyrosinase, Melan-A and S100 cDNA. Tissue slices were maintained on 3.0 µm PET membrane cell culture inserts (BD Biosciences).
Statistical analyses. Statistical analyses were performed using GraphPad Prism software (version 5.04; GraphPad Software, La Jolla, CA). Tumor volumes, FACS, and ELISA data were analyzed by one-way ANOVA and Tukey's multiple comparison test. Survival curves were analyzed by log rank (Mantel-Cox) test. P values below 0.05 were considered statistically significant.
SUPPLEMENTARY MATERIAL Figure S1. Targeted Infection of CD20-positive cells. Figure S2. PD-L1 Expression in B16-CD20 Cells. Figure S3. Growth Kinetics in Vero and Vero-αHis. Figure S4. Replication of MV in primary melanoma tissue.
Acknowledgments
We thank S Hamzaoui-Nord, C Lay-Mees, J Albert, and B Hoyler for their valuable technical assistance. A Lieber and SJ Russell are acknowledged for providing plasmids and cell lines. C.E.E. is a fellow of the MD/PhD Program of the Medical Faculty and the Faculty of Biosciences at the University of Heidelberg. This work was supported by Deutsche Krebshilfe (German Cancer Aid), Max Eder Program No. 108307 (to G.U.). The authors declare no conflict of interest. This work was performed in Heidelberg, Germany.
Supplementary Material
References
- Miest TS, Cattaneo R. New viruses for cancer therapy: meeting clinical needs. Nat Rev Microbiol. 2014;12:23–34. doi: 10.1038/nrmicro3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol Ther. 2011;19:1008–1016. doi: 10.1038/mt.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik JD, Twelves CJ, Selby PJ, Vile RG, Chester JD. Immune recruitment and therapeutic synergy: keys to optimizing oncolytic viral therapy. Clin Cancer Res. 2011;17:4214–4224. doi: 10.1158/1078-0432.CCR-10-2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossardt C, Engeland CE, Bossow S, Halama N, Zaoui K, Leber MF, et al. Granulocyte-macrophage colony-stimulating factor-armed oncolytic measles virus is an effective therapeutic cancer vaccine. Hum Gene Ther. 2013;24:644–654. doi: 10.1089/hum.2012.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Peng KW, Dingli D, Kratzke RA, Russell SJ. Oncolytic measles viruses encoding interferon beta and the thyroidal sodium iodide symporter gene for mesothelioma virotherapy. Cancer Gene Ther. 2010;17:550–558. doi: 10.1038/cgt.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote D, Cattaneo R, Fielding AK. Neutrophils contribute to the measles virus-induced antitumor effect: enhancement by granulocyte macrophage colony-stimulating factor expression. Cancer Res. 2003;63:6463–6468. [PubMed] [Google Scholar]
- Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19:329–336. doi: 10.1038/nm.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G, et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol. 2009;27:5763–5771. doi: 10.1200/JCO.2009.24.3675. [DOI] [PubMed] [Google Scholar]
- Quezada SA, Peggs KS. Exploiting CTLA-4, PD-1 and PD-L1 to reactivate the host immune response against cancer. Br J Cancer. 2013;108:1560–1565. doi: 10.1038/bjc.2013.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo R. Paramyxovirus entry and targeted vectors for cancer therapy. PLoS Pathog. 2010;6:e1000973. doi: 10.1371/journal.ppat.1000973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias JD, Hemminki O, Diaconu I, Hirvinen M, Bonetti A, Guse K, et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012;19:988–998. doi: 10.1038/gt.2011.176. [DOI] [PubMed] [Google Scholar]
- Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009;229:12–26. doi: 10.1111/j.1600-065X.2009.00770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bour-Jordan H, Esensten JH, Martinez-Llordella M, Penaranda C, Stumpf M, Bluestone JA. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/B7 family. Immunol Rev. 2011;241:180–205. doi: 10.1111/j.1600-065X.2011.01011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24:207–212. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Thomas L, Bondarenko I, O'Day S, M D JW, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–2526. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365–1366. doi: 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- Ungerechts G, Springfeld C, Frenzke ME, Lampe J, Parker WB, Sorscher EJ, et al. An immunocompetent murine model for oncolysis with an armed and targeted measles virus. Mol Ther. 2007;15:1991–1997. doi: 10.1038/sj.mt.6300291. [DOI] [PubMed] [Google Scholar]
- Tuve S, Chen BM, Liu Y, Cheng TL, Touré P, Sow PS, et al. Combination of tumor site-located CTL-associated antigen-4 blockade and systemic regulatory T-cell depletion induces tumor-destructive immune responses. Cancer Res. 2007;67:5929–5939. doi: 10.1158/0008-5472.CAN-06-4296. [DOI] [PubMed] [Google Scholar]
- Ungerechts G, Springfeld C, Frenzke ME, Lampe J, Johnston PB, Parker WB, et al. Lymphoma chemovirotherapy: CD20-targeted and convertase-armed measles virus can synergize with fludarabine. Cancer Res. 2007;67:10939–10947. doi: 10.1158/0008-5472.CAN-07-1252. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Peng KW, Harvey M, Greiner S, Lorimer IA, James CD, et al. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat Biotechnol. 2005;23:209–214. doi: 10.1038/nbt1060. [DOI] [PubMed] [Google Scholar]
- Kaufmann JK, Bossow S, Grossardt C, Sawall S, Kupsch J, Erbs P, et al. Chemovirotherapy of malignant melanoma with a targeted and armed oncolytic measles virus. J Invest Dermatol. 2013;133:1034–1042. doi: 10.1038/jid.2012.459. [DOI] [PubMed] [Google Scholar]
- Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dötsch C, et al. Rescue of measles viruses from cloned DNA. EMBO J. 1995;14:5773–5784. doi: 10.1002/j.1460-2075.1995.tb00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.