

Abstract
The phenotype of vascular smooth muscle cells (vSMC) is dynamically regulated in response to various stimuli. In a cellular process known as “phenotype switching”, vSMC alternate between a contractile and synthetic phenotype state. Deregulation of phenotype switching is associated with vascular disorders such as atherosclerosis, restenosis after angioplasty, and pulmonary hypertension. An important role for microRNAs (miRNAs) in vascular smooth muscle cell development and phenotype switching has been recently uncovered. Individual miRNAs are involved in promoting both contractile and synthetic vSMC phenotype. In this review we summarize recent advances in the understanding of miRNA function in the regulation of vSMC phenotype regulation.
Introduction
vSMC are a highly differentiated cell type present within the medial region of arteries and arterioles. vSMC express proteins that are important for contractility, ion channels and signaling molecules that allow these cells to regulate systemic blood pressure through the modulation of vascular tone1. Unlike the majority of differentiated cells, vSMC maintain phenotypic plasticity. vSMC can switch between a ‘differentiated’ (also termed “contractile”) state and a ‘dedifferentiated’ (also termed “synthetic”) phenotype in response to extracellular cues1 (Fig. 1A). Differentiated smooth muscle phenotype is characterized by high levels of contractile gene expression and low rates of proliferation, migration and extracellular matrix synthesis. Conversely dedifferentiated vSMC have increased rates of proliferation, migration and production of extracellular matrix, as well as reduced expression of contractile genes1 (Fig. 1A). vSMC phenotype modulation is critically important for the resolution of vascular injury. Immediately after an insult, vSMC dedifferentiate to promote repair of the vessel; however, once the injury is resolved, healthy vSMC return to a non-proliferative, contractile phenotype. Although important for normal homeostasis, the plasticity of vSMC phenotype makes these cells particularly susceptible to both physiological and non-physiological stimuli. Deregulation of vSMC phenotype switching contributes to the development and progression of vascular pathologies. In particular, abnormal vSMC phenotype is considered to play an important role in the progression of proliferative and obstructive vascular diseases, such as atherosclerosis, post-angioplasty restenosis, lymphangioleiomyomatosis (LAM) and pulmonary arteriole hypertension (PAH)1-2.
Fig 1. Regulation of vSMC phenotype.
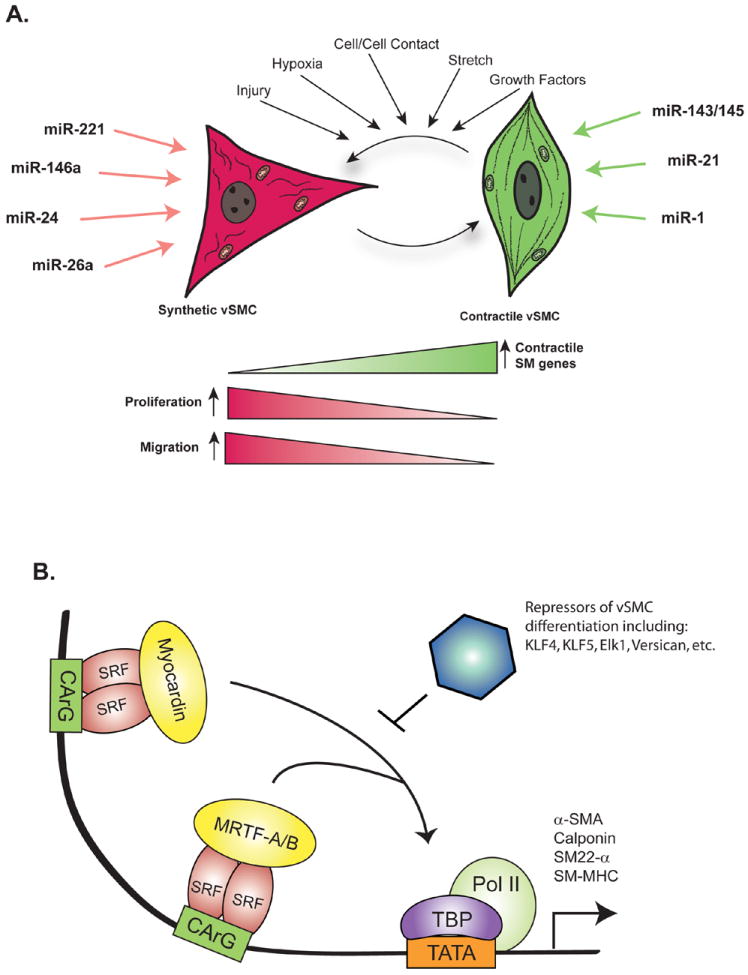
A. In response to a variety of stimuli, vSMC switch between contractile phenotype and synthetic phenotype. Although not mutually exclusive, contractile phenotype is characterized by high expression of contractile genes and low rates of proliferation and migration. Conversely, synthetic vSMC express low levels of contractile genes and have increased rates of proliferation and migration. microRNAs reported to promote contractile and synthetic phenotype are indicated. B. Transcriptional regulation of vSMC contractile gene expression involves binding of SRF and either Myocardin or MRTF-A/B to CArG box elements located within the promoter of contractile genes. KLF4 is a potent repressor of Myocardin/MRTF-A/B activity.
vSMC phenotype is determined through the integration of numerous environmental cues, including cytokines, cell-cell contact, cell adhesions, extracellular matrix interactions, injury stimuli and mechanical force1 (Fig. 1A). In particular, growth factor/cytokine signaling can dramatically affect the differentiation status of vSMC. Platelet Derived Growth Factor (PDGF) promotes multiple aspects of the synthetic vSMC phenotype, including reduced expression of contractile genes and increased rate of proliferation and migration3. Conversely, Transforming Growth Factor-β (TGF-β) and its related family member; Bone Morphogenetic Protein 4 (BMP4) reduce vSMC proliferation and migration and promote increased expression of vSMC contractile genes2, 4. The importance of TGF-β and BMP in promoting differentiated vSMC phenotype is confirmed by the identification of mutations in these signaling cascades in patients with the vascular disorders Hereditary Hemorrhagic Telangiectasia (HHT) and PAH2.
Transcriptional control of vSMC contractile gene expression
Significant progress has been made in uncovering the basic mechanisms that regulate transcription of vSMC contractile gene expression. The transcriptional regulation of smooth muscle cell markers involves a complex combination of cis-acting elements located within the promoters of vSMC genes and trans-acting factors1 (Fig. 1B). In particular, a cis-acting element found in the promoters of contractile genes termed the ‘CArG box’ [CC(AT)6GG] is critical for the regulation of vSMC gene expression1. The MADS box transcription factor Serum Response Factor (SRF) binds to the CArG box as a homodimer and activates gene transcription. The affinity of SRF for the CArG boxes of vSMC contractile genes is strongly enhanced by the cofactors Myocardin and Myocardin-Related Transcription Factors A and B (MRTF-A and MRTF-B)1 (Fig. 1B). Moreover, transcription of contractile genes can be negatively regulated by Krϋpple-like factor 4 (KLF4). KLF4 is a potent repressor of contractile genes and functions through multiple mechanisms including remodeling the chromatin of CArG box-containing promoters, sequestering of SRF, and reducing Myocardin expression1 (Fig. 1B). Diverse mechanisms have been identified to regulate contractile gene expression and other facets of vSMC phenotype. A more complete understanding of the mechanisms utilized by cytokines and other stimuli to alter vSMC phenotype may allow the development of novel therapeutic approaches for vascular proliferative disorders. The recent identification of miRNAs as critical regulators of gene expression in a variety of tissues suggested that miRNAs could be involved in the regulation of vSMC phenotype.
miRNA
miRNAs are a class of evolutionarily conserved, ~22 nucleotides (nt) non-coding RNAs that control diverse biological functions through the repression of target genes during normal development as well as during pathological responses5. To date, more than 1,400 miRNA transcripts have been identified in the human genome according to miBase annotation6. The mechanism of miRNA biosynthesis is evolutionarily conserved and is initiated by the transcription of a long (several kb) capped and poly-adenylated primary miRNA (pri-miRNA) transcript7-8. The RNAse III enzyme Drosha cleaves the pri-miRNA into a ~60-100 nt hairpin structure termed the precursor-miRNA (pre-miRNA)8. Following cleavage, the pre-miRNA is transported out of the nucleus through interaction with exportin-5 and Ran-GTP8. The pre-miRNA then undergoes a second round of processing catalyzed by Dicer. This cleavage event gives rise to a double-stranded (ds) ~22 nt product comprised of the mature miRNA guide strand and the miRNA* passenger strand. The final step of miRNA biogenesis involves loading of the mature miRNA into a large protein complex termed the RNA induced silencing complex (RISC) while the passenger strand is degraded8. The mature miRNA is then active to promote the association of the RISC with specific regions in the 3’Untranslated Region (UTR) of target genes.
Selection of miRNA targets is mediated by imperfect base pairing between the miRNA and miRNA binding sites present in the 3’UTR of the target mRNA. The imperfect nature of the miRNA:mRNA interaction means that a single miRNA can target 10s to 100s of mRNAs9. Association of the miRNA-RISC results in the repression of the target gene by promoting mRNA degradation and/or translational inhibition10. Through the repression of targets, miRNAs elicit important changes in gene expression programs which contribute to both normal development and disease.
miRNA expression is often tissue specific and developmentally regulated; aberrant expression of miRNAs has been linked to developmental abnormalities and human diseases, including cancer and cardiovascular disorders5. Pri-miRNAs are located in diverse regions of the genome and can be transcribed from an independent promoter or, when the miRNA is located in the intron of a protein-coding mRNA, under the control of the host gene promoter7-8. Nucleosome positioning and chromatin immunoprecipitation-on-genomic DNA microarray chip (or ChIP-on-chip) analysis suggests that the promoter structure of miRNA genes is nearly identical to that of mRNAs11. Therefore, the DNA binding factors that regulate miRNA transcription largely overlap with those that regulate mRNA expression.
Although transcription of the miRNA gene is the first step of miRNA biogenesis and is critically regulated by various stimuli, it has been recently uncovered that post-transcriptional regulation of miRNA processing also plays an important role in the regulation of miRNA expression7. In particular, several additional cofactors, such as the RNA helicases p68 (DDX5) and p72 (DDX17), are identified to associate with Drosha and modulate Drosha-dependent miRNA processing12. We recently identified that the signal transducers of the TGF-β and BMP4 signaling cascades, the Smads, associate with p68 in the Drosha complex and promote the processing of a subset of miRNAs, including miR-2113-14. Interestingly, although the Smads are bona fide transcription factors which bind to a specific DNA sequence, TGF-β or BMP4 treatment results in induction of the mature levels of several miRNAs in the absence of induction of the pri-miRNA13. In addition to the Smads, recent studies have shown that two other transcription factors, p53 and Estrogen Receptor-α (ER-α), also modulate miRNA processing through association with p68 and Drosha15-16. Upon activation by DNA damage, p53 facilitates the pri- to pre-miRNA processing of a subset of miRNAs by interacting with p68 and Drosha on the pri-miRNA15, similarly to the role of the Smads. Conversely, upon Estradiol (E2) association, ER-α attenuates miRNA processing by preventing the association of p68/Drosha with pri-miRNAs16. Altogether, transcriptional and post-transcriptional regulatory mechanisms allow the levels of mature miRNAs to be tightly regulated in response to cellular cues.
miRNAs are required for vSMC differentiation: implications from Dicer knockout
One way to interrogate the overall function of miRNAs during development or pathologic processes is to examine the effect of genetic deletion of the miRNA processing enzymes, such as Dicer. Targeted disruption of dicer results in a complete ablation of miRNAs. Consistent with the essential role of miRNAs in a wide variety of functions, homozygous disruption of Dicer in mouse results in early embryonic lethality at day E8.517. Tissue-specific and inducible disruption of Dicer indicates that miRNAs play a critical role in the differentiation and survival of a multitude of cell types including stem cells, cardiomyocytes, neurons, glia, and skin cells17. An important role for miRNAs in vSMC is supported by the observation that vSMC-specific inactivation of Dicer results in embryonic lethality at day E16-E17 due to severe vascular abnormalities and extensive hemorrhage18. The vessels in vSMC-specific homozygous Dicer deficient mice are dilated and thin-walled due to reduced vSMC proliferation. Furthermore, the arteries of these mice exhibit impaired active and passive force contractility and reduced expression of vSMC contractile genes18. Inducible disruption of Dicer in the adult also results in dramatic loss of vSMC function, including decreased vessel contractility, vSMC numbers, and vSMC contractile gene expression19. Altogether, these results strongly suggest that miRNAs are essential not only for the differentiation of vSMC during early development but also for the maintenance of vSMC in the adult. Although dicer knockout mice exhibit a phenotype indicating positive roles of miRNAs in vSMC differentiation and/or maintenance, it is likely that some miRNAs may also play an inhibitory role in vSMC differentiation and/or maintenance. Thus, it is important to interrogate the function of individual miRNAs in vSMC phenotype regulation.
miR-143/145: critical regulators of vSMC differentiation
miR-143 and miR-145 are very well characterized in the vasculature and known as critical regulators of vSMC differentiation.
Function of miR-143/145
In vitro analysis indicates that miR-143 and miR-145 promote multiple aspects of vSMC contractile phenotype (Fig. 1A). In particular, the expression of contractile genes is elevated following over-expression of miR-143 and miR-14520-22. Multiple targets of miR-143 and miR-145 have been identified including KLF4, KLF5, ELK1, Versican, several actin remodeling proteins, and angiotensin-converting enzyme23 (Fig. 2). Interestingly, all of these proteins are antagonistic to the vSMC differentiation, thus repression of these targets by miR-143/145 facilitates vSMC differentiation21-24. In particular, miR-145-mediated repression of KLF4 is required for BMP4- or TGF-β-mediated induction of contractile gene expression20-21. As more targets of miR-143/145 are identified, it will be crucial to examine how downregulation of such targets affects vSMC phenotype.
Fig 2. Summary of miR-143/145 regulation.

Diverse extracellular signals promote vSMC differentiation through regulation of miR-143/145 expression. Conversely, PDGF inhibits miR-143/145 expression both transcriptionally and post-transcriptionally at the Drosha processing step. Following transcription, pri-miR-145 is processed by Drosha and exported out of the nucleus by exportin-5. Dicer promotes the final cleavage step to generate mature miR-143 and miR-145 in the cytoplasm, which then promotes vSMC differentiation through the inhibition of pro-synthetic factors, such as KLF4.
The role of miR-143/145 in vSMC phenotype is further supported by in vivo analysis. To date, several mouse models of miR-143/145 deletion have been generated22, 24-27. Despite different transgenic strategies, all studies have found that loss of miR-143 and miR-145 significantly compromises vSMC contractile phenotype. The blood vessels of miR-143/145 knockout animals are thinner and contain fewer vSMCs when compared to control animals. Furthermore, knockout animals exhibited reduced systolic and diastolic blood pressures, supporting an important role for miR-143/145 in the maintenance of vascular tone27.
Regulation of miR-143/145 expression
miR-143 and miR-145 are encoded in close proximity on chromosome 5 and are transcribed as a bicistronic transcript from a common promoter27 (Fig. 2). In vivo analysis in mouse indicates that the region ~0.9 kb upstream of miR-143 is sufficient for regulated expression during development22, however, additional regulatory regions may also play a role in the expression of miR-143/145 in response to various stimuli. The miR-143/145 promoter contains several highly conserved cis-acting elements which represent potential binding sites for Nkx2.522 and SRF22, 24 Indeed, the SRF co-factors Myocardin and MRTF-A/B strongly activate the miR-143/145 promoter both in vivo and in vitro22, 24.
The expression of miR-143/145 is regulated by cytokines that control vSMC differentiation. For example, pri-miR-143/145 is dramatically elevated by BMP4 and TGF-β20. While MRTF-A is required for BMP-4 mediated induction of miR-143/145, Myocardin is required for induction by TGF-β20 (Fig. 2). BMP4 enhances MRTF-A nuclear localization by activating Rho signaling and actin polymerization and promotes transcription of pri-miR-143/1454, 20. Conversely, TGF-β elicits the rapid transcriptional induction of Myocardin20 (Fig. 2). Interestingly, time-course analysis showed that TGF-β induction of miR-143/145 is more rapid than that for MRTF-A. The miR-143/145 promoter additionally contains a Smad response element which can be directly activated in response to TGF-β signal21. Thus, at early time points following TGF-β, Smads induce both miR-143/145 as well as Myocardin, while sustained elevation of miR-143/145 requires Myocardin induction. Pri-miR-143/145 transcription is also regulated through activation of Notch by Jagged-1 (Jag-1)28 (Fig. 2). Similarly to BMP and TGF-β, Jag-1 promotes differentiation of vSMC through activation of Notch receptors. Activation of Notch results in CBF1 binding to the miR-143/145 promoter and transcriptional activation of miR-143/14528 (Fig. 2). Importantly, induction of miR-143 and miR-145 is required for the pro-differentiation activities of Jag-128.
In addition to positive regulation by pro-differentiation factors, miR-143/145 are negatively regulated in response to pro-synthetic stimuli, such as PDGF26. PDGF-BB promotes activation of Src, which in turn inhibits p5329. As p53 promotes miR-143/145 expression through both transcriptional29 and post-transcriptional15mechanisms, reduced p53 activity leads to loss of miR-143/145 expression (Fig. 2). Altogether, these results position miR-143/145 as a central target of multiple growth factor signaling pathways that regulate vSMC phenotype.
Clinical Relevance of miR-143/145
The expression of miR-143/145 is dramatically reduced in several models of vascular disease, including carotid artery ligation injury in mouse, carotid balloon injury in rat, and ApoE knockout mice22, 25, 27. In each of these studies miR-143/145 was dramatically downregulated in the experimental condition compared to controls22, 25, 27. Importantly, overexpression of miR-145 reduced neointimal formation following balloon injury, suggesting that modulation of miRNA expression may represent a viable therapeutic option for vSMC proliferative disorders 26. The significance of miR-143/145 in human vascular diseases is suggested by the observation that miR-143/145 levels are decreased in aorta from patients with aortic aneurism 27. Furthermore, lower levels of circulating miR-145 are detected in the serum of patients with coronary artery disease 30. This finding suggests the exciting possibility that the level of miR-145 in circulation may serve as a biomarker for coronary artery disease and potentially other vascular disorders. Unlike vSMC-specific dicer knockout mice, miR-143/145 knockout animals are viable through adulthood22, 25, 27. Therefore, although miR-143/145 may be critical for the homeostasis of vSMC, additional miRNAs are required to provide robust control of vSMC differentiation during development and in response to cellular stimuli.
Additional miRNAs promote vSMC differentiation
miR-1
Similarly to miR-143/145, Myocardin is also reported to regulate the expression of miR-1 in vSMC31. Overexpression of Myocardin in human aortic SMC strongly induces the expression of miR-1 and inhibits SMC proliferation31. Myocardin-induced inhibition of SMC proliferation is partially reduced when miR-1 is repressed by a miRNA inhibitor against miR-1, indicating that Myocardin-induced inhibition of SMC proliferation is in part mediated by miR-131. Moreover, Pim-1, a serine/threonine kinase shown to promote SMC proliferation, is found to be a direct target of miR-131. Interestingly, compared to uninjured controls, the expression of Myocardin and miR-1 is significantly reduced in neointimal lesions induced by carotid artery ligation in mice, while the expression of Pim-1 is upregulated31. Together, these observations suggest that induction of miR-1 by Myocardin results in downregulation of Pim-1, which leads to an inhibition of vSMC proliferation. Furthermore, miR-1 also targets KLF4 during all-trans retinoic acid-induced differentiation of SMC in mouse embryonic stem cells32. The observation that both miR-1 and miR-145 repress KLF4 during vSMC differentiation indicates that repression of inhibitory pathways by multiple miRNAs is critical for vSMC differentiation.
miR-21
BMP and TGF-β have long been appreciated to promote vSMC differentiation and prevent vSMC switching to synthetic phenotype2. In addition to inducing the transcription of miR-143/145, BMP4 and TGF-β promote increased expression of miR-21 post-transcriptionally. Induction of miR-21 is essential for the differentiation of vSMC by BMP4 and TGF-β. One target of miR-21, which is critical for this process, is Programmed Cell Death Protein 4 (PDCD4)13. Although the precise mechanism is unclear, downregulation of PDCD4 is required for BMP- and TGF-β-mediated vSMC differentiation. Similarly, TGF-β treatment of pulmonary fibroblasts elevates miR-21 and enhanced conversion to myofibroblasts33, suggesting that miR-21 mediated regulation of contractile genes plays a role in multiple cell types. The relevance of miR-21 in regulating vSMC differentiation in response to BMP signaling is confirmed by the observation that miR-21 levels are decreased in vSMC from IPAH patients34.
It is interesting to note that miR-21 has been investigated extensively in tissues outside of the vasculature; these studies suggest that the function of miR-21 is likely to be complex and highly context-dependent. For example, miR-21 is highly expressed in a large variety of solid tumors relative to normal cells and has been shown to both promote and inhibit cell proliferation35. Furthermore, miR-21 is increased in the heart following transaortic banding; however, miR-21 has been reported to both increase and decrease the hypertrophic response of cardiomyocytes35. Similarly, miR-21 may play opposing functions within vSMC. In addition to promoting contractile gene expression in response to BMP4 or TGF-β, miR-21 is found to promote vSMC proliferation and reduce apoptosis36. Moreover, knockdown of miR-21 using antisense oligonucleotides in the rat reduced vascular remodeling following balloon injury in carotid arteries36. Although differentiation processes are typically coupled to a decrease in proliferation, this is not necessarily the case in vSMC. The dual action of miR-21 in promoting both differentiation and proliferation may reflect the interesting possibility that miR-21 could target diverse set of target genes and mediate differential biological outcomes depending on the cellular context.
miRNAs promote vSMC dedifferentiation and proliferation
miR-221
In response to PDGF-BB, vSMC switch from a contractile to a synthetic phenotype1. One aspect of PDGF-mediated vSMC phenotype switch is mediated by induction of miR-22137-38. In response to PDGF treatment, the transcription of pri-miR-221 is rapidly elevated leading to increased mature miR-221 levels within 3 hours37. Mature miR-221 elicits multiple effects on the phenotype of vSMC including increased proliferation and migration, as well as reduced expression of contractile genes. miR-221 promotes vSMC proliferation through the repression of the cyclin dependent kinase (CDK) inhibitor p27Kip137. Inhibition of miR-221 prevented PDGF-mediated reduction of p27Kip1, as well as vSMC proliferation37. Elevation of miR-221 levels is also required for PDGF-mediated repression of contractile genes37. However, this effect is independent of the regulation of p27Kip1 as knockdown of p27Kip1 by small inhibitory RNA (siRNA) had no effect on contractile gene expression37. These results indicate that the expression of contractile genes and cell growth are not coupled, but instead can be regulated by distinct mechanisms. Overexpression of miR-221 is accompanied by dramatic reduction of Myocardin expression37. Interestingly, miR-221 did not target Myocardin directly. This is due to the downregulation of the tyrosine kinase c-Kit which positively regulates Myocardin expression. Similarly to the effect of PDGF-BB, miR-221 overexpression also increased vSMC migration37; however, siRNA-mediated knockdown of p27Kip1 or c-Kit does not recapitulate the PDGF or miR-221-like pro-migratory effect, suggesting that a yet-unknown target of miR-221 is responsible for this aspect of vSMC phenotype switch. Altogether, these findings provide an example of the potency of a single miRNA in mediating diverse cellular outcomes by regulating multiple targets.
It is of note that miR-221 and miR-222 are clustered on the X-chromosome and share a common seed sequence, and some reports indicate that they are transcribed from a common promoter39. The importance of these miRNAs in regulating vSMC switching is further emphasized by the finding that miR-221 and miR-222 are strongly elevated in vivo in vSMC following balloon injury of the vessel38. Importantly, targeted knockdown of miR-221 and miR-222 in the vessel reduced vSMC proliferation and intimal thickening in response to vascular injury. This response is due to elevated expression of p27Kip1, c-Kit and p57Kip2 in response to miR-221/222 knockdown38 which results in inhibition of CDKs. These findings suggest the exciting possibility that modulation of miR-221 and miR-222 may provide an effective treatment in vascular proliferative disorders, such as restenosis following angioplasty.
miR-146a
Similarly to miR-221, miRNA microarray analysis indicates that the expression of miR-146a is elevated in rat balloon-injured arteries compared to uninjured controls36. In addition, the expression of miR-146a was elevated in proliferative vSMC40. Overexpression of miR-146a increased vSMC proliferation, while knockdown of miR-146a attenuated PDGF-mediated increase of vSMC growth. The relevance of miR-146a in vivo is supported by the observation that treatment of balloon-injured rat carotid arteries with antisense oligonucleotides (antagomir) against miR-146a results in reduced neointima formation and vSMC proliferation40. Interestingly, a validated target of miR-146a is KLF440. This is not consistent with the observation that KLF4 promotes synthetic phenotype and targeted by miRNAs with pro-contractile activity, such as miR-14522,24 and miR-132. It is possible that additional targets of miR-146a may be responsible for miR-146a-mediated vSMC proliferation.
Regulation of BMP/TGF-β signal by miRNAs
miR-24
As discussed above, BMP and TGF-β promote and maintain vSMC contractile phenotype. Consistently, inhibition of BMP and/or TGF-β signaling promotes PDGF-mediated induction of synthetic phenotype. One mechanism of inhibition of BMP signaling is mediated by miR-24. In response to PDGF-BB, the expression of miR-24 is elevated and the miR-24 target, Tribbles-like protein 3 (Trb3), is decreased41. Trb3 promotes the degradation of the Smad ubiquitin ligase Smurf1. Reduced expression of Trb3 in response to miR-24 thus increases Smurf1, leading to decreased Smad1 expression and reduced BMP signal41-42. Knockdown and overexpression analysis indicate that miR-24-mediated repression of Trb3 is required for PDGF-mediated induction of synthetic phenotype including induction of vSMC proliferation and repression of contractile gene expression41.
miR-26a
miR-26a promotes vSMC proliferation and attenuates serum starvation-induced vSMC differentiation43. Interestingly, the signal transducers of the BMP signaling pathway; Smad1 and Smad4 are both identified as direct targets of miR-26a and knockdown of miR-26a results in elevation of expression of Smad1 and Smad443. Consistent with inhibition of the Smads, miR-26a overexpression reduces transcription of a promoter containing consensus Smad binding sites43. Together, these studies indicate miRNAs regulate key pathways involved in vSMC differentiation and suggest that disruption of miRNA function may result in disorders associated with altered BMP and TGF-β signaling, such as PAH.
Clinical applications of miRNAs
Modulation of miRNA levels represents an exciting opportunity for the development of novel therapeutics for cardiovascular disorders. It has been just over a decade since the discovery of miRNAs in Humans, however, the first miRNA-based therapy is already in phase II clinical trials 30, 44. Identification of miRNAs that modulate vSMC phenotype in vitro and in vivo is a critical step forward for the use of this technology for the treatment of cardiovascular diseases. Although modulating miRNA expression levels appears to be promising in animal models, much work still needs to be done before miRNA-related therapies can be brought to the clinic. It will be important to find ways to safely and efficiently deliver miRNA mimics or antagomirs to the tissue of interest45. Additionally, since a single miRNA has the potential to target hundreds of genes, it will be critical to better define the targeting potential of miRNAs to reduce the potential of detrimental effects due to unknown targets45. Despite these challenges, the prospect of using miRNAs as therapeutics and diagnostics for vascular diseases in the near future is an exciting possibility. Finally, recent findings that altered levels of miRNA(s) are found in plasma or sera of patients with specific condition may provide a novel approach for the blood-based detection of cardiovascular disorders 30, 46.
Future prospects
miRNAs represent a critical layer of regulation in the control of vSMC phenotype and miRNA expression is often deregulated in vascular disorders, including PAH, post-angioplasty restenosis, and atherosclerosis. Through the regulation of diverse targets, miRNAs regulate gene expression and vSMC differentiation processes. The identification of miRNAs that regulate vSMC phenotypes may provide novel therapeutic targets for the treatment of cardiovascular disease. In addition to modulating the expression of microRNAs themselves, therapeutics which directly modulate expression of miRNA target genes may be useful for the development of novel cardiovascular disorders. Although great progress has been made in the understanding of vSMC regulation by miRNAs, much exciting work remains.
Acknowledgments
We apologize to the many authors in the field whose work is not cited due to page limitations. This work was supported by grants from the National Institute of Health: HD042149 and HL082854 to A.H., T32 HL069770 training grant to B.D-D., LeDucq Foundation to A.H., and the American Heart Association: 0940095N to A.H.
References
- 1.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 2.ten Dijke P, Arthur HM. Extracellular control of TGFbeta signalling in vascular development and disease. Nat Rev Mol Cell Biol. 2007;8:857–869. doi: 10.1038/nrm2262. [DOI] [PubMed] [Google Scholar]
- 3.Tallquist M, Kazlauskas A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004;15:205–213. doi: 10.1016/j.cytogfr.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 4.Lagna G, Ku MM, Nguyen PH, Neuman NA, Davis BN, Hata A. Control of phenotypic plasticity of smooth muscle cells by BMP signaling through the myocardin-related transcription factors. J Biol Chem. 2007;282:37244–37255. doi: 10.1074/jbc.M708137200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441–450. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis BN, Hata A. Regulation of MicroRNA Biogenesis: A miRiad of mechanisms. Cell Commun Signal. 2009;7:18. doi: 10.1186/1478-811X-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- 9.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936. [DOI] [PubMed] [Google Scholar]
- 11.Ozsolak F, Poling LL, Wang Z, Liu H, Liu XS, Roeder RG, Zhang X, Song JS, Fisher DE. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008;22:3172–3183. doi: 10.1101/gad.1706508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fukuda T, Yamagata K, Fujiyama S, Matsumoto T, Koshida I, Yoshimura K, Mihara M, Naitou M, Endoh H, Nakamura T, Akimoto C, Yamamoto Y, Katagiri T, Foulds C, Takezawa S, Kitagawa H, Takeyama K, O’Malley BW, Kato S. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nat Cell Biol. 2007;9:604–611. doi: 10.1038/ncb1577. [DOI] [PubMed] [Google Scholar]
- 13.Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by Drosha. Mol Cell. 2010;39:373–384. doi: 10.1016/j.molcel.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460:529–533. doi: 10.1038/nature08199. [DOI] [PubMed] [Google Scholar]
- 16.Yamagata K, Fujiyama S, Ito S, Ueda T, Murata T, Naitou M, Takeyama K, Minami Y, O’Malley BW, Kato S. Maturation of MicroRNA Is Hormonally Regulated by a Nuclear Receptor. Mol Cell. 2009;36:340–347. doi: 10.1016/j.molcel.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 17.Blakaj A, Lin H. Piecing together the mosaic of early mammalian development through microRNAs. J Biol Chem. 2008;283:9505–9508. doi: 10.1074/jbc.R800002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Albinsson S, Suarez Y, Skoura A, Offermanns S, Miano JM, Sessa WC. MicroRNAs are necessary for vascular smooth muscle growth, differentiation, and function. Arterioscler Thromb Vasc Biol. 2010;30:1118–1126. doi: 10.1161/ATVBAHA.109.200873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Albinsson S, Skoura A, Yu J, DiLorenzo A, Fernandez-Hernando C, Offermanns S, Miano JM, Sessa WC. Smooth muscle miRNAs are critical for post-natal regulation of blood pressure and vascular function. PLoS ONE. 2011;6:e18869. doi: 10.1371/journal.pone.0018869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis-Dusenbery BN, Chan MC, Reno KE, Weisman AS, Layne MD, Lagna G, Hata A. Downregulation of KLF4 by MIR-143/145 is critical for modulation of vascular smooth muscle cell phenotype by TGF-{beta} and BMP. J Biol Chem. 2011 doi: 10.1074/jbc.M111.236950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Long X, Miano JM. TGF{beta}1 utilizes distinct pathways for the transcriptional activation of microRNA 143/145 in human coronary artery smooth muscle cells. J Biol Chem. 2011 doi: 10.1074/jbc.M111.258814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–710. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rangrez AY, Massy ZA, Metzinger-Le Meuth V, Metzinger L. miR-143 and miR-145: Molecular keys to switch the phenotype of vascular smooth muscle cells. Circ Cardiovasc Genet. 2011;4:197–205. doi: 10.1161/CIRCGENETICS.110.958702. [DOI] [PubMed] [Google Scholar]
- 24.Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel-Duby R, Olson EN. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009;23:2166–2178. doi: 10.1101/gad.1842409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boettger T, Beetz N, Kostin S, Schneider J, Kruger M, Hein L, Braun T. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest. 2009;119:2634–2647. doi: 10.1172/JCI38864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009;105:158–166. doi: 10.1161/CIRCRESAHA.109.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV, Peterson KL, Indolfi C, Catalucci D, Chen J, Courtneidge SA, Condorelli G. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ. 2009;16:1590–1598. doi: 10.1038/cdd.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boucher JM, Peterson SM, Urs S, Zhang C, Liaw L. The Mir-143/145 Cluster Is a Novel Transcriptional Target of Jagged-1/Notch Signaling in Vascular Smooth Muscle Cells. J Biol Chem. 2011 doi: 10.1074/jbc.M111.221945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quintavalle M, Elia L, Condorelli G, Courtneidge SA. MicroRNA control of podosome formation in vascular smooth muscle cells in vivo and in vitro. J Cell Biol. 2010;189:13–22. doi: 10.1083/jcb.200912096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fichtlscherer S, De Rosa S, Fox H, Schwietz T, Fischer A, Liebetrau C, Weber M, Hamm CW, Roxe T, Muller-Ardogan M, Bonauer A, Zeiher AM, Dimmeler S. Circulating microRNAs in patients with coronary artery disease. Circ Res. 2010;107:677–684. doi: 10.1161/CIRCRESAHA.109.215566. [DOI] [PubMed] [Google Scholar]
- 31.Chen J, Yin H, Jiang Y, Radhakrishnan SK, Huang ZP, Li J, Shi Z, Kilsdonk EP, Gui Y, Wang DZ, Zheng XL. Induction of microRNA-1 by myocardin in smooth muscle cells inhibits cell proliferation. Arterioscler Thromb Vasc Biol. 2011;31:368–375. doi: 10.1161/ATVBAHA.110.218149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xie C, Huang H, Sun X, Guo Y, Hamblin M, Ritchie RP, Garcia-Barrio MT, Zhang J, Chen YE. MicroRNA-1 regulates smooth muscle cell differentiation by repressing Kruppel-like factor 4. Stem Cells Dev. 2011;20:205–210. doi: 10.1089/scd.2010.0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, Kaminski N, Abraham E. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010;207:1589–1597. doi: 10.1084/jem.20100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caruso P, MacLean MR, Khanin R, McClure J, Soon E, Southgate M, MacDonald RA, Greig JA, Robertson KE, Masson R, Denby L, Dempsie Y, Long L, Morrell NW, Baker AH. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler Thromb Vasc Biol. 2010;30:716–723. doi: 10.1161/ATVBAHA.109.202028. [DOI] [PubMed] [Google Scholar]
- 35.Krichevsky AM, Gabriely G. miR-21: a small multi-faceted RNA. J Cell Mol Med. 2009;13:39–53. doi: 10.1111/j.1582-4934.2008.00556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ Res. 2007;100:1579–1588. doi: 10.1161/CIRCRESAHA.106.141986. [DOI] [PubMed] [Google Scholar]
- 37.Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Induction of microRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J Biol Chem. 2009;284:3728–3738. doi: 10.1074/jbc.M808788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res. 2009;104:476–487. doi: 10.1161/CIRCRESAHA.108.185363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A, Anile C, Maira G, Mercatelli N, Ciafre SA, Farace MG, Agami R. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26:3699–3708. doi: 10.1038/sj.emboj.7601790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun SG, Zheng B, Han M, Fang XM, Li HX, Miao SB, Su M, Han Y, Shi HJ, Wen JK. miR-146a and Kruppel-like factor 4 form a feedback loop to participate in vascular smooth muscle cell proliferation. EMBO Rep. 2011;12:56–62. doi: 10.1038/embor.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan MC, Hilyard AC, Wu C, Davis BN, Hill NS, Lal A, Lieberman J, Lagna G, Hata A. Molecular basis for antagonism between PDGF and the TGFbeta family of signalling pathways by control of miR-24 expression. EMBO J. 2010;29:559–573. doi: 10.1038/emboj.2009.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan MC, Nguyen PH, Davis BN, Ohoka N, Hayashi H, Du K, Lagna G, Hata A. A novel regulatory mechanism of the bone morphogenetic protein (BMP) signaling pathway involving the carboxyl-terminal tail domain of BMP type II receptor. Mol Cell Biol. 2007;27:5776–5789. doi: 10.1128/MCB.00218-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leeper NJ, Raiesdana A, Kojima Y, Chun HJ, Azuma J, Maegdefessel L, Kundu RK, Quertermous T, Tsao PS, Spin JM. MicroRNA-26a is a novel regulator of vascular smooth muscle cell function. J Cell Physiol. 2011;226:1035–1043. doi: 10.1002/jcp.22422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haussecker D, Kay MA. miR-122 continues to blaze the trail for microRNA therapeutics. Mol Ther. 2010;18:240–242. doi: 10.1038/mt.2009.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li T, Cao H, Zhuang J, Wan J, Guan M, Yu B, Li X, Zhang W. Identification of miR-130a, miR-27b and miR-210 as serum biomarkers for atherosclerosis obliterans. Clin Chim Acta. 2011;412:66–70. doi: 10.1016/j.cca.2010.09.029. [DOI] [PubMed] [Google Scholar]
- 46.Scalbert E, Bril A. Implication of microRNAs in the cardiovascular system. Curr Opin Pharmacol. 2008;8:181–188. doi: 10.1016/j.coph.2007.12.013. [DOI] [PubMed] [Google Scholar]