

Abstract
The recruitment and organization of clathrin at endocytic sites first to form coated pits and then clathrin-coated vesicles depend on interactions between the clathrin N-terminal domain (TD) and multiple clathrin binding sequences on the cargo adaptor and accessory proteins that are concentrated at such sites. Up to four distinct protein binding sites have been proposed to be present on the clathrin TD, with each site proposed to interact with a distinct clathrin binding motif. However, an understanding of how such interactions contribute to clathrin coat assembly must take into account observations that any three of these four sites on clathrin TD can be mutationally ablated without causing loss of clathrin-mediated endocytosis. To take an unbiased approach to mapping binding sites for clathrin-box motifs on clathrin TD, we used isothermal titration calorimetry (ITC) and nuclear magnetic resonance spectroscopy. Our ITC experiments revealed that a canonical clathrin-box motif peptide from the AP-2 adaptor binds to clathrin TD with a stoichiometry of 3:1. Assignment of 90% of the total visible amide resonances in the TROSY-HSQC spectrum of 13C-, 2H-, and 15N-labeled TD40 allowed us to map these three binding sites by analyzing the chemical shift changes as clathrin-box motif peptides were titrated into clathrin TD. We found that three different clathrin-box motif peptides can each simultaneously bind not only to the previously characterized clathrin-box site but also to the W-box site and the β-arrestin splice loop site on a single TD. The promiscuity of these binding sites can help explain why their mutation does not lead to larger effects on clathrin function and suggests a mechanism by which clathrin may be transferred between different proteins during the course of an endocytic event.
The major pathway of cellular endocytosis, as well as intracellular vesicle trafficking, involves clathrin, a protein comprised of three clathrin heavy chains (CHCs) and three light chains (CLCs) that associate to form a molecule with the shape of a triskelion. Clathrin triskelia are recruited to endocytic sites where they associate with each other to form lattices that dynamically reorganize as vesicles bud and pinch off of the plasma membrane, so that the vesicle that is ultimately released from the membrane ends up covered by a clathrin shell or coat.1 Clathrin coat formation involves interactions between membrane-associated adaptor or accessory proteins and binding sites located predominately on the clathrin N-terminal β-propeller domain (TD), which is a member of the “WD-40” family of protein interaction modules.2 Crystal structures of complexes between clathrin TD and peptides derived from the adaptors β-arrestin 2 or the AP-3 β-subunit revealed these peptides binding with 1:1 stoichiometry in a groove between propeller blades 1 and 2 (site 1).3 The peptides used in these studies contain single “clathrin-box” motifs [consensus sequence LΦXΦ(DE), where Φ is a hydrophobe and X is any residue]. A distinct TD binding sequence was identified in amphiphysin and dubbed the “W-box” because of the conservation of two tryptophans in the consensus sequence, PWXXW, where P is a polar residue.4 The amphiphysin W-box peptide was observed to bind to a site (site 2) different from that of the clathrin-box peptides, in a deep pocket in the center of the TD. A third peptide binding site on the TD, between propeller blades 4 and 5, was identified for a β-arrestin 1 splice loop (SL) variant from which a consensus sequence of (LI)(LI)GXL was derived.5 Despite the detailed information that these peptide–TD crystal structures provide, the exact role of these interactions in coat assembly has been difficult to define.
In particular, studies showing that mutations in TD that abrogate peptide binding have little to no effect on the recruitment of clathrin to membranes or clathrin-mediated endocytosis (CME) in vivo(6,7) represent a significant challenge to understanding the role of these interactions in clathrin function. Even clathrin in which all three crystallographically defined peptide binding sites were mutationally ablated has been shown to support CME of transferrin receptors at near-WT levels.6 The simplest conclusion from this result, that the TD is not important for endocytosis, was, however, contradicted by the observation that deletion of the TD did disrupt the recruitment of clathrin to membranes and endocytosis6 (a different study concluded that TD deletion disrupted endocytosis, but not clathrin recruitment,8 but the discrepancy in these results has not been resolved). These results suggested that other protein binding sites on the TD existed and led to the identification of a fourth potential protein binding site.6 It was found that mutation of all three of the crystallographically defined peptide binding sites as well as this fourth site was required to disrupt CME, while the presence of a single functional site in TD was sufficient to support endocytosis at near-WT levels.6 This could indicate a high level of redundancy in these interactions, but this conclusion is potentially contradicted by other studies. Small molecules (“pitstops”) that bind to TD site 1 and were shown to inhibit binding of a number of adaptor or accessory proteins to clathrin were shown to disrupt CME in vivo, suggesting that blocking only site 1 on TD was enough to block clathrin function.8 To reconcile these contradictory results, it was subsequently proposed that pitstops could be less specific than believed, and that inhibition of CME by pitstops might be due to their binding to other sites on TD or to other proteins,2 though direct evidence of this has not been obtained.
In an effort to clarify some of these questions, we took an unbiased, solution-based approach to study the interaction of clathrin TD with clathrin-box peptides derived from the AP2 adaptor and accessory protein AP180. We find that these peptides can simultaneously bind not only to clathrin-box site 1 but also to sites 2 (the “W-box” site) and 3 (the β-arrestin splice loop site) on a single TD. The high promiscuity and stoichiometry of binding of peptide to these multiple sites on TD may underlie the functional redundancy of these sites and may be important for the dynamic reorganization of clathrin TD–protein interactions during coat assembly on membranes.
Experimental Procedures
Plasmids and Peptides
A construct of bovine clathrin TD (residues 1–363) fused to GST was kindly provided by L. Traub.9 Three clathrin-box peptides were synthesized commercially (GenScript USA Inc., AnaSpec, Inc.). AP180 peptide 1 (CSPAKAESSGVIDLFGDAFGSGASETQ) (residues 627–652 of mouse AP180) and AP180 peptide 2 (CPQAVASSSASADLLAGFGGSFMAPS) (residues 655–679 of mouse AP180) were selected from the M5 fragment of the AP180 C-terminal domain (residues 623–680 of mouse mAP180),10 and the AP2 peptide (CQVIPSQGDLLGDLLNLDLGPPVNVPQ) was designed from the hinge of the AP2 β2 subunit (residues 619–644).11 An AP2 peptide containing three changes in its clathrin-box motif to greatly reduce the level of TD binding was also produced (CQVIPSQGDLLGAAANLDLGPPVNVPQ).
Expression and Purification of the Clathrin Terminal Domain
GST-clathrin TD was expressed in freshly transformed Escherichia coli BL21(DE3) pLysS host cells (Stratagene) selected on LB plates containing 25 μg/mL carbenicillin and 17 μg/mL chloramphenicol. Cells were cultured in 2xYT at 30 °C containing 50 μg/mL carbenicillin. Protein expression was induced as described previously.10 Cells were cultured in 4 L of LB until the OD600 reached 0.7–0.8. Cells were pelleted at 5000g and 4 °C for 6 min prior to being transferred into 1 L of M9 minimal medium containing 50 μg/mL carbenicillin, 1 g/L [15N]NH4Cl, and 3 g/L [13C]glucose for preparation of a partially deuterated sample, or [2H,13C]glucose for preparation of a perdeuterated sample. All reagents added to the growth medium were prepared in 99.99% 2H2O. After incubation for 1 h at 30 °C, expression was induced by adding 1 mL of 1 M IPTG. Expression of GST-clathrin TD with selective amino acid labeling was performed similarly in 1 L of M9 minimal medium containing 50 μg/mL carbenicillin, 1 g/L NH4Cl, 3 g/L glucose, and a mixture of unlabeled l-amino acids (42 mg of alanine, 126.5 mg of arginine, 42 mg of asparagine, 50 mg of aspartate, 36.5 mg of cysteine, 730 mg of glutamine, 850 mg of glutamate, 10 mg of glycine, 230 mg of histidine, 39.5 mg of isoleucine, 39.5 mg of leucine, 55 mg of lysine, 45 mg of methionine, 50 mg of phenylalanine, 230 mg of proline, 420 mg of serine, 35.5 mg of threonine, 20.5 mg of tryptophan, 18 mg of tyrosine, and 35 mg of valine, plus 67.5 mg of adenine, 40 mg of cytosine, 45.5 mg of guanine, 40.5 mg of thymine, and 11.2 mg of uracil), where the desired amino acid was replaced with the 15N-labeled form (leucine, lysine, tyrosine, tryptophan, or phenylalanine was used). Cells were harvested after 28–30 h and frozen at −80 °C. Cells were resuspended in 40 mL of lysis buffer (phosphate-buffered saline containing 100 mM EDTA, 3 mM DTT, 1 mM PMSF, 1 mM benzamidine, 10 μM leupeptin, and 1 μM pepstatin). After sonication, 40 mL of lysis buffer and 4 mL of 20% Triton X-100 were added to the lysate and centrifuged at 125000g for 30 min to remove cellular debris. An 8 mL bed volume of glutathione-Sepharose 4B resin was equilibrated with lysis buffer prior to loading clarified lysate. The resin-bound protein was successively washed with lysis buffer, PBS with 3 mM DTT, and then cleavage buffer [50 mM Tris (pH 8.3), 150 mM NaCl, and 3 mM DTT]. For on-resin cleavage, the resin was equilibrated overnight with cleavage buffer containing 0.2 mg/mL thrombin at 4 °C. The cleaved protein was eluted, and the cleavage reaction was stopped with 1 mM PMSF. The eluted protein was dialyzed against a Tris buffer [20 mM Tris (pH 8.0) and 3 mM DTT] and further purified on a 6.5 mL Q-Sepharose ion-exchange column with a 0 to 600 mM NaCl gradient. The purified proteins were either dialyzed into storage buffer [10 mM Tris (pH 8.0), 3 mM DTT, and 50% glycerol] and kept at −20 °C or dialyzed directly into a nuclear magnetic resonance (NMR) buffer [60 mM NaCl, 30 mM Na2HPO4 (pH 7.5), 0.024% NaN3, and 6 mM [2H]DTT] and concentrated using Centricon 10 at 5000g (Millipore, Billerica, MA).
Isothermal Titration Calorimetry (ITC)
ITC experiments were performed in a Microcal VP-ITC instrument (GE Healthcare). AP2 peptide (14.6 mM) was titrated into 0.48 mM TD40 in 5 μL increments in 25 mM Na2HPO4 (pH 7.5), 50 mM NaCl, and 2 mM β-mercaptoethanol at 25 °C. Data were fit to an N-identical binding sites model with Microcal modified Origin 7 software, as well as processed with NITPIC12 and fit with SEDPHAT13,14 to macroscopic one-, two-, and three-site models. In any given SEDPHAT model, statistical factors relating KD’s of multiple sites were constrained such that the underlying microscopic constants were equal across all sites. Plots were generated using GUSSI.15
NMR Spectroscopy
Assignment of Backbone Amide Resonance Peaks
All NMR experiments were performed at 300 K in buffer containing 25 mM Na2HPO4 (pH 7.5), 50 mM NaCl, 25 mM [2H]DTT, 0.02% NaN3, 10% 2H2O, and 10% [2H]glycerol. NMR spectra were acquired using either a Bruker 700 MHz spectrometer equipped with a cryogenically cooled 5 mm 1H probe equipped with a 13C and 15N decoupler and pulsed-field gradient coils at the University of Texas Health Science Center at San Antonio Biomolecular NMR Core or a Bruker 900 MHz spectrometer equipped with the same cryogenically cooled probe at the University of California (Berkeley, CA). The spectra were processed with NMRPipe and analyzed with Sparky and CcpNmr.16−18 The majority of the backbone assignments of clathrin TD were obtained by analyzing triple-resonance data sets, including TROSY-based HNCA, HNCACB, HNCACB (CB only), HN(CA)CO, HNCO, HN(CO)CA, HN(CO)CACB, and HN(CO)CACB (CB only), recorded at 700 MHz with partially deuterated 15N- and 13C-labeled TD. These were further augmented by analyzing a TROSY-based HNCACB spectrum recorded at 900 MHz with a perdeuterated [13C,15N]TD sample.19−21 Samples of clathrin TD singly labeled with [15N]leucine, -lysine, -tyrosine, -tryptophan, or -phenylalanine were used to facilitate and confirm assignments.
Titration of Clathrin-Box Peptides into 15N-Labeled Clathrin TD
Each peptide was titrated into a solution of 15N-labeled clathrin TD at the indicated concentrations spanning a range of peptide:TD molar ratios of 0–9 (Figure 1S of the Supporting Information). Two-dimensional TROSY-HSQC spectra were collected for each peptide concentration. The weighted average chemical shift changes of the backbone amide 1HN and 15N of each residue in TD were determined by the equation22,23
| 1 |
The weighted average chemical shift change reaches a maximum value, Δδmax, when all the available binding sites on TD are bound with a peptide and can be expressed as
| 2 |
where [pep]bound is the concentration of peptide bound to labeled TD, [TD]total is the total concentration of labeled TD, and n is the number of binding sites on TD. For the reaction of a protein with n independent sites, with the same dissociation constant, the concentration of bound ligand is related to the concentration of unbound peptide by the following equation:24,25
| 3 |
Using these relationships,26,27 the dissociation constants for the clathrin TD–peptide interactions were determined by using OriginPro 9.1 software to perform least-squares nonlinear curve fitting of the weighted average of the peptide-induced chemical shift changes of TD residues with the following equation for n binding sites with identical KD values:
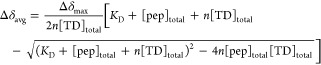 |
4 |
The KD, Δδmax, and n values of TD with each of the clathrin-box peptides were determined independently by globally fitting the residues with chemical shift changes at least two standard deviations above the mean at the end point of the titration.
Results
Each Clathrin N-Terminal Domain Binds Three Clathrin-Box Peptides with Low Affinity
To measure the stoichiometry and affinity of binding of a clathrin-box peptide to TD, we used ITC and a peptide derived from the clathrin binding hinge11 of the β2 subunit of the AP2 adaptor (AP2 peptide, amino acids 619–644) that contains a single canonical clathrin box and has been previously shown to acutely perturb CME upon being injected into nerve terminals.28,29 The displayed thermogram was integrated (Figure 1a) and analyzed (Figure 1b) with an N-identical binding site model using Microcal Origin software, which reported a KD of 420 ± 4 μM, and a stoichiometry of 3.01 ± 0.01 AP2 peptides binding per clathrin TD. Similar results were obtained in three independent experiments, with an average KD of 474 ± 140 μM and a stoichiometry of 3.0 ± 0.2 sites. We obtained statistical support for this model by two approaches. In the original fit, we allowed the stoichiometry N to float as a parameter. Therefore, first we performed a series of fits in which we fixed the stoichiometry N to either 1, 2, 3, 4, or 5 (Figure 2S of the Supporting Information). The only statistical values reported by this software is a χ2/DoF value, and the fits for all the stoichiometries other than 3 were considerably higher than the value obtained when N either floated to 3 or was fixed at 3. The specific increase in χ2/DoF above the value at N = 3 was 4210-fold (N = 1), 208-fold (N = 2), 582-fold (N = 4), and 1963-fold (N = 5). To gain additional statistical support for the model used, we also analyzed the data in SEDPHAT,13 which allows us to take advantage of F statistics. First, the raw data were exported to NITPIC12 (Figure 1c), which was used to integrate the thermogram (Figure 1d). Then the data were fit in SEDPHAT to macroscopic one-, two-, and three-site models (Figure 1d–f). In any given model, statistical factors relating KD’s of multiple sites were constrained such that the underlying microscopic constants were equal across all sites. These microscopic constants were extracted from the macroscopic ones and are reported on the figure, together with the other thermodynamic parameters, the residuals, and the reduced χ2 values for each fit. The three-site model displayed the lowest χ2 and residual values. We then calculated the critical reduced χ2 at a 95% confidence level for the three-site fit using F statistics in SEDPHAT and found it to be 0.00196. The reduced χ2 values of the fits given by the one- and two-site models are both more than 60-fold above this value, indicating that the three-site model is the best fit. The thermodynamic parameters found by the SEDPHAT three-site fit are very similar to those found by the Microcal Origin N-identical site fit. Taken together, the case is strong that each clathrin N-terminal domain binds three clathrin-box peptides with a KD of 480 μM.
Figure 1.

ITC indicates that the clathrin N-terminal domain (TD) binds three clathrin-box AP2 peptides with low affinity. (a) Heat evolution as a function of adding increasing amounts of AP2 peptide to TD. The heats of dilution were measured separately and found to be <1% of the signal at the start of titration so were not considered further in the analysis. (b) Fitting the ITC data with Microcal-modified Origin 7 software to an N-identical site model indicates that each TD binds three AP2 peptides with a KD of 420 ± 4 μM. The fit (red) is overlaid on the data (black). Values shown on the figure are from the fit of the displayed data set. The average and standard deviations of three independent experiments gave the following values: KD = 474 ± 140 μM, N = 3.0 ± 0.2 sites, ΔH = −2.8 ± 0.2 kcal/mol, and TΔS = 1.8 ± 0.4 kcal/mol. (c) The same data set shown in panel a was processed with NITPIC to produce the displayed thermogram. (d) The thermogram shown in panel c was integrated with NITPIC to produce the displayed isotherm. The error bars (blue) come from examining the noise in the interinjection periods. The data were then fit in SEDPHAT to a three-site model. The fit (red) is overlaid on the data (black), with the residual displayed below each isotherm (red). (e) The isotherm shown in panel c was fit in SEDPHAT to a two-site model. (f) The isotherm shown in panel d was fit to a one-site model. Notice the poor quality of the fits to the one- and two-site models, compared to the fit to the three-site models.
NMR Chemical Shifts Identify Three Peptide Binding Sites on the Clathrin TD
To determine where on TD the AP2 peptide was binding, we used NMR chemical shift analysis. To do so, we prepared triply labeled TD (13C, 2H, and 15N) by expressing the protein in minimal 2H2O medium with 15NH4Cl and [2H,13C]glucose as nitrogen and carbon sources, respectively. In addition, TDs selectively labeled with leucine, lysine, tyrosine, tryptophan, or phenylalanine were each prepared to simplify spectra and facilitate assignment of overlapped peaks. Despite its relatively large size (40 kDa), the TROSY-HSQC spectrum of the TD was well-resolved (Figure 2), and fully 90% of the non-proline residues in the protein were assigned by combining TROSY-based triple-resonance data sets acquired with perdeuterated uniformly 13C- and 15N-labeled samples and selectively 15N-labeled samples. Labeled TD was then titrated with increasing amounts of AP2 peptide, as well as with clathrin-box peptides derived from AP180 (AP180 peptide 1, amino acids 627–652; AP180 peptide 2, amino acids 655–679). The AP180 peptides correspond to sequences whose affinities for TD have been previously characterized by AUC and in NMR experiments using unlabeled TD and labeled peptide.10 Addition of any of these peptides led to significant chemical shift changes and/or broadening of multiple TD amide resonances, with the magnitude of the shifts increasing with peptide concentration (Figure 3). When we highlighted the location of the residues that either broaden or show large chemical shift changes (at least two standard deviations above the mean of CSC changes at the end point of the titration) on the TD structure, it was observed that these residues cluster in three patches centered on the three peptide binding sites previously mapped by crystallography (Figure 4). While NMR chemical shift data alone are insufficient to precisely map protein binding sites, our ITC data (Figure 1), together with the available crystallographic data, allowed us to map the chemical shift changes to defined locations on clathrin TD.
Figure 2.

Backbone sequential resonance assignments of clathrin TD. (a) Two-dimensional 1H–15N TROSY-HSQC spectrum and assignments of the amide resonances. The assignable peaks are labeled by residue number; 90% of the total visible peaks could be assigned. (b) Enlarged view of the area boxed in panel a.
Figure 3.

Mapping of chemical shifts induced by peptide binding. (a) TROSY-HSQC spectrum of 15N-labeled TD alone (black) overlaid with spectra of TD in the presence of increasing concentrations of unlabeled AP2 peptide. (b–d) Expanded views of the boxed area selected from panel a. TROSY-HSQC spectra of labeled TD titrated with (b) AP2 peptide, (c) AP180 peptide 1, and (d) AP180 peptide 2. In all panels, black spectra are 15N-labeled TD only, while red, orange, green, blue, and purple spectra correspond to ligand:protein ratios of 1:1, 2:1, 3:1, 5:1, and 9:1, respectively. Arrows indicate the directions of peak shifts upon addition of peptide.
Figure 4.

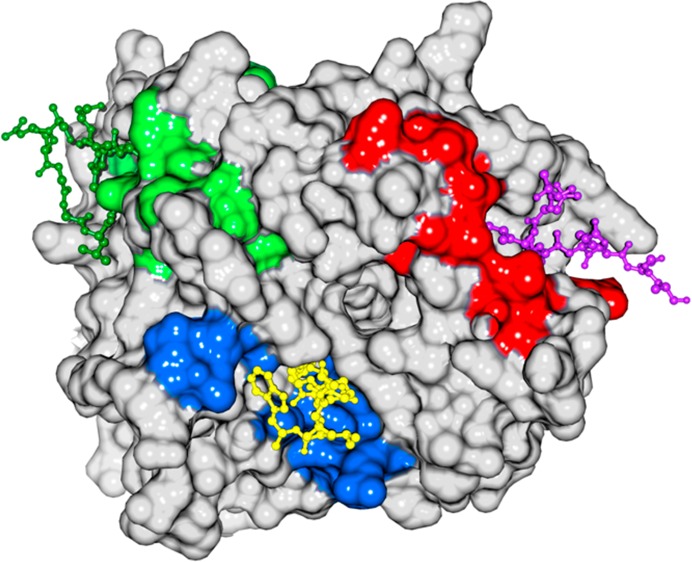
Locations of shifted and broadened residues define three peptide binding sites on TD coincident with those previously defined by crystallography and mutational analyses. (a) Surface representation of TD structure in white with residue peaks that were either broadened or shifted by at least two standard deviations above the mean by the AP2 peptide in either red (site 1), blue (site 2), or green (site 3). (b) As in panel a, but for AP180 peptide 1. (c) As in panel a, but for AP180 peptide 2. (d) Mutations shown to affect binding to β-arrestin 2 (Q89, F91, K96, and K98),33 amphiphysin (F27 and Q152),4 and the β-arrestin 1 splice loop (R188)34 are highlighted on the TD structure in red, blue, and green, respectively. In all panels, a peptide from the β3 subunit of AP-3 [purple, Protein Data Bank (PDB) entry 1C9I], amphiphysin (yellow, PDB entry 1UTC), and the β-arrestin 1 splice loop (green, PDB entry 3GC3) are shown as they occur in the crystal structures of the respective TD–peptide complexes.
To determine if there were residues outside of the three mapped peptide binding sites whose peaks were shifted, we assigned every shifted peak to either site 1, 2, or 3 based on whether it was within 7 Å of the clathrin box,3 W-box,4 or splice loop peptide,5 respectively, in the cocrystal structures of these complexes. Using this criterion, all peaks that were broadened or shifted at least two standard deviations above the mean could be assigned to one of the three sites. If we also examined all peaks shifted at least one standard deviation above the mean, then all but one peak could be assigned to either site 1 (amino acids 40–115), 2 (amino acids 151–153, 171, 172, and 302–305), or 3 (amino acids 185–194 and 232–246), with no overlap between the sites. The only peak that could not be assigned to one of these sites (N355 with AP2Pep) was outside the β-propeller domain and in the helical leg, very near the C-terminus of this construct. The putative fourth protein binding site on TD is defined only by the location of a single mutation at E11, but there were no residues within 7 Å of E11 that were broadened or shifted by at least one standard deviation upon interaction with any of the peptides. While there were some differences in the number and specific residues within each binding site region that were either broadened or shifted by each of the three peptide sequences, these data indicate that all three peptides are occupying the three crystallographically mapped peptide binding sites on the TD, with no detectable binding to any other sites on the β-propeller.
All Three Peptides Bind All Three Sites in TD with Similar, Low Affinities
To assess the affinities of each of the three peptides for the three sites in TD, we plotted the extent of the chemical shifts as a function of peptide:TD ratio and globally fit the data using equations modified from those used previously to account for stoichiometries potentially higher than 1:1 to measure KD values and stoichiometries for binding of labeled peptides to unlabeled TD10 (see Experimental Procedures for details). The values obtained are estimates, because the n-equivalent binding site model is the simplest analytical model that can be used to fit the data. To identify residues to use for fitting, we selected those that exhibited chemical shift changes more than two standard deviations above the mean with each peptide, while eliminating those that were extensively broadened or cases in which the extent of the shift could not be precisely determined because of peak overlap. Peaks were assigned to site 1, 2, or 3 as described above. To have a minimum of three peaks for each site for each peptide in the fitting, we also included V305 (AP2 peptide, AP180 peptide 1, and AP2 mutant peptide) or I302 and I303 (AP180 peptide 2) in the data even though the chemical shifts for these residues were between one and two standard deviations above the mean.
The KD values estimated from these fits were in the range of 800–900 μM, with two to four peptides estimated to bind to each TD molecule (Figure 5). Though the standard error ranges for the KD values were large and it is unlikely that 2–3-fold affinity differences between the different sites would have been detected, inspection of the plots revealed that residues in all three sites shifted similarly in response to increasing peptide concentration and did not justify fitting with separate KD values for each site. Binding was specific as, when we used an AP2 peptide with a mutated clathrin box, the induced chemical shifts at the same residues averaged ∼6-fold smaller than the shifts obtained with the WT peptide and the estimated KD for the mutant was at least 6-fold larger (compare panels a and d in Figure 5).
Figure 5.

KD determinations indicate that all three peptides bind all three sites on TD with similar, low affinities. (a) Chemical shift changes (at least two standard deviations above the mean) induced by AP2 peptide plotted vs peptide:TD ratio and globally fit as described in Experimental Procedures. Fit lines colored red, blue, and green correspond to residues in peptide binding sites 1–3, respectively: KD = 867 ± 246 μM, n = 2.6 ± 0.4, r2 = 0.992, and reduced χ2 = 3.8 × 10–5. (b) As in panel a, but for AP180 peptide 1: KD = 829 ± 221 μM, n = 3.9 ± 0.3, r2 = 0.997, and reduced χ2 = 2.0 × 10–5. (c) As in panel a, but for AP180 peptide 2: KD = 899 ± 529 μM, n = 3.7 ± 0.6, r2 = 0.986, and reduced χ2 = 2.8 × 10–5. (d) As in panel a, but for an AP2 mutant peptide in which the DLL residues at positions 630–632 of the clathrin box were mutated to AAA. Residues and n were as in panel a to match the values of the WT peptide from which this mutant was derived: KD = 5371 ± 1602 μM, r2 = 0.972, and reduced χ2 = 2.1 × 10–5.
Discussion
Crystal structures have suggested that clathrin-box,3 W-box,4 and arrestin splice loop5 peptides bind uniquely to sites 1–3, respectively, on the TD. However, there has been evidence that this conclusion represents an oversimplification. For example, yeast epsin Ent2p is able to bind a TD in which both sites 1 and 2 have been mutated. Deletion of the Ent2p C-terminal clathrin-box sequence eliminates this binding.7 Together, these results indicate that clathrin-box sequences can bind the TD at a site(s) distinct from site 1 or 2. Furthermore, data indicate that the extent of the degeneracy in the sequences known to bind clathrin is even greater than that defined by the already loose consensus motif for these sequences. For example, the AP180 accessory protein contains as many as 12 “clathrin-box” motif sequences that usually diverge from the canonical clathrin-box sequence, LΦPΦP, at positions 3–5 where they frequently have small hydrophobic, polar, and hydrophobic residues, respectively. These AP180 sequences also show a high degree of conservation of a polar residue, most frequently an Asp, preceding the first well-conserved leucine of the motif.30 Despite this divergence, these sequences have been demonstrated to be bona fide clathrin TD binders as determined by AUC, NMR chemical shift, and mutational analyses, which have shown that the most critical residues are the D, L, and Φ at positions −1, 1, and 2, respectively, as defined with respect to the canonical clathrin box.10 As the polar (P) or otherwise unconserved (X) residues in the consensus sequence of the clathrin binding motifs are usually negatively charged in the specific sequences observed in clathrin binding proteins, the common feature of almost all of the clathrin TD binding sequences is the presence of large hydrophobic and negatively charged residues.2
Our results indicate that all three of the crystallographically defined peptide binding sites on TD can bind all of the peptides used in this study, and that they all do so with similar, low affinity, though it is unlikely that our data would have revealed differences in binding affinity of less than a fewfold. If the affinity differences of the different TD sites for a peptide did, however, vary by an order of magnitude or more, then we would have observed residues in the different sites shifting differently in response to peptide concentration; i.e., high-affinity site residues would have shifted at peptide concentrations lower than those of low-affinity site residues. This was not the case, as residues in all three sites were seen to shift similarly in response to increasing peptide concentration. The KD values we measure by ITC or chemical shift analyses are in the range of 400–900 μM and are modestly higher than the values of 200–400 μM measured previously for binding of TD to peptides containing the AP180 peptide 1 and AP180 peptide 2 sequences.10,28 The relatively small differences with these previously determined values may represent differences in the methods used to measure binding. However, significantly tighter binding was seen for binding of an amphiphysin clathrin-box peptide to the TD, where a KD of 22 μM was measured.4 The latter binding studies were, however, conducted in 20 mM Tris with no additional ionic components, while our studies were conducted in 25 mM Na2HPO4, 50 mM NaCl, and 25 mM DTT (for NMR; high DTT concentrations were required to ensure complete sample reduction during extended data collection) or 25 mM Na2HPO4, 50 mM NaCl, and 2 mM β-mercaptoethanol (for ITC), as we found that, at the concentrations required for NMR or ITC, the TD would aggregate under low-ionic strength conditions. Given the ionic contribution to TD–clathrin-box peptide binding (two of the five residues in the amphiphysin clathrin-box element are aspartates), it is simplest to conclude that the tighter binding reported for this peptide reflects the low ionic strength of the buffer used.
The weakness of individual peptide–TD interactions means that multiple interactions are required for stable association with clathrin, as shown by experiments in which the ability of clathrin-box peptides to precipitate clathrin depends on coupling of high densities of such peptides to beads.9,31 Even if clathrin-box peptides bound only to site 1 on TD, each trimeric clathrin molecule would still display three binding sites for each peptide. However, our data indicate that each TD can bind up to three such peptides, which amplifies the potential avidity of the interaction by 3-fold and can help explain why individual binding sites on the TD can be mutationally ablated without significantly compromising CME.2
It has also been proposed that the weakness of individual TD–peptide interactions facilitates the dynamic reorganization of the clathrin as the lattice assembles on the membrane and then morphs into a more spherical structure.10 We can extend these ideas by considering that this reorganization appears to be temporally regulated and involves the transfer of clathrin between different adaptor and accessory proteins during the course of the endocytic event.1,32 Initial recruitment of clathrin to endocytic sites, for example, appears to depend on interactions with Eps15, intersectins, and FCHo proteins; however, subsequently, clathrin interactions are transferred to adaptors and accessory proteins like AP2 and AP180, while Eps15 is displaced to the edge of the endocytic site where it may more effectively recruit more clathrin.1 Differences in the affinity and number of clathrin binding sequences in a protein could be important in guiding this transfer: a protein with tighter binding or more numerous clathrin binding sequences could displace one with weaker or fewer binding elements. Conceivably, such mechanisms could account for how pitstops inhibit CME, even though they are reported to bind to only TD site 1, which can be mutationally ablated with only minimal effects on endocytosis.8 For example, it is possible that TD site 1 binds first to a (relatively) high-affinity clathrin binding element in one protein that is subsequently displaced by weaker binding but more numerous sites on another protein (or vice versa). Pitstops could kinetically impede such transfers and cause endocytosis to stop or slow at certain steps. This is exactly the effect seen for these small molecule inhibitors, which were shown to greatly decrease mobility and increase the lifetime of clathrin and the FCHo2 protein at endocytic puncta.8 In contrast, mutationally ablating TD site 1 could have a weaker effect if simple loss of this site is less detrimental than interfering with its transfer between proteins. Such competitive displacement mechanisms would depend on different proteins competing for the same, multiple binding sites on TD and would be consistent with the promiscuous binding that we report here.
Acknowledgments
We thank Drs. Peter Schuck (National Institutes of Health, Bethesda, MD) and Chad Brautigam (University of Texas Southwestern Medical Center, Dallas, TX) for helpful discussions about data fitting and the implementation of NITPIC, SEDPHAT, and GUSSI for the analysis of ITC data. We would also like to thank Dr. Jeff Pelton (UC Berkeley), who assisted us in the collection of Trosy-based triple-resonance data at the Central California 900 MHz NMR facility.
Glossary
Abbreviations
- AUC
analytical ultracentrifugation
- CHC
clathrin heavy chain
- CLC
clathrin light chain
- CME
clathrin-mediated endocytosis
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- HSQC
heteronuclear single-quantum coherence
- IPTG
isopropyl β-d-galactopyranoside
- ITC
isothermal titration calorimetry
- NMR
magnetic resonance spectroscopy
- PBS
phosphate-buffered saline
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PMSF
phenylmethanesulfonyl fluoride
- SL
β-arrestin 1 splice loop
- TD
N-terminal domain of the clathrin heavy chain (amino acids 1–363)
- WT
wild type.
Supporting Information Available
Peptide and TD concentrations used for each step of the four NMR titrations performed in the study (AP2 peptide, AP180 peptide 1, AP180 peptide 2, and AP2 mutant peptide) (Figure S1) and analysis of ITC data for the titration of AP2 peptide into clathrin TD (Figure S2). This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The clathrin TD chemical shift assignments have been deposited in the Biological Magnetic Resonance Data Bank as entry 25403.
Author Contributions
Y.Z. and K.E.C. contributed equally to this work.
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS029051 to E.M.L. We also gratefully acknowledge the support of the University of Texas Health Science Center at San Antonio (UTHSCSA) Biomolecular NMR Core, the UTHSCSA Center for Macromolecular Interactions, and the UTHSCSA Mass Spectrometry Laboratory, which are supported by the Cancer Therapy and Research Center through the National Cancer Institute P30 Grant CA054174, CPRIT RP120867, and Texas State funds provided through the Office of the Vice President for Research of the UTHSCSA. The 900 MHz NMR data were collected at the Central California 900 MHz NMR facility which is supported by NIH grant GM68933.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- McMahon H. T.; Boucrot E. (2011) Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. 12, 517–533. [DOI] [PubMed] [Google Scholar]
- Lemmon S. K.; Traub L. M. (2012) Getting in touch with the clathrin terminal domain. Traffic 13, 511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Haar E.; Harrison S. C.; Kirchhausen T. (2000) Peptide-in-groove interactions link target proteins to the β-propeller of clathrin. Proc. Natl. Acad. Sci. U.S.A. 97, 1096–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele A. E.; Watson P. J.; Evans P. R.; Traub L. M.; Owen D. J. (2004) Two distinct interaction motifs in amphiphysin bind two independent sites on the clathrin terminal domain β-propeller. Nat. Struct. Mol. Biol. 11, 242–248. [DOI] [PubMed] [Google Scholar]
- Kang D. S.; Kern R. C.; Puthenveedu M. A.; von Zastrow M.; Williams J. C.; Benovic J. L. (2009) Structure of an arrestin2-clathrin complex reveals a novel clathrin binding domain that modulates receptor trafficking. J. Biol. Chem. 284, 29860–29872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willox A. K.; Royle S. J. (2012) Functional analysis of interaction sites on the N-terminal domain of clathrin heavy chain. Traffic 13, 70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collette J. R.; Chi R. J.; Boettner D. R.; Fernandez-Golbano I. M.; Plemel R.; Merz A. J.; Geli M. I.; Traub L. M.; Lemmon S. K. (2009) Clathrin functions in the absence of the terminal domain binding site for adaptor-associated clathrin-box motifs. Mol. Biol. Cell 20, 3401–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kleist L.; Stahlschmidt W.; Bulut H.; Gromova K.; Puchkov D.; Robertson M. J.; MacGregor K. A.; Tomilin N.; Pechstein A.; Chau N.; Chircop M.; Sakoff J.; von Kries J. P.; Saenger W.; Krausslich H. G.; Shupliakov O.; Robinson P. J.; McCluskey A.; Haucke V. (2011) Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 146, 471–484. [DOI] [PubMed] [Google Scholar]
- Drake M. T.; Traub L. M. (2001) Interaction of two structurally distinct sequence types with the clathrin terminal domain β-propeller. J. Biol. Chem. 276, 28700–28709. [DOI] [PubMed] [Google Scholar]
- Zhuo Y.; Ilangovan U.; Schirf V.; Demeler B.; Sousa R.; Hinck A. P.; Lafer E. M. (2010) Dynamic interactions between clathrin and locally structured elements in a disordered protein mediate clathrin lattice assembly. J. Mol. Biol. 404, 274–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih W.; Gallusser A.; Kirchhausen T. (1995) A clathrin-binding site in the hinge of the β 2 chain of mammalian AP-2 complexes. J. Biol. Chem. 270, 31083–31090. [DOI] [PubMed] [Google Scholar]
- Keller S.; Vargas C.; Zhao H.; Piszczek G.; Brautigam C. A.; Schuck P. (2012) High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 84, 5066–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.; Piszczek G.; Schuck P. (2015) SEDPHAT: A platform for global ITC analysis and global multi-method analysis of molecular interactions. Methods 76, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigam C. A. (2015) Fitting two- and three-site binding models to isothermal titration calorimetric data. Methods 76, 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H., Brautigam C. A., Ghirlando R., and Schuck P. (2013) Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. In Current protocols in protein science, Chapter 20, Unit 20, 12, Wiley, New York. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Goddard T. D., and Kneller D. G. (2010) Sparky 3, University of California, San Francisco. [Google Scholar]
- Vranken W. F.; Boucher W.; Stevens T. J.; Fogh R. H.; Pajon A.; Llinas M.; Ulrich E. L.; Markley J. L.; Ionides J.; Laue E. D. (2005) The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 59, 687–696. [DOI] [PubMed] [Google Scholar]
- Grzesiek S.; Bax A. (1992) Correlating Backbone Amide and Side-Chain Resonances in Larger Proteins by Multiple Relayed Triple Resonance NMR. J. Am. Chem. Soc. 114, 6291–6293. [Google Scholar]
- Yamazaki T.; Lee W.; Arrowsmith C. H.; Muhandiram D. R.; Kay L. E. (1994) A Suite of Triple Resonance NMR Experiments for the Backbone Assignment of 15N, 13C, 2H Labeled Proteins with High Sensitivity. J. Am. Chem. Soc. 116, 11655–11666. [Google Scholar]
- Yu J.; Simplaceanu V.; Tjandra N. L.; Cottam P. F.; Lukin J. A.; Ho C. (1997) 1H, 13C, and 15N NMR backbone assignments and chemical-shift-derived secondary structure of glutamine-binding protein of Escherichia coli. J. Biomol. NMR 9, 167–180. [DOI] [PubMed] [Google Scholar]
- Grzesiek S.; Bax A.; Clore G. M.; Gronenborn A. M.; Hu J. S.; Kaufman J.; Palmer I.; Stahl S. J.; Wingfield P. T. (1996) The solution structure of HIV-1 Nef reveals an unexpected fold and permits delineation of the binding surface for the SH3 domain of Hck tyrosine protein kinase. Nat. Struct. Biol. 3, 340–345. [DOI] [PubMed] [Google Scholar]
- Garrett D. S.; Seok Y. J.; Peterkofsky A.; Clore G. M.; Gronenborn A. M. (1997) Identification by NMR of the binding surface for the histidine-containing phosphocarrier protein HPr on the N-terminal domain of enzyme I of the Escherichia coli phosphotransferase system. Biochemistry 36, 4393–4398. [DOI] [PubMed] [Google Scholar]
- Klotz I. M.; Hunston D. L. (1971) Properties of graphical representations of multiple classes of binding sites. Biochemistry 10, 3065–3069. [DOI] [PubMed] [Google Scholar]
- Hunston D. L. (1975) Two Techniques for Evaluating Small Molecule: Macromolecule Binding in Complex Systems. Anal. Biochem. 63, 99–109. [Google Scholar]
- Belshaw N. J.; Williamson G. (1993) Specificity of the binding domain of glucoamylase 1. Eur. J. Biochem. 211, 717–724. [DOI] [PubMed] [Google Scholar]
- Fielding L. (2007) NMR methods for the determination of protein-ligand dissociation constants. Prog. Nucl. Magn. Reson. Spectrosc. 51, 219–242. [Google Scholar]
- Morgan J. R.; Prasad K.; Hao W.; Augustine G. J.; Lafer E. M. (2000) A conserved clathrin assembly motif essential for synaptic vesicle endocytosis. J. Neurosci. 20, 8667–8676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine G. J.; Morgan J. R.; Villalba-Galea C. A.; Jin S.; Prasad K.; Lafer E. M. (2006) Clathrin and synaptic vesicle endocytosis: Studies at the squid giant synapse. Biochem. Soc. Trans. 34, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafer E. M. (2002) Clathrin-protein interactions. Traffic 3, 513–520. [DOI] [PubMed] [Google Scholar]
- Drake M. T.; Downs M. A.; Traub L. M. (2000) Epsin binds to clathrin by associating directly with the clathrin-terminal domain. Evidence for cooperative binding through two discrete sites. J. Biol. Chem. 275, 6479–6489. [DOI] [PubMed] [Google Scholar]
- Boettner D. R.; Chi R. J.; Lemmon S. K. (2012) Lessons from yeast for clathrin-mediated endocytosis. Nat. Cell Biol. 14, 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman O. B. Jr.; Krupnick J. G.; Gurevich V. V.; Benovic J. L.; Keen J. H. (1997) Arrestin/clathrin interaction. Localization of the arrestin binding locus to the clathrin terminal domain. J. Biol. Chem. 272, 15017–15022. [DOI] [PubMed] [Google Scholar]
- Kang Y. S.; Zhao X.; Lovaas J.; Eisenberg E.; Greene L. E. (2009) Clathrin-independent internalization of normal cellular prion protein in neuroblastoma cells is associated with the Arf6 pathway. J. Cell Sci. 122, 4062–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.