

Abstract
We describe design parameters for the synthesis and analytical application of a label-free RNA molecular beacon, termed Spinach.ST. The RNA aptamer Spinach fluoresces upon binding the small-molecule fluorophore DFHBI ((Z)-4-(3,5-difluoro-4-hydroxybenzylidene)-1,2-dimethyl-1H-imidazol-5(4H)-one). Spinach has been reengineered by extending its 5′- and 3′-ends to create Spinach.ST, which is predicted to fold into an inactive conformation that fails to bind DHFBI. Hybridization of a trigger oligonucleotide to a designed toehold on Spinach.ST initiates toehold-mediated strand displacement and restores the DFHBI-binding, fluorescence-enhancing conformation of Spinach. The versatile Spinach.ST sensor can detect DNA or RNA trigger sequences and can readily distinguish single-nucleotide mismatches in the trigger toehold. Primer design techniques are described that augment amplicons produced by enzymatic amplification with Spinach.ST triggers. Interaction between these triggers and Spinach.ST molecular beacons leads to the real-time, sequence-specific quantitation of these amplicons. The use of Spinach.ST with isothermal amplification reactions such as nucleic acid sequence-based amplification (NASBA) may enable point-of-care applications. The same design principles could also be used to adapt Spinach reporters to the assay of nonnucleic acid analytes in trans.
1. INTRODUCTION
Nucleic acid probes generated through the covalent conjugation of fluorophores and quenchers have become indispensable for sequence-specific detection and quantitation of nucleic acid molecules and amplicons. Molecular beacons are widely used for real-time, sequence-specific quantitation of nucleic acids (Tyagi & Kramer, 1996) and consist of a central, target-specific, single-stranded loop flanked by a stretch of five to seven complementary nucleotides that can base-pair to form a stem that terminates in a paired fluorophore and quencher. In the absence of a specific sequence target, fluorescence remains quenched due to the proximity of the fluorophore and quencher at the 5′- and 3′-ends of the molecular beacon. Binding of a complementary oligonucleotide to the loop leads to a conformational change that forces the beacon stem to unpair. The ensuing separation of the fluorophore from the quencher results in sequence-specific fluorescence enhancement.
While the widespread use of labeled nucleic acid probes in vitro is well documented, their application as in vivo reporters remains less than ideal due to the expense of chemical synthesis as well as the inefficiency of intracellular delivery. As one possible alternative, several RNA aptamers (Ellington & Szostak, 1990; Tuerk & Gold, 1990) have been described that bind and enhance the fluorescence of small-molecule dyes such as the Hoechst 33258 derivative 2,6-di-tert-butyl-4-[5-(4-methylpiperazin-1-yl)-1H,1′H-2,5′-bibenzo[d]imidazol-2′-yl]phenol (Sando, Narita, Hayami, & Aoyama, 2008); triphenylmethane dyes including malachite green, patent blue VF, and patent blue violet (Babendure, Adams, & Tsien, 2003; Grate & Wilson, 1999; Holeman, Robinson, Szostak, & Wilson, 1998); cyanine dyes such as dimethylindole red (Constantin et al., 2008) and thiazole orange conjugates (Pei, Rothman, Xie, & Stojanovic, 2009), in addition to photoinduced N-(p-methoxyphenyl)piperazine derivatives of 2′,7′-dichlorofluorescein (Sparano & Koide, 2005, 2007). Some of these aptamers have been adapted to function as biosensors whose fluorescence enhancement is triggered by analytes such as nucleotides, theophylline (Furutani, Shinomiya, Aoyama, Yamada, & Sando, 2010; Stojanovic & Kolpashchikov, 2004), and nucleic acid sequences (Afonin, Danilov, Novikova, & Leontis, 2008; Kolpashchikov, 2005).
Recently, another RNA aptamer, termed Spinach, was selected to bind DFHBI ((Z)-4-(3,5-difluoro-4-hydroxybenzylidene)-1,2-dimethyl-1H-imidazol-5(4H)-one), a fluorophore that resembles those found in fluorescent proteins such as green fluorescent protein. A large increase in the green fluorescence emission of DFHBI occurs upon its binding to the aptamer (Paige, Wu, & Jaffrey, 2011; Strack, Disney, & Jaffrey, 2013).
Theoretically, any of these fluorescent RNA aptamers could be adapted to function as in vivo reporters via transcription and dye uptake. In particular, Spinach has been adapted to act as a genetically encoded sensor for RNA transcription in cells (Pothoulakis, Ceroni, Reeve, & Ellis, 2013), as an imaging agent, as a sensor for the intracellular detection of small-molecule analytes such as adenosine 5′-diphosphate (ADP) and S-adenosylmethionine (SAM) (Paige, Nguyen-Duc, Song, & Jaffrey, 2012), and as a sensor for proteins such as streptavidin, thrombin, and the MS2 coat protein (Song, Strack, & Jaffrey, 2013).
To create a more versatile, trans-acting reporter module, we reengineered Spinach to act as a conformational switch that can be triggered by toehold-mediated nucleic acid strand displacement (Bhadra & Ellington, 2014a; Zhang & Seelig, 2011). Similar programmable nucleic acid strand displacement reactions underlie multiple nucleic acid assays and devices, including molecular logic circuits and motors, catalytic amplifiers, and reconfigurable self-assembled nanostructures (Andersen et al., 2009; Chen & Ellington, 2010; Han, Pal, Liu, & Yan, 2010; Li, Ellington, & Chen, 2011; Qian & Winfree, 2011; Qian, Winfree, & Bruck, 2011; Seelig, Soloveichik, Zhang, & Winfree, 2006; Yin, Choi, Calvert, & Pierce, 2008; Yurke, Turberfield, Mills, Simmel, & Neumann, 2000; Zhang & Seelig, 2011). The resultant sequence-dependent Spinach (Spinach.ST) is the aptamer-based equivalent of a molecular beacon that fluoresces only upon hybridization with specific target sequences. However, this reagent no longer relies on the chemical conjugation of fluorophore and quencher moieties.
The sequence specificity of Spinach.ST can be readily altered by in silico design and cotranscriptional folding algorithms, and thus new sensors can be quickly built and then directly transcribed either in vitro or potentially in vivo. Spinach.ST can be readily inserted into layered nucleic acid circuits to integrate and report function. As an example, we have used Spinach.ST for the real-time quantitation of the output from a catalyzed hairpin assembly (CHA) circuit in which the circuit input, processors, and output are all composed of transcribed RNA. We have also developed novel in vitro one-pot enzymatic isothermal nucleic acid assay systems in which Spinach.ST molecular beacons are transcriptionally generated in situ and then report the accumulation of target amplicons in real-time. Single-nucleotide mismatches can also be distinguished within the target amplicons.
In the following sections, we elaborate the design principles for engineering Spinach.ST molecular beacons and their triggers. We also describe techniques to synthesize and measure the activity of Spinach.ST beacons in vitro. Finally, we detail the application of Spinach.ST molecular beacons for sequence-specific signal transduction of isothermal nucleic acid sequence-based amplification (NASBA).
2. HOW TO ENGINEER SPINACH MOLECULAR BEACONS TRIGGERED BY TOEHOLD-MEDIATED STRAND DISPLACEMENT
The 80-nucleotide minimized Spinach aptamer (24-2-min) (Paige et al., 2011) can be reengineered into a molecular beacon (Spinach.ST) by placing oligonucleotide extensions at its 5′- and 3′-ends, forming a nonbinding conformation. In the presence of a trigger nucleic acid, toehold-mediated strand displacement will lead to the formation of a binding conformation and aptamer fluorescence (Figs. 1 and 2).
Figure 1.

Engineering the Spinach.ST molecular beacon. (A) The minimum free energy (MFE) structure of the minimized 80-mer RNA aptamer Spinach 24-2-min as predicted by NUPACK. Nine nucleotides at the 5′- and the 3′-ends of the aptamer (designated as domains 6* and 6.1, respectively) hybridize to form the basal stem (highlighted in gray). (B) The minimized Spinach aptamer was extended at its 3′-end with a duplicate domain 6.1 (designated as domain 6.2) and is predicted to fold into an alternate MFE conformation by NUPACK. Domain 6.2 hybridizes with domain 6* and disrupts the basal stem of the Spinach aptamer. (C) The NUPACK-predicted MFE conformation of Spinach when extended with an additional domain 5* at its 5′-end and with domains 2 and 5 at its 3′-end. The Spinach.ST1 sequence is depicted. The aptamer domains 6* and 6.1 that form the Spinach 24-2-min basal stem are unable to interact. The extended domains 5 and 6.2 (outlined) hybridize with complementary domains 5* and 6* to form a long stem that leaves the unpaired domain 2 free to act as a toehold. (D–F) The Spinach.ST molecular beacon is stabilized by flanking it with tRNA sequences that fold into hairpins. Structures of Saccharomyces cerevisiae tRNAtrp flanks (D) and human tRNALys flanks (E) as predicted by NUPACK are depicted. Structures of Saccharomyces cerevisiae tRNAtrp flanks generated by mFold are depicted in (E).
Figure 2.
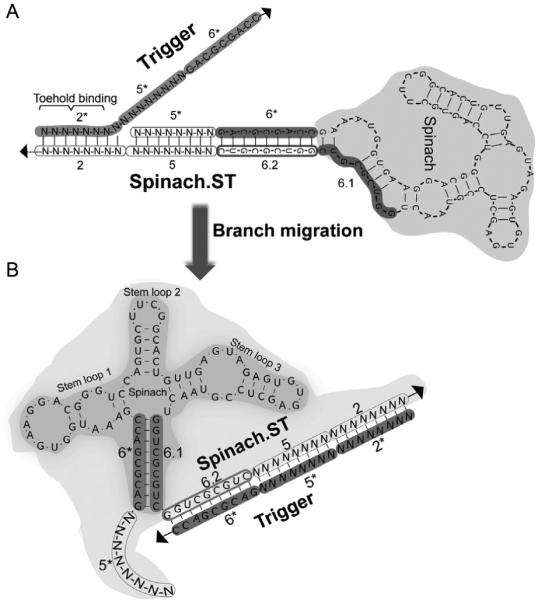
Sequence-specific activation of Spinach.ST molecular beacon by toehold-mediated strand displacement. Sequences of the invariant (aptamer-derived) domains 6*, 6.1, and 6.2 are shown, while the variable (target-derived) domains 2 and 5 (and their complementary domains 2* and 5*) are depicted as strings of “N.” (A) Trigger toehold domain 2* initiates strand displacement upon binding to its complementary domain 2 in Spinach.ST. (B) Toehold hybridization initiates branch migration leading to contiguous hybridization of the trigger domains 2*, 5*, and 6* to the Spinach.ST 3′-end extended domains 2, 5, and 6.2, respectively. The ensuing conformational rearrangement restores the Spinach 24-2-min aptamer structure, including the basal stem formed by base pairing between domain 6* and 6.1. All structures were generated using NUPACK.
2.1 Engineering conformations and sequence modules for Spinach beacons
In its predicted minimum free energy (MFE) conformation at 37 °C, the minimized Spinach aptamer has a predicted folding free energy of −30.4 kcal/mol and bears three internal stem loops and a basal stem that is formed by base pairing between the nine terminal nucleotides at its 5′- and 3′-ends (Fig. 1A). While stem loops 1 and 3 have sequence-specific roles in inducing DFHBI fluorescence, stem 2 appears to play a structural role in aptamer function (Paige et al., 2012).
For simplicity, we designate the terminal nine-nucleotide-long complementary sections that form the basal stem of the aptamer as domains 6* (at the 5′-end) and 6.1 (at the 3′-end) (Fig. 1A). In nucleic acid devices and circuits driven by toehold-mediated strand displacement, the term “domain” is typically used for referring to short stretches of sequences that act as a unit in hybridization, branch migration, dissociation, structure, or function (Zhang, 2010). To create Spinach.ST, we appended a duplicate domain 6.1 (now designated as domain 6.2) to the 3′-end of the aptamer (Fig. 1B). This molecule is predicted to assume a conformation (with a free energy of −37.4 kcal/mol) in which the 5′-domain 6* hybridizes with the duplicate domain 6.2 and forces the aptamer to misfold such that stem loops 1 and 3 are disrupted and stem loop 2 is altered. To favor this misfolding pathway, we also appended eight-nucleotide-long complementary domains 5* and 5 to the 5′- and 3′-ends of the aptamer, respectively. The resulting continuously base-paired stretch of domains 5* and 6* lends additional thermodynamic stability to the misfolded state of Spinach.ST (Fig. 1C).
Disruption of the stem loop organization in Spinach.ST should render it unable to enhance the fluorescence of DFHBI. To allow sequence-specific activation of Spinach.ST, we further extended the molecular beacon at its 3′-end with a unique eight-nucleotide-long sequence termed domain 2 (Fig. 1C). This domain should remain unpaired in the predicted secondary structures of Spinach.ST (Fig. 1C). This single-stranded domain 2 can thus act as a toehold for binding complementary domain 2* in trigger oligonucleotides. A contiguous arrangement of domains 2*-5*-6* should, in turn, lead to the initiation and propagation of branch migration (Fig. 2A), ultimately releasing domains 5* and 6* at the 5′-end of Spinach.ST while sequestering domains 2, 5, and 6.2 at the 3′-end. The newly exposed 6* domain can potentially pair with domain 6.1, leading to conformational rearrangement of Spinach into an active structure that enhances DFHBI fluorescence (Fig. 2B).
It should be noted that the Spinach.ST domain numbering is a user-defined appellation. We have numbered the Spinach.ST domains discontinuously starting with domain 2 instead of 1 to maintain constancy with our previous publications. The prototype Spinach.ST was designed as a signal transducer for an upstream RNA circuit that included sequence domains 1 through 6 (Bhadra & Ellington, 2014a). The Spinach.ST domains that allowed it to interact with the circuit were accordingly designated as domains 2, 5, and 6.2.
As detailed above, there are two critical structural constraints for engineering Spinach.ST molecular beacons: (i) the toehold domain should remain fully exposed and (ii) the branch migration domains should sequester aptamer domain 6* by hybridization with the duplicate domain 6.2 (Fig. 1C). However, the design principles are broadly applicable and the sequence space available for designing the aptamer extension domains is expected to be large. We have designed three versions of Spinach.ST that are exclusively activated by different trigger sequences (Bhadra & Ellington, 2014b). The lengths and sequences of the toehold (domain 2) and branch migration (domains 5 and 6.2) domains appended to the minimal Spinach sequence were chosen based on previously described kinetic and thermodynamic considerations (Bhadra & Ellington, 2014a; Li et al., 2011). The effective rate constant of strand displacement is known to rise exponentially with increasing toehold length, reaching a plateau somewhere between 5- and 10-nucleotide toeholds (Yurke & Mills, 2003; Zhang & Winfree, 2009). In general, toehold binding should be strong enough to initiate strand displacement effectively. We commonly use eight-nucleotide-long toeholds (with 37–50% GC content and −10 to −13 kcal/mol free energy of binding) at 37–52 °C for operation of toehold-mediated strand displacement circuits composed of RNA under physiological as well as other salt concentrations.
The duplex structures of the branch migration domains of Spinach.ST should be strong enough to prevent unwanted intra- or intermolecular hybridizations. Furthermore, the total number of base pairs that can form between the cognate trigger and Spinach.ST (a combination of the toehold domain 2 and branch migration domains 5 and 6.2) should be long enough to discriminate effectively against nontarget sequences. Unnecessarily long extensions would likely slow down the conformational switching and/or frustrate the folding pathway for the Spinach beacon. The sequence and length (nine nucleotides) of the appended domain 6.2 is fixed in advance because it is designed to be complementary to the Spinach aptamer domain 6*. The other appended branch migration domains (5 and its complement 5*) are typically designed to be eight-nucleotides long and have a near 50% GC ratio. Thus, the effective trigger length is 25 nucleotides (eight-nucleotide-long toehold domain 2* combined with eight-nucleotide-long branch migration domain 5* and the nine-nucleotide-long branch migration domain 6*), which should ensure a very high degree of sequence specificity in Spinach.ST activation. We have also successfully created Spinach.ST designs for probing longer trigger sequences by including an additional eight-nucleotide-long branch migration domain between domains 5 and 6.2 (Bhadra & Ellington, 2014b).
The branch migration domains of Spinach.ST may be designed to include mismatches that create bubbles within the stem; such mismatches can impact both the thermodynamics and kinetics of reporter function. We have tested molecules containing two mismatches, one located at position 6 of domain 6.2 and the other at position 3 of domain 5 (Fig. 3A). Such designs might be more suitable for favoring strand displacement of longer stem domains by perfectly matched trigger oligonucleotides (especially DNA triggers). Also, periodic interruption of long RNA duplex regions with unpaired, bulged out nucleotides provides protection from RNase III-mediated cleavage in bacteria and disfavors the activation of RNA-activated protein kinase (PKR), a vital component of the mammalian innate immune response (Court, 1993; Heinicke, Nallagatla, Hull, & Bevilacqua, 2011; Hjalt & Wagner, 1995). We have found that trigger sequences that preserve these mismatches in the branch migration domains are still capable of activating cognate Spinach.ST molecules (Figs. 3B and 6A).
Figure 3.

Engineering Spinach.ST with mismatched nucleotides. (A) Single mismatched bases introduced into the Spinach.ST1 domains 5 and 6.2 are depicted. The resulting molecule is still predicted to fold into the characteristic Spinach.ST conformation, but with two bulges within the extended duplex. (B) Spinach.ST trigger oligonucleotides harboring single mismatched bases in domains 5* and 6* are predicted to bind Spinach.ST and trigger its conformational rearrangement. All structures were generated using NUPACK.
Figure 6.

Sequence-specific activation of Spinach.ST molecular beacons. Spinach.ST.2m (A) (for structure, see Fig. 3) and Spinach.ST (B) (for structure, see Fig. 1) flanked by tRNA and embedded within larger transcripts (for structure, see Fig. 4) were incubated with cognate or scrambled trigger oligonucleotides that were similarly flanked by tRNA and expressed as a portions of larger transcripts. Spinach molecular beacons were activated only by cognate trigger-containing transcripts and led to the accumulation of DFHBI fluorescence over time.
2.2 Using NUPACK and KineFold for design
The sequences of the Spinach extension domains were determined by in silico conformation modeling. We have designed Spinach.ST molecular beacons by an iterative process of manual sequence design guided by in silico thermodynamic modeling of RNA MFE structures and probabilities of intermolecular hybridization, and by stochastic folding simulations of cotranscriptional RNA folding kinetics. The goal of these design and modeling iterations was to generate sequences that should display the desired Spinach.ST MFE nonbinding structure (depicted in Fig. 1) and also to ensure that this structure will be kinetically favored along the cotranscriptional RNA folding pathway.
We typically use the NUPACK Web server (accessible at http://www.nupack.org/) for modeling MFE conformations of individual RNA sequences and for determining the structure and probability of complex formation with an interacting RNA (Dirks, Bois, Schaeffer, Winfree, & Pierce, 2007; Dirks & Pierce, 2003, 2004; Zadeh et al., 2011). All RNA analyses are performed in the “Analysis” mode of NUPACK with the following parameters. RNA energy parameters can currently be toggled between “Serra and Turner (1995)” and “Mathews, Sabina, Zuker, & Turner (1999),” and we typically perform Spinach and Spinach.ST folding and interaction analyses at 37 °C using the Serra and Turner RNA folding energy parameters (Serra & Turner, 1995). Three options (None, Some, All) are available on the NUPACK Web server for factoring in the effect of unpaired (dangling) bases on the folding landscape. We use the default “Some” dangle treatment option, in which dangle energy is incorporated for each unpaired base flanking a duplex. In addition, the NUPACK Web server does not allow the incorporation of pseudoknots into RNA structures of sequences longer than 100 nucleotides. Since most of the engineered Spinach sequences are greater than 100 nucleotides, we do not allow pseudoknots during the NUPACK analysis.
Both kinetic and thermodynamic folding considerations must be taken into account during design. Spinach.ST and its RNA trigger sequences will eventually be synthesized by enzymatic transcription of duplex DNA templates. RNA is known to undergo sequential or cotranscriptional folding that often involves the formation of transient structures that guide the folding pathway toward kinetically trapped secondary structures (Kramer & Mills, 1981). It is possible that the thermodynamically predicted, non-binding conformations of the Spinach.ST molecular beacons will be disfavored during cotranscriptional folding of RNA. Therefore, it is critical to guide the iterative design process by modeling the cotranscriptional folding of the engineered RNA sequences. To this end, we typically use the KineFold Web server (accessible at http://kinefold.curie.fr/) to model cotranscriptional folding (Xayaphoummine, Bucher, & Isambert, 2005). Since we use T7 RNA polymerase for in vitro transcription of Spinach.ST, the cotranscriptional folding of the molecular beacons was simulated at the transcription speed of T7 RNA polymerase.
The end product of the design process is RNA sequences that are predicted (by both NUPACK and KineFold) to fold into the Spinach.ST molecular beacon conformation as depicted in Fig. 1. The interaction of these newly designed Spinach.ST molecular beacons with their complementary trigger sequences (with the domain organization of 2*-5*-6*) can then be modeled using NUPACK to ensure that the Spinach molecular beacon will bind its trigger sequence and result in structural reorganization of the Spinach.ST beacon into a binding, fluorescent conformation (Fig. 2). For analyzing intermolecular interactions using NUPACK, set the “Number of strand species to 2”: Spinach.ST and its trigger. Also set the maximum number of interacting species in NUPACK to 2. It is best to model the interaction between Spinach.ST and its trigger at various stoichiometries, starting with equimolar conditions and potentially at different concentrations. Under all conditions tested, the trigger should form a complex with its cognate Spinach.ST molecular beacon with near 100% probability (as predicted by NUPACK), indicating activation of the molecular beacon. However, since NUPACK does not take kinetic traps into account when predicting intermolecular complexes, it is critical to examine the predicted structures visually. For example, if the toeholds in the trigger sequence or Spinach.ST molecular beacon were occluded, NUPACK would still predict complete hybridization between the two molecules, although of course, in reality, the absence of an exposed toehold will prevent any such productive interactions. To evaluate whether and how the Spinach molecular beacon might be nonspecifically triggered, scrambled or mismatched trigger sequences can be substituted for the cognate trigger during modeling.
2.3 Promoters for Spinach.ST expression
The speed of transcription is known to influence RNA folding pathways (Meyer & Miklos, 2004; Xayaphoummine et al., 2005). Slowing down or speeding up transcription might yield inactive transcripts. Thus far, we have expressed Spinach.ST using T7 RNA polymerase from the canonical 17-mer T7 promoter “TAATACGACTCACTATA” (Dunn & Studier, 1983; Martin & Coleman, 1987; Milligan, Groebe, Witherell, & Uhlenbeck, 1987). We have most commonly employed “GGAAGC” and to a lesser extent “GGGTGG” as the +1 to +6 initiation sequences (part of the 5′-end tRNA scaffold, see Section 2.4). Because of the potential impact of transcription kinetics on folding, users should always examine the effects of altering the promoter and/or initiation sequences on Spinach.ST function.
2.4 Stabilization of Spinach.ST within a tRNA scaffold
We have embedded the designed Spinach.ST molecular beacon within a tRNA scaffold to stabilize the RNA (Iioka, Loiselle, Haystead, & Macara, 2011). The tRNA flanks are predicted to form stable hairpins at the 5′- and 3′-ends of Spinach.ST (Fig. 1D–F). Such scaffolds may improve the structural stability of aptamers in vitro as well as improve the life span of RNA in vivo (Paige et al., 2011; Ponchon & Dardel, 2007). We have used sequences derived from Saccharomyces cerevisiae tRNAtrp (Fig. 1D and F) as well as human tRNALys (Fig. 1E) to flank various Spinach.ST molecules. It should be noted that while the human tRNALys is predicted by NUPACK to fold into the expected tRNA conformation (Fig. 1E), the MFE structure of the S. cerevisiae tRNAtrp flanks that is predicted by NUPACK does not conform to the tRNA fold (Fig. 1D). In contrast, mFold depicts a normal tRNA conformation for the S. cerevisiae tRNAtrp flanks of Spinach.ST1 (Fig. 1F) and predicts an overall free energy that is about 10 kcal/mol lower than that predicted by NUPACK. This discrepancy is atypical and does not significantly impact the design of modular folding units. NUPACK remains the most versatile tool available for modeling (and designing) base pairing probabilities in ordered complexes.
2.5 Programming triggers
The choice of the trigger will also impact Spinach.ST signaling. In general, trigger conformation should also be analyzed using NUPACK to ensure that it is not prohibitively structured and that its toehold is readily accessible for binding to Spinach.ST and for initiating branch migration. Beyond that, triggers may be configured for various applications. For instance, single-nucleotide mismatches in target sequences may be distinguished by positioning the mismatched nucleotide within the toehold domains of trigger oligonucleotides, thus increasing the ΔG of toehold binding and slowing the kinetics of Spinach.ST activation (detailed in Section 2.5.1). Since the magnitude of Spinach.ST activation is dependent on the trigger concentration, trigger sequences can also potentially be quantitated, allowing the measurement of nucleic acid circuits (see Section 2.5.2) and amplicons (see Section 5) with Spinach.ST. The simultaneous presence of two different target sequences, including device outputs, may be logically processed using Spinach.ST by restricting the generation of a functional trigger in association with two targets (detailed in Section 2.5.3).
2.5.1 Designing triggers to discriminate single-nucleotide mismatches
It has previously been shown that toehold design can significantly modulate the kinetics of strand displacement (Zhang, Chen, & Yin, 2012; Zhang & Winfree, 2009). We have demonstrated that mismatches at the first or second nucleotide position of the toehold (trigger domain 2*) do not significantly disrupt Spinach.ST strand displacement (Bhadra & Ellington, 2014b). However, toehold mismatches occurring at nucleotide positions 3, 4, or 5 almost completely destroy the ability of a trigger to activate Spinach.ST1, resulting in near-zero initial rates similar to those observed with an unrelated trigger.
In general, to achieve maximal distinction among single-nucleotide polymorphisms (SNPs), mismatches are localized to trigger toehold domain 2* at positions that result in maximal increase in the ΔG of toehold binding (Fig. 4B). However, SNPs located deep in the branch migration domain are not expected to be able to distinguish triggers; previous work with RNA strand displacement circuits has demonstrated that branch migration can readily jump over two contiguous mismatches (Bhadra & Ellington, 2014a). Also, as depicted in Fig. 6A, trigger sequences with mismatches designed in the branch migration domains are capable of activating Spinach.ST.
Figure 4.

Design options for Spinach.ST trigger sequences. (A) Generation of Spinach.ST trigger sequences by associative toehold activation. The trigger toehold domain 2* is placed at the 5′-end of one oligonucleotide, while the trigger branch migration domains 5* and 6* are placed at the 3′-end of a second oligonucleotide. Neither oligonucleotide alone is capable of activating Spinach.ST. However, when brought together by hybridization between the complementary domains D and D*, the toehold and branch migration domains activate Spinach.ST. (B) Presence of a single mismatched nucleotide (M) at position 3, 4, or 5 within the trigger toehold domain 2* significantly decreases the efficiency of Spinach.ST activation. The fully complementary toehold is denoted as a string of “N” with nucleotide positions numbered above. (C) Detection of RNA CHA circuit output using Spinach.ST. Toehold-mediated strand displacement by a catalyst oligonucleotide leads to the formation of H1:H2 from the kinetically trapped CHA hairpins H1 and H2. The Spinach.ST trigger is appended to H1 such that the toehold domain 2* remains sequestered within the H1 stem, and hence is unable to initiate activation of the Spinach molecular beacon. Upon formation of the H1:H2 circuit output complex, the trigger toehold 2* is exposed and can be quantitated by measuring the fluorescence accumulation of Spinach.ST molecules that undergo conformational activation.
2.5.2 Designing triggers to function in the context of nucleic acid circuits
Nucleic acid devices, circuits, and sensors can be engineered to undergo programmed intra- or intermolecular conformational rearrangements that lead to the exposure of previously sequestered trigger sequences. Spinach.ST activation can potentially be used as a reporter for such conformational changes. Trigger sequestration may be achieved by ensconcing trigger sequences in base pairing interactions with the functional nucleic acid such that the single-stranded toehold is unavailable for initiating nucleic acid strand displacement and thus activation of Spinach.ST. Triggers may be either covalently appended to (design detailed below in the context of a CHA circuit) or intermolecularly annealed to the functional nucleic acid. Release of the single-stranded trigger (or its toehold domain) occurs upon input-dependent operation of the functional nucleic acid. For instance, we have used Spinach.ST for measuring the kinetic development of a CHA circuit (Bhadra & Ellington, 2014a). In CHA, two partially complementary nucleic acid oligonucleotides are ensconced within hairpin structures (H1 and H2) leading to kinetic trapping that prevents them from interacting with one another (Fig. 4C). The Spinach.ST trigger sequence appended to one of the hairpins cannot activate Spinach.ST, since the toehold domain 2* is sequestered within the double-stranded hairpin stem. A short, single-stranded oligonucleotide “catalyst” that initiates toehold-mediated strand displacement of one of the hairpins in turn leads to revelation of sequences that can interact with the other hairpin, the formation of a double-stranded product, and the recycling of the catalyst. The trigger toehold 2* that is exposed in the double-stranded circuit output (H1:H2) can be quantitated by measuring fluorescence accumulation of trigger-activated Spinach.ST molecules. Using this approach, we have created completely RNA-based circuits, wherein the circuit input, processor, output, and sensor molecules are all composed of transcribed RNA (Bhadra & Ellington, 2014a).
Similar design strategies can be employed to adapt Spinach.ST to the detection of nonnucleic acid analytes. Structure-switching aptamers can be engineered to undergo analyte-induced intra- or intermolecular rearrangements that lead to the display of sequestered Spinach.ST trigger sequences or toeholds. These exposed sequences would in turn activate standalone Spinach.ST (Cho, Lee, & Ellington, 2009; Nutiu & Li, 2003). Structure-switching aptamers have already been widely used in developing aptamer-based optical (Li & Ho, 2008) and electrochemical sensors (Lubin & Plaxco, 2010) for analytes such as nucleotides, cocaine, and thrombin (Liu & Lu, 2006; Tang et al., 2008), and their adaptation to Spinach.ST signaling should be straightforward.
Another strategy that has been previously described for Spinach-based ligand-sensing relies on interruption of the Spinach sequence with ligand-specific aptamer domains (Paige et al., 2012; Strack, Song, & Jaffrey, 2014). In this approach, unique Spinach biosensors must be designed for each analyte, wherein (a) the stem loop 2 of the Spinach aptamer is replaced with the ligand-binding aptamer sequence and (b) Spinach is forced to misfold into a nonfluorescent conformation. Upon binding of the cognate analyte, a “communication module” (Koizumi, Soukup, Kerr, & Breaker, 1999) between the aptamer and Spinach promotes a conformational change of the Spinach aptamer into its fluorescent state.
2.5.3 Associative toehold triggers
The trigger need not be a contiguous sequence. Indeed, in some instances it is advantageous to disrupt the trigger or spread it out over multiple nucleic acids. Spinach.ST activation should then occur only upon the programmed assembly of the separate trigger units into one complex that would be jointly capable of toehold-mediated strand displacement. Such discontinuous triggers should prove useful for diverse applications such as nucleic acid amplicon validation (Section 5), determining the coincident presence or expression of multiple nucleic acid sequences and logical processing of nucleic acid circuit outputs.
Discontinuous trigger molecules may be built by “associative toehold activation” wherein the toehold and branch migration domains become connected via the hybridization of auxiliary domains, and then allow strand displacement across a three-way junction (Chen, 2012). In one instantiation, the trigger may be split between two separate nucleic acids such that the toehold domain 2* is presented by oligonucleotide 1, while the branch migration domains 5* and 6* are located on oligonucleotide 2 (Fig. 4A). Either oligonucleotide alone will fail to initiate Spinach.ST strand displacement. One of the ways to facilitate association between the two oligonucleotides would be to append domain D at the 3′-end of the toehold-bearing oligonucleotide 1 and to append the complementary domain D* at the 5′-end of oligonucleotide 2 that bears the branch migration domains. Hybridization of domains D and D* will juxtapose the toehold and branch migration domains and allow activation of Spinach.ST (Fig. 4A).
We have previously used random 20- to 28-nucleotide-long domains D and D* with approximately 50% GC contents and free energies (at 37 °C) ranging between −37 and −53 kcal/mol to create associative toeholds. However, the choice of length and sequence should be flexible as long as hybridization probability and thermodynamic stability under assay conditions remains high. The presence of two bulged thymidine bases at the three-way junction of associative DNA toeholds is known to accelerate strand displacement substantially, especially in reactions mediated by reversible toehold binding (Chen, 2012). Although the eight-nucleotide-trigger toehold is expected to bind reversibly (as is evident from the operation of multiple turnover RNA CHA circuits using eight-nucleotide toeholds; Bhadra & Ellington, 2014a), we have found it unnecessary to include bulged thymidines for stabilization of the RNA three-way junctions. All domain structures and interactions should be verified using NUPACK prior to experimentation.
2.6 Spinach.ST function within larger RNA contexts
The conformational rearrangements required for Spinach.ST function can occur within the context of larger RNAs (Figs. 5 and 6). The ability of Spinach.ST to function when it and/or its trigger is embedded within a larger RNA is not only important for in vitro applications such as signal transduction from nucleic acid amplicons (see Section 5) but also might be of particular importance for in vivo applications. Potential examples include detection of RNA localization and/or interaction using programmed Spinach.ST molecules as probes and monitoring RNA splicing by detecting associative trigger sequences whose toehold and branch migration domains become juxtaposed only upon splicing. We embedded the tRNA–Spinach.ST–tRNA cassette within a larger transcript such that it was flanked by 35 nucleotides on the left and 55 nucleotides on the right followed by a 42-nucleotide-long T7 terminator sequence (Fig. 5A). The Spinach.ST beacon in this transcript fluoresced only in the presence of cognate trigger oligonucleotides (Fig. 6B). The trigger sequence could also be successfully embedded in a larger transcript (Fig. 5B). Embedded scrambled sequences failed to activate the Spinach.ST molecular beacon. Intracellularly generated Spinach.ST transcripts may contain upstream or downstream sequences (for example, promoter or terminator regions) that might prevent folding and function. Such flanking sequences can potentially be removed by appending ribozyme cleavases at each end (Bhadra & Ellington, 2014a).
Figure 5.

Spinach.ST and trigger modules embedded in larger sequence contexts. Spinach.ST (A) and its trigger oligonucleotide (B) (both highlighted in gray) flanked with tRNA sequences and expressed as part of a larger transcript. All structures were generated using NUPACK.
3. HOW TO SYNTHESIZE SPINACH.ST MOLECULAR BEACONS ENZYMATICALLY
3.1 DNA oligonucleotides
We have generally purchased all oligonucleotides from Integrated DNA Technologies (IDT, Coralville, IA, USA). Resuspend oligonucleotides at a final concentration of 100 μM in TE (10:0.1) buffer (10 mM Tris–HCl, pH 7.5, 0.1 mM EDTA, pH 8.0), and measure the concentration of all nucleic acid solutions by UV spectrophotometry. We typically use a Nanodrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA) for this purpose. Store dissolved oligonucleotides at −20 °C.
3.2 How to generate transcription templates
We typically use fully double-stranded DNA as templates for T7 RNA polymerase-driven transcription. Short transcription templates (≤60 bp) can be prepared by annealing two complementary synthetic oligonucleotides that have been mixed at an equimolar concentration in TE (10:0.1) buffer containing 50 mM NaCl. Mixtures of oligonucleotides are denatured for 5 min in a thermocycler at 95 °C and then annealed by slow cooling (0.1 °C/s) to 25 °C. Annealed oligonucleotide templates should be quantitated by UV spectrophotometry and directly used for in vitro transcription reactions.
Longer transcription templates may be purchased from IDT as double-stranded gBlocks or single-stranded Ultramers and then amplified by the polymerase chain reaction (PCR). Longer templates may also be sequentially assembled from sets of shorter, overlapping oligonucleotides by oligonucleotide annealing, primer extension, and PCR. The design of overlapping oligonucleotides can be automated using tools such as DNAWorks (http://helixweb.nih.gov/dnaworks/). However, since Spinach.ST contains numerous secondary structures, it is important that the 3′-ends of the overlapping oligonucleotides are sufficiently single-stranded to allow base pairing and polymerase-mediated extension. We usually perform all enzymatic amplification reactions using high fidelity DNA polymerases such as the Phusion DNA polymerase (New England Biolabs (NEB), Ipswich, MA, USA).
While longer transcription templates can be used directly for transcription, we typically prefer to first clone them into vectors. Cloning creates a convenient repository and a homogenous, error-free source of synthetic transcription templates for continued use. Enzymatically synthesized transcription templates (as above) should first be purified by agarose gel electrophoresis, and then extracted from the gel using Wizard SV clean-up columns (Promega, Madison, WI, USA). Purified transcription templates may then be inserted into vectors such as pCR2.1 (Life Technologies) by TOPO-TA cloning (Life Technologies, Grand Island, NY, USA) or by Gibson cloning (NEB).
Cloned transcription templates should be amplified from previously sequence-verified plasmids by PCR using Phusion DNA polymerase. We typically amplify a Spinach.ST expression cassette using primers that reach into the flanking plasmid sequence at the 5′-end and primers specific to the 3′-end sequence of Spinach.ST, creating a template for runoff transcription. A small aliquot of the amplification reaction should be analyzed by agarose gel electrophoresis prior to transcription, to ensure that PCR has yielded a product of the appropriate size and purity. The remainder of the amplification reaction should be purified using the Wizard SV Clean-up system (Promega) and quantitated by UV spectrophotometry prior to setting up the in vitro transcription reaction.
4. HOW TO PERFORM FUNCTIONAL ASSAYS OF SPINACH.ST MOLECULAR BEACONS
In our experience, Spinach.ST molecules are readily activated upon posttranscriptional incubation with cognate RNA or DNA trigger oligonucleotides (Bhadra & Ellington, 2014b) (Section 4.2). Spinach.ST molecules can also be activated by triggers that are cotranscribed along with them (Section 4.3). However, the performance of each new construct should of course be evaluated.
4.1 In vitro transcription reactions
We typically transcribe some 100–500 ng of double-stranded DNA template using 100 units of T7 RNA polymerase (NEB) in a 50 μl reaction containing 40 mM Tris–HCl, pH 7.9, 30 mM MgCl2, 10 mM DTT, 2 mM spermidine, 4 mM of each ribonucleotide (rNTPs), and 20 units of the recombinant ribonuclease inhibitor RNaseOUT (Life Technologies). Transcription reactions are incubated at 42 °C for 120 min.
KineFold predicts that initial base pairing during transcription would occur between domain 6* and the first domain 6.1, but as transcription proceeds the molecule should rearrange into the nonfluorescent, trapped conformation. Our experimental observations have also verified that the Spinach.ST molecular beacons cotranscriptionally fold into their inactive conformation. Thus, it is unnecessary to perform posttranscriptional thermal equilibration of Spinach.ST prior to use. Upon completion of transcription, the Spinach.ST RNA molecules may be used directly or they can be purified by filtration through Sephadex G25 prior to use. We commonly employ the Illustra MicroSpin G-25 columns (GE Healthcare, Piscataway, NJ, USA) for crude transcript purification. For applications requiring even higher purity, Spinach.ST can be isolated by denaturing polyacrylamide gel electrophoresis followed by gel extraction. Unused portions of Spinach.ST transcripts may be stored frozen at −20 or −80 °C and reused immediately upon thawing.
4.2 Reactions with added trigger and control sequences
Activation assays can be set up as 15 μl reactions in 1 × TNaK buffer (20 mM Tris–HCl, pH 7.5, 140 mM NaCl, 5 mM KCl). The Spinach.ST system is compatible with buffers such as those used for transcription and enzymatic nucleic acid amplification reactions (see Sections 4.3 and 5.2), with physiological buffers such as phosphate buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.2) with or without 3 mM MgCl2, and in buffers that mimic intracellular salt concentrations such as the IC buffer (20 mM HEPES, 140 mM KCl, 10 mM NaCl, 2 mM MgCl2, 5 mM KH2PO4, pH 7.2) (Lodish & Darnell, 1995). However, we do not recommend using the Spinach aptamer selection buffer (40 mM HEPES, pH 7.4, 125 mM KCl, 5 mM MgCl2, and 5% DMSO), since the Spinach molecular beacon shows significant background with this buffer. This is likely due to increased folding of Spinach.ST into the native Spinach conformation even in the absence of the trigger oligonucleotide.
The reaction also contains 70 μM DFHBI, 20 units of RNaseOUT, and some 3–6 μl aliquots of the 50 μl G25-filtered Spinach.ST transcripts. DFHBI may be synthesized using the published pathway (Paige et al., 2011) or it may be purchased from Lucerna, Inc. (New York, NY, USA). Prepare a 10 mM solution of DFHBI in dimethyl sulfoxide and store in the dark at −20 °C.
Trial activation reactions should include 0.5 μM single-stranded trigger DNA oligonucleotides and 3–6 μl of G25 Sephadex-purified Spinach.ST transcripts, prepared as described above. The Spinach.ST molecular beacons can be assayed by incubation with equimolar trigger oligonucleotides. Increasing trigger concentrations should increase the rate and amplitude of Spinach.ST activation. Reactions containing no and nonspecific trigger oligonucleotides should also be included as controls. Reactions without Spinach.ST transcripts or trigger oligonucleotides will provide values for subtracting the baseline fluorescence of DFHBI.
Reactions can be assembled in 0.2-ml PCR tubes and then transferred to alternate wells of a Nunc™ 384-well flat-bottom black plate (Thermo Scientific) covered with a MicroAmp® optical adhesive film (Life Technologies). Spinach.ST activation is then monitored by measuring fluorescence accumulation as a function of time at 37 °C. The Spinach.ST molecular beacons at micromolar concentrations typically require approximately 30–60 min to develop maximal fluorescence in response to cognate trigger oligonucleotides. We perform fluorimetry as a function of time using the TECAN Safire, a monochromator-based microplate reader, and set to 469 nm excitation and 501 nm emission wavelengths.
While the fluorescence quantum yield of unmodified Spinach–DFHBI complex is reported to be over a 1000-fold greater than DFHBI alone, activated Spinach biosensors are typically not as bright (Kellenberger, Wilson, Sales-Lee, & Hammond, 2013). The fluorescence intensity of Spinach.ST rises with increasing trigger concentration, typically demonstrating a 4- to 70-fold increment over the fluorescence of DFHBI alone. This level of activation is similar to the approximately 4- to 32-fold fluorescence reported for aptamer-based Spinach-based biosensors when incubated with 103- to 104-fold excess of small-molecule ligands such as adenosine, ADP, SAM, guanine, guanosine 5′-triphosphate (GTP), cyclic di-GMP, and cyclic AMP–GMP (Kellenberger et al., 2013; Paige et al., 2012).
4.3 Real-time cotranscriptional functional assays with Spinach.ST
Sequence-dependent Spinach.ST activation can also be measured by transcribing the beacon in parallel with trigger sequences. Cotranscriptions can be performed in vitro in 25 μl reaction volumes in Nunc™ 384-well flat-bottom black plates. As an example, some 500 ng of double-stranded Spinach.ST and trigger transcription templates can be transcribed using 100 units of T7 RNA polymerase in a buffer composed of 40 mM Tris–HCl, pH 7.9, 6 mM MgCl2, 10 mM DTT, 2 mM spermidine, 50 mM NaCl, 4.8 mM rNTPs, 40 μM DFHBI, and 20 units of RNaseOUT. The plate is then covered with an optical film and incubated at 37–39 °C in a TECAN Safire plate reader, and real-time fluorescence measurements are recorded at regular intervals. Specificity of the cotranscriptional activation can be determined by substitution of the cognate trigger transcription templates with templates that generate scrambled trigger RNAs. Control reactions with only trigger or Spinach.ST transcription templates should be included to determine background and nonspecific activation. Significant accumulation of Spinach.ST fluorescence should only be observed when Spinach.ST is cotranscribed with its cognate trigger.
5. APPLICATION: REAL-TIME SPINACH.ST-BASED DETECTION OF NASBA
We have previously used nucleic acid circuits for transducing signals from the amplification reactions that underlie molecular diagnostics (Jiang, Li, Milligan, Bhadra, & Ellington, 2013; Li, Chen, & Ellington, 2012). In one such amplification reaction, NASBA (Compton, 1991), RNA templates are reverse transcribed into double-stranded DNA using forward and reverse primers, one of which carries the T7 RNA polymerase promoter sequence. The resulting double-stranded DNA is then transcribed by T7 RNA polymerase to generate additional RNA amplicons, which can in turn continuously be converted into additional double-stranded DNA templates. The application of Spinach.ST for real-time, sequence-specific signal transduction of NASBA amplicons is especially appealing, because the Spinach.ST molecular beacons can be enzymatically synthesized in situ by simply including their double-stranded transcription templates in one-pot NASBA reactions.
5.1 Including Spinach.ST trigger components in NASBA primers
Choose a relatively structure-free region of the target for amplification and design a Spinach.ST molecular beacon with domains 2 and 5 derived from a continuous stretch of target sequence. We routinely model the RNA amplicon structure at the amplification reaction temperature using NUPACK. The amplicon should not present major structural challenges to primer binding, polymerase progression, primer-driven amplicon structural organization (see below), or toehold accessibility. Conformations and probabilities of complex formation between NASBA amplicons, primers, and Spinach.ST should be verified using NUPACK.
Guidelines commonly used for designing PCR primers should also be used to design the target-binding domains (PBS-F and PBS-R*, respectively) of the NASBA forward (NF) and reverse (NR) primers (Fig. 7). For clarity, we provide example sequences of a model NASBA template, primers, transcription templates, and the resulting RNA amplicons in Fig. 8. The NR primer-binding site (PBS-R*) should be downstream of amplicon domain 2. The NF binding site (PBS-F) should reside at a position at least 20-nucleotide upstream of amplicon domain 5. Extend the NR primer by adding the 17-mer T7 promoter sequence “TAATACGACTCACTATA” to its 5′-end. If the adjoining PBS-R* domain does not begin with two guanine nucleotides, then these should be appended immediately following the T7 promoter sequence; for example, the PBS-R domain of the model NASBA template sequence depicted in Fig. 8 does not end with two cytosine (CC) groups at its 3′-end. Therefore, to allow more efficient transcription, two guanine residues were inserted in the NR primer immediately prior to the complementary PBS-R* domain. We also typically include a sequence of four additional nucleotides (such as “ATGA”) upstream of the promoter to enhance the transcription efficiency (Fig. 8).
Figure 7.

Schematic of trigger-generating primers for real-time signal transduction with NASBA. Regions on the amplicon that are involved in primer binding (PBS-F and PBS-R), associative toehold formation (domain D), and partial Spinach.ST trigger generation (domains 2 and 5) are depicted as dark boxes. Hybridization is depicted using vertical slashes, while the potential hybridization of the PBS-F domain of the NF primer to the complementary strand is indicated with colons. Dashed regions in the target, double-stranded DNA transcription template and RNA transcript denote target sequences that are not involved in priming or in Spinach.ST activation.
Figure 8.

Examples of nucleic acid sequence-based amplification (NASBA) primer and template design. Sequences complementary to the desired trigger domains 2* and 5* are designated in the templates. Sequences complementary to the trigger domain 6* are appended at the 5′-end of the NF primer. The resulting NASBA RNA amplicons contain the trigger domains 2* and 5* (transcribed from the template) and domain 6* (transcribed from the NF primer). The domain D* is complementary to a stretch of template sequence, termed D, that is inserted in the NF primer. Hybridization of D and D* juxtaposes the otherwise distributed trigger domains via hairpin formation (see Fig. 7). Sequences for domains 2*, 5*, and 6* are derived from the trigger sequence for Spinach.ST2 molecular beacon with the sequence GGA AGC GGT GGC TCA ATG GTA GAG CTT TCG ACG ACA TCT GAC GCG ACC GAA ATG GTG AAG GAC GGG TCC AGT GCT TCG GCA CTG TTG AGT AGA GTG TGA GCT CCG TAA CTG GTC GCG TCG GTC GCG TCA GAT GTC GGT CAG GTC TCG AAG GGT TGC AGG TTC AAT TCC TGT CCG TTT C (Bhadra & Ellington, 2014b). For the synthetic target primer-binding sites (PBS-F and PBS-R) with approximately 50% GC content were used; this was also the case for the hybridization domains (D and its complement D*). The strong 17-mer T7 RNA polymerase promoter sequence (including the two guanine nucleotides at the transcription initiation site) was obtained from literature (Dunn & Studier, 1983; Martin & Coleman, 1987; Milligan et al., 1987). An additional four nucleotide sequence was included at the 5′-end of the promoter based on reports that suggested that T7 polymerase contacts DNA through the −21 position (Baklanov, Golikova, & Malygin, 1996; Ikeda & Richardson, 1986).
To allow hybridization of Spinach.ST to the NASBA amplicon the NF primer should be extended according to the following guidelines. Although Spinach.ST can be designed to accept many different sequences via trigger domains 2* and 5*, domain 6* should remain invariant as it is a part of the aptamer basal stem (Fig. 1A). Only domains 2* and 5* of the trigger should originate from target sequences (Fig. 7). The nine-nucleotide invariant complementary domain 6 should be appended to the 5′-end of the NF primer such that all resulting RNA products will contain the requisite trigger domain 6*. To facilitate understanding the design process, an example of template sequence and the corresponding NASBA primers that facilitate Spinach.ST-based detection is depicted in Fig. 8.
As few as eight intervening, nonpaired nucleotides in trigger sequences render them incapable of activating Spinach.ST (Bhadra & Ellington, 2014b). Therefore, the gap between the RNA amplicon trigger domains 6* and 5* (composed of PBS-F* and the adjoining 20 nucleotides) must be bridged by what we term “associative toehold activation” (see Section 2.5.3). The associative toeholds will be juxtaposed by hairpin formation during transcription (Fig. 7). In the example NASBA system, we show in Fig. 8, hairpin formation is achieved by inserting domain D* in the NF primer between domains 6 and PBS-F. Domain D* has been designed to be complementary to a template sequence that lies immediately upstream of template domain 5 (Fig. 8). The resultant NASBA-generated RNA amplicons should therefore have the following domain organization: 5′-PBS-R*—2*—5*—D*—PBS-F*—D—6*-3′ (Figs. 7 and 8). Hybridization during transcription between the complementary domains D* and D will lead to sequestration of intervening sequences into a stem–loop structure and will also bring domains 5* and 6* into apposition, thereby generating the associative toehold that should lead to the Spinach.ST activation. In our designs, we typically use a 20-nucleotide-long domain D* for intramolecular base pairing with a 20-nucleotide-long domain D leading to formation of a double-stranded stem whose loop is composed of a 21-nucleotide-long PBS-F domain. Since the PBS-F domain acts as the primer, its length should at least be 18 nucleotides and have a predicted melting temperature that is greater than the isothermal amplification reaction temperature to maintain target specificity. The lengths of domains D and D* may also be varied as long as the probability of their intramolecular hybridization remains close to 100%. Unnecessarily long domain lengths should be avoided as these might frustrate the folding pathways.
Strand displacement across the three-way RNA junction formed by a transcribed NASBA amplicon not only activates the Spinach.ST reporter but can also allow SNPs located in the middle of the toehold domain to be readily distinguished in real-time. Similar to the substantial disruption in Spinach.ST activation that is observed with standalone trigger oligonucleotides harboring mismatches within the toehold (see Section 2.5.1 and Fig. 6B), we have achieved significant discrimination between wild-type and SNP-containing amplicons by placing the mismatched nucleotide at position 4 of amplicon toehold domain 2* (Bhadra & Ellington, 2014b). Spinach.ST molecules that perfectly matched the wild-type or the SNP amplicons could detect their targets with a seven- to ninefold signal-to-noise ratio. In contrast, the signal-to-noise ratio was less than 2 for mismatched amplicons.
A single Spinach.ST molecular beacon can potentially be used as a universal reporter for a wide variety of amplicons (Fig. 9). In one approach the constant Spinach.ST trigger is split between the two NASBA primers such that the complementary branch migration domains 6 and 5 are appended at the 5′-end of the NF primer, while the toehold domain 2* is inserted at the 5′-end of the NR primer, immediately downstream of the T7 RNA polymerase promoter (Fig. 9). Associative toehold activation via template sequence-dependent apposition of the toehold and branch migration domains of the trigger can then be facilitated by engineering the formation of two adjacent intramolecular stem–loop structures within each RNA amplicon. Two stretches of RNA sequence that are flanked by the PBS-F and PBS-R primer-binding domains are designated as hybridization domains D1 and D2. As D1 and D2 are transcribed, they can form duplexes with transcribed D1* and D2* that are inserted into the NF and NR primers, respectively (Fig. 9). The resulting NASBA RNA amplicons will have the domain organization: 5′—2*—D1—PBS-R*—D1*—D2*— PBS-F*—D2—5*—6*—3′, and hybridization of D1:D1* and D2:D2* should lead to formation of two adjoining stem loops that juxtapose the trigger domains 2*, 5*, and 6* that would otherwise be separated on either end of the RNA amplicons. The conformation of the junction between 2* and 5* can potentially be adjusted with single-nucleotide bulges or base pairs to optimize signaling. Of course, the entire trigger sequence could be appended to one primer; however, given the propensity for spurious amplification by isothermal processes, we prefer the target sequence-dependent assembly of the Spinach.ST triggers.
Figure 9.

Potential real-time nucleic acid sequence-based amplification (NASBA) amplicon detection using universal Spinach.ST molecular beacons. A scheme for generating NASBA primers that upon transcription creates an intramolecular RNA fold that can activate a universal Spinach.ST trigger is depicted. Transcribed, primer-derived sequences are shown as black lines. Transcribed, target-derived sequences are shown as dashed lines. Regions D1 and D2 that are involved in intramolecular RNA stem–loop formation are highlighted as thick gray blocks. Hybridization is depicted using vertical slashes. The dark bars at the junctions of domains 2* and D1 and domains D2 and 5* designate possible complementary base pairs that might stabilize the associative trigger.
5.2 Detecting NASBA amplification with Spinach.ST
NASBA reactions can be set up starting from either ssDNA or RNA templates that have been freshly diluted into TE (10:0.1) buffer containing 1 μM oligo dT17. We typically set up the NASBA reactions in two steps. First, increasing concentrations of template molecules are mixed with 24 mM MgCl2, 20 mM KCl, 50 mM NaCl, 100 nM each of forward and reverse primers, and 500 μM dNTP mix in a total volume of 15 μl. This solution is heated to 65 °C for 5 min followed by incubation on ice for 2 min to allow primer annealing. Subsequently, a 10 μl aliquot containing the remaining reaction components including 100–138 ng of Spinach.ST dsDNA transcription templates, 1 × RNAPol reaction buffer (NEB; 40 mM Tris–HCl, 6 mM MgCl2, 10 mM DTT, 2 mM spermidine), 6 mM of each rNTP (NEB), 100 units of MMLV reverse transcriptase (NEB), 50 units of T7 RNA polymerase, 20 units of RNaseOUT, and 40 μM DFHBI are added (final volume=25 μl). The reactions are immediately transferred to NUNC™ 384-well flat-bottom black plates and covered with an optical film. The plate is incubated for 6–8 h in a TECAN Safire microplate reader at 37 °C, and real-time Spinach–DFHBI fluorescence measurements are recorded at regular intervals. Allelic discrimination may be improved by incubation at 42 °C instead of 37 °C.
To determine the specificity of NASBA RNA amplicon-mediated activation of Spinach.ST molecular beacons, control reactions that generate nonspecific NASBA amplicons should be also be carried out. The most convenient way to create nonspecific amplicons is to place the T7 promoter at the 5′-end of the NF primer instead of the NR primer. RNA amplicons generated using such a primer pair would harbor domains 6, 5, and 2 instead of the complementary domains 2*, 5*, and 6*. As an additional control, remove 1–2 μl aliquots at the completion of NASBA reactions and analyze these via denaturing polyacrylamide gel electrophoresis. This will ensure that amplification has actually occurred, although all NASBA reactions (containing either specific or nonspecific RNA amplicons) will likely display similar amounts of Spinach.ST transcripts. Visualization by gel electrophoresis is also a very intuitive way of observing how Spinach.ST is useful for discriminating true from artifactual amplicons.
6. CONCLUSIONS
We have described the design principles for engineering the fluorescent RNA aptamer Spinach to be a sequence-dependent molecular beacon that is trapped during transcription into a nonfluorescent conformation and is then switched to an active, fluorescent state upon sequence-specific, toehold-mediated strand displacement. Spinach.ST reporters and their triggers are very amenable to in silico engineering since their experimentally determined behavior has proven to be consistent with the predictions of design and cotranscriptional modeling algorithms.
Overall, the potential utility of Spinach.ST as a signal transducer for detecting DNA and RNA sequences in vitro is based on its simplicity relative to competing methods. A variety of other fluorescent probes have been described for detection of specific nucleic acid sequences in vitro. These include single as well as dual-labeled probes such as the molecular beacons (Tyagi & Kramer, 1996), assimilation probes (Kubota et al., 2013), molecular zippers (Yi, Zhang, & Zhang, 2006), TaqMan® probes (Holland, Abramson, Watson, & Gelfand, 1991), cycling probes (Duck, Alvarado-Urbina, Burdick, & Collier, 1990), adjacent hybridization probes (Didenko, 2001), scorpion primers (Whitcombe, Theaker, Guy, Brown, & Little, 1999), double-stranded hybridization probes (Didenko, 2001), eclipse probes (Afonina, Reed, Lusby, Shishkina, & Belousov, 2002), LUX primers (Kusser, 2006), and the Qzyme probes (Mokany, Todd, Fuery, & Applegate, 2006). Many of these detection methods require prior synthesis, and new probes must be resynthesized for every new target. Cotranscriptional generation of Spinach.ST should be comparatively cost-effective, including in terms of sequence redesign. This is especially true because Spinach.ST can fold and function correctly without requiring purification of transcripts, unlike most synthetic probes that require postsynthetic purification. This capability not only reduces the cost of probe generation but also potentially enables novel applications involving in situ probe synthesis.
We have demonstrated several permutations for how different Spinach.ST trigger sequences can enable Spinach.ST molecular beacons to act as reporters in varied applications such as sequence validation of enzymatic isothermal nucleic acid amplification reactions, SNP distinction, and nucleic acid circuit output measurement. Even in the complex milieu of NASBA or other isothermal amplification reactions, Spinach.ST beacons fold cotranscriptionally into stable, inactive conformations that are only activated in the presence of cognate trigger RNA sequences. The sensor is able to readily distinguish single-nucleotide mismatches within NASBA-generated sequence targets.
Into the future, since Spinach.ST molecular beacons operate well in physiological buffers, we anticipate that they should be useful tools for intracellular sensing as well. It is conceivable that Spinach.ST might be used as an information processor in a more complex nucleic acid strand displacement circuit. Spinach.ST could both detect a target sequence and also transmit this information to other portions of a circuit via the newly exposed domain 5* (Fig. 2).
ACKNOWLEDGMENTS
This work was supported by the Welch Foundation (F1654), by the National Institutes of Health in conjunction with the Boston University (1U54EB015403), and by the Gates Foundation (OPP1028808).
REFERENCES
- Afonin KA, Danilov EO, Novikova IV, Leontis NB. TokenRNA: A new type of sequence-specific, label-free fluorescent biosensor for folded RNA molecules. Chembiochem. 2008;9(12):1902–1905. doi: 10.1002/cbic.200800183. http://dx.doi.org/10.1002/cbic.200800183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonina IA, Reed MW, Lusby E, Shishkina IG, Belousov YS. Minor groove binder-conjugated DNA probes for quantitative DNA detection by hybridization-triggered fluorescence. Biotechniques. 2002;32(4):940–944. doi: 10.2144/02324pf01. 946–949. [DOI] [PubMed] [Google Scholar]
- Andersen ES, Dong M, Nielsen MM, Jahn K, Subramani R, Mamdouh W, et al. Self-assembly of a nanoscale DNA box with a controllable lid. Nature. 2009;459(7243):73–76. doi: 10.1038/nature07971. http://dx.doi.org/10.1038/Nature07971. [DOI] [PubMed] [Google Scholar]
- Babendure JR, Adams SR, Tsien RY. Aptamers switch on fluorescence of triphenylmethane dyes. Journal of the American Chemical Society. 2003;125(48):14716–14717. doi: 10.1021/ja037994o. http://dx.doi.org/10.1021/Ja037994o. [DOI] [PubMed] [Google Scholar]
- Baklanov MM, Golikova LN, Malygin EG. Effect on DNA transcription of nucleotide sequences upstream to T7 promoter. Nucleic Acids Research. 1996;24(18):3659–3660. doi: 10.1093/nar/24.18.3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhadra S, Ellington AD. Design and application of cotranscriptional nonenzymatic RNA circuits and signal transducers. Nucleic Acids Research. 2014a;42(7):e58. doi: 10.1093/nar/gku074. http://dx.doi.org/10.1093/nar/gku074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhadra S, Ellington AD. A Spinach molecular beacon triggered by strand displacement. RNA. 2014b;20(8):1183–1194. doi: 10.1261/rna.045047.114. http://dx.doi.org/10.1261/rna.045047.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. Expanding the rule set of DNA circuitry with associative toehold activation. Journal of the American Chemical Society. 2012;134(1):263–271. doi: 10.1021/ja206690a. http://dx.doi.org/10.1021/ja206690a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ellington AD. Shaping up nucleic acid computation. Current Opinion in Biotechnology. 2010;21(4):392–400. doi: 10.1016/j.copbio.2010.05.003. http://dx.doi.org/10.1016/j.copbio.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho EJ, Lee JW, Ellington AD. Applications of aptamers as sensors. Annual Review of Analytical Chemistry. 2009;2:241–264. doi: 10.1146/annurev.anchem.1.031207.112851. http://dx.doi.org/10.1146/annurev.anchem.1.031207.112851. [DOI] [PubMed] [Google Scholar]
- Compton J. Nucleic acid sequence-based amplification. Nature. 1991;350(6313):91–92. doi: 10.1038/350091a0. http://dx.doi.org/10.1038/350091a0. [DOI] [PubMed] [Google Scholar]
- Constantin TP, Silva GL, Robertson KL, Hamilton TP, Fague K, Waggoner AS, et al. Synthesis of new fluorogenic cyanine dyes and incorporation into RNA fluoromodules. Organic Letters. 2008;10(8):1561–1564. doi: 10.1021/ol702920e. http://dx.doi.org/10.1021/Ol702920e. [DOI] [PubMed] [Google Scholar]
- Court D. RNA processing and degradation by RNase III. Academic Press; New York: 1993. [Google Scholar]
- Didenko VV. DNA probes using fluorescence resonance energy transfer (FRET): Designs and applications. Biotechniques. 2001;31(5):1106–1116. doi: 10.2144/01315rv02. 1118, 1120–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks RM, Bois JS, Schaeffer JM, Winfree E, Pierce NA. Thermodynamic analysis of interacting nucleic acid strands. SIAM Review. 2007;49(1):65–88. http://dx.doi.org/10.1137/060651100. [Google Scholar]
- Dirks RM, Pierce NA. A partition function algorithm for nucleic acid secondary structure including pseudoknots. Journal of Computational Chemistry. 2003;24(13):1664–1677. doi: 10.1002/jcc.10296. http://dx.doi.org/10.1002/jcc.10296. [DOI] [PubMed] [Google Scholar]
- Dirks RM, Pierce NA. An algorithm for computing nucleic acid base-pairing probabilities including pseudoknots. Journal of Computational Chemistry. 2004;25(10):1295–1304. doi: 10.1002/jcc.20057. http://dx.doi.org/10.1002/jcc.20057. [DOI] [PubMed] [Google Scholar]
- Duck P, Alvarado-Urbina G, Burdick B, Collier B. Probe amplifier system based on chimeric cycling oligonucleotides. Biotechniques. 1990;9(2):142–148. [PubMed] [Google Scholar]
- Dunn JJ, Studier FW. Complete nucleotide-sequence of bacteriophage-T7 DNA and the locations of T7 genetic elements. Journal of Molecular Biology. 1983;166(4):477–535. doi: 10.1016/s0022-2836(83)80282-4. http://dx.doi.org/10.1016/S0022-2836(83)80282-4. [DOI] [PubMed] [Google Scholar]
- Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346(6287):818–822. doi: 10.1038/346818a0. http://dx.doi.org/10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Furutani C, Shinomiya K, Aoyama Y, Yamada K, Sando S. Modular blue fluorescent RNA sensors for label-free detection of target molecules. Molecular Bio-Systems. 2010;6(9):1569–1571. doi: 10.1039/c001230k. http://dx.doi.org/10.1039/c001230k. [DOI] [PubMed] [Google Scholar]
- Grate D, Wilson C. Laser-mediated, site-specific inactivation of RNA transcripts. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(11):6131–6136. doi: 10.1073/pnas.96.11.6131. http://dx.doi.org/10.1073/pnas.96.11.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han DR, Pal S, Liu Y, Yan H. Folding and cutting DNA into reconfigurable topological nanostructures. Nature Nanotechnology. 2010;5(10):712–717. doi: 10.1038/nnano.2010.193. http://dx.doi.org/10.1038/nnano.2010.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinicke LA, Nallagatla SR, Hull CM, Bevilacqua PC. RNA helical imperfections regulate activation of the protein kinase PKR: Effects of bulge position, size, and geometry. RNA. 2011;17(5):957–966. doi: 10.1261/rna.2636911. http://dx.doi.org/10.1261/rna.2636911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjalt TA, Wagner EG. Bulged-out nucleotides protect an antisense RNA from RNase III cleavage. Nucleic Acids Research. 1995;23(4):571–579. doi: 10.1093/nar/23.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holeman LA, Robinson SL, Szostak JW, Wilson C. Isolation and characterization of fluorophore-binding RNA aptamers. Folding and Design. 1998;3(6):423–431. doi: 10.1016/S1359-0278(98)00059-5. http://dx.doi.org/10.1016/S1359-0278(98)00059-5. [DOI] [PubMed] [Google Scholar]
- Holland PM, Abramson RD, Watson R, Gelfand DH. Detection of specific polymerase chain reaction product by utilizing the 5′—3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(16):7276–7280. doi: 10.1073/pnas.88.16.7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iioka H, Loiselle D, Haystead TA, Macara IG. Efficient detection of RNA–protein interactions using tethered RNAs. Nucleic Acids Research. 2011;39(8):e53. doi: 10.1093/nar/gkq1316. http://dx.doi.org/10.1093/nar/gkq1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda RA, Richardson CC. Interactions of the RNA polymerase of bacteriophage T7 with its promoter during binding and initiation of transcription. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(11):3614–3618. doi: 10.1073/pnas.83.11.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YS, Li B, Milligan JN, Bhadra S, Ellington AD. Real-time detection of isothermal amplification reactions with thermostable catalytic hairpin assembly. Journal of the American Chemical Society. 2013;135(20):7430–7433. doi: 10.1021/ja4023978. http://dx.doi.org/10.1021/ja4023978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger CA, Wilson SC, Sales-Lee J, Hammond MC. RNA-based fluorescent biosensors for live cell imaging of second messengers cyclic di-GMP and cyclic AMP-GMP. Journal of the American Chemical Society. 2013;135(13):4906–4909. doi: 10.1021/ja311960g. http://dx.doi.org/10.1021/ja311960g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi M, Soukup GA, Kerr JN, Breaker RR. Allosteric selection of ribozymes that respond to the second messengers cGMP and cAMP. Nature Structural Biology. 1999;6(11):1062–1071. doi: 10.1038/14947. http://dx.doi.org/10.1038/14947. [DOI] [PubMed] [Google Scholar]
- Kolpashchikov DM. Binary malachite green aptamer for fluorescent detection of nucleic acids. Journal of the American Chemical Society. 2005;127(36):12442–12443. doi: 10.1021/ja0529788. http://dx.doi.org/10.1021/Ja0529788. [DOI] [PubMed] [Google Scholar]
- Kramer FR, Mills DR. Secondary structure formation during RNA synthesis. Nucleic Acids Research. 1981;9(19):5109–5124. doi: 10.1093/nar/9.19.5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota R, Labarre P, Weigl B, Li Y, Haydock P, Jenkins D. Molecular diagnostics in a teacup: Non-instrumented nucleic acid amplification (NINA) for rapid, low cost detection of Salmonella enterica. Chinese Science Bulletin. 2013;58(10):1162–1168. doi: 10.1007/s11434-012-5634-9. http://dx.doi.org/10.1007/s11434-012-5634-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusser W. Use of self-quenched, fluorogenic LUX primers for gene expression profiling. Methods in Molecular Biology. 2006;335:115–133. doi: 10.1385/1-59745-069-3:115. http://dx.doi.org/10.1385/1-59745-069-3:115. [DOI] [PubMed] [Google Scholar]
- Li B, Chen X, Ellington AD. Adapting enzyme-free DNA circuits to the detection of loop-mediated isothermal amplification reactions. Analytical Chemistry. 2012;84(19):8371–8377. doi: 10.1021/ac301944v. http://dx.doi.org/10.1021/ac301944v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Ellington AD, Chen X. Rational, modular adaptation of enzyme-free DNA circuits to multiple detection methods. Nucleic Acids Research. 2011;39(16):e110. doi: 10.1093/nar/gkr504. http://dx.doi.org/10.1093/nar/gkr504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Ho CM. Aptamer-based optical probes with separated molecular recognition and signal transduction modules. Journal of the American Chemical Society. 2008;130(8):2380–2381. doi: 10.1021/ja076787b. http://dx.doi.org/10.1021/ja076787b. [DOI] [PubMed] [Google Scholar]
- Liu JW, Lu Y. Fast colorimetric sensing of adenosine and cocaine based on a general sensor design involving aptamers and nanoparticles. Angewandte Chemie-International Edition. 2006;45(1):90–94. doi: 10.1002/anie.200502589. http://dx.doi.org/10.1002/anie.200502589. [DOI] [PubMed] [Google Scholar]
- Lodish HF, Darnell JE. Molecular cell biology. 3rd Scientific American Books: Distributed by W.H. Freeman and Co; New York: 1995. [Google Scholar]
- Lubin AA, Plaxco KW. Folding-based electrochemical biosensors: The case for responsive nucleic acid architectures. Accounts of Chemical Research. 2010;43(4):496–505. doi: 10.1021/ar900165x. http://dx.doi.org/10.1021/Ar900165x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CT, Coleman JE. Kinetic analysis of T7 RNA polymerase–promoter interactions with small synthetic promoters. Biochemistry. 1987;26(10):2690–2696. doi: 10.1021/bi00384a006. [DOI] [PubMed] [Google Scholar]
- Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. Journal of Molecular Biology. 1999;288(5):911–940. doi: 10.1006/jmbi.1999.2700. http://dx.doi.org/10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- Meyer IM, Miklos I. Co-transcriptional folding is encoded within RNA genes. BMC Molecular Biology. 2004;5:10. doi: 10.1186/1471-2199-5-10. http://dx.doi.org/10.1186/1471-2199-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Research. 1987;15(21):8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokany E, Todd AV, Fuery CJ, Applegate TL. Diagnosis and monitoring of PML-RARalpha-positive acute promyelocytic leukemia by quantitative RT-PCR. Methods in Molecular Medicine. 2006;125:127–147. doi: 10.1385/1-59745-017-0:127. [DOI] [PubMed] [Google Scholar]
- Nutiu R, Li Y. Structure-switching signaling aptamers. Journal of the American Chemical Society. 2003;125(16):4771–4778. doi: 10.1021/ja028962o. http://dx.doi.org/10.1021/ja028962o. [DOI] [PubMed] [Google Scholar]
- Paige JS, Nguyen-Duc T, Song W, Jaffrey SR. Fluorescence imaging of cellular metabolites with RNA. Science. 2012;335(6073):1194. doi: 10.1126/science.1218298. http://dx.doi.org/10.1126/science.1218298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige JS, Wu KY, Jaffrey SR. RNA mimics of green fluorescent protein. Science. 2011;333(6042):642–646. doi: 10.1126/science.1207339. http://dx.doi.org/10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei R, Rothman J, Xie YL, Stojanovic MN. Light-up properties of complexes between thiazole orange-small molecule conjugates and aptamers. Nucleic Acids Research. 2009;37(8):e59. doi: 10.1093/nar/gkp154. http://dx.doi.org/10.1093/nar/gkp154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponchon L, Dardel F. Recombinant RNA technology: The tRNA scaffold. Nature Methods. 2007;4(7):571–576. doi: 10.1038/nmeth1058. http://dx.doi.org/10.1038/nmeth1058. [DOI] [PubMed] [Google Scholar]
- Pothoulakis G, Ceroni F, Reeve B, Ellis T. The Spinach RNA aptamer as a characterization tool for synthetic biology. ACS Synthetic Biology. 2013;3(3):182–187. doi: 10.1021/sb400089c. http://dx.doi.org/10.1021/sb400089c. [DOI] [PubMed] [Google Scholar]
- Qian L, Winfree E. Scaling up digital circuit computation with DNA strand displacement cascades. Science. 2011;332(6034):1196–1201. doi: 10.1126/science.1200520. http://dx.doi.org/10.1126/science.1200520. [DOI] [PubMed] [Google Scholar]
- Qian L, Winfree E, Bruck J. Neural network computation with DNA strand displacement cascades. Nature. 2011;475(7356):368–372. doi: 10.1038/nature10262. http://dx.doi.org/10.1038/nature10262. [DOI] [PubMed] [Google Scholar]
- Sando S, Narita A, Hayami M, Aoyama Y. Transcription monitoring using fused RNA with a dye-binding light-up aptamer as a tag: A blue fluorescent RNA. Chemical Communications. 2008;September(33):3858–3860. doi: 10.1039/b808449a. http://dx.doi.org/10.1039/B808449a. [DOI] [PubMed] [Google Scholar]
- Seelig G, Soloveichik D, Zhang DY, Winfree E. Enzyme-free nucleic acid logic circuits. Science. 2006;314(5805):1585–1588. doi: 10.1126/science.1132493. http://dx.doi.org/10.1126/science.1132493. [DOI] [PubMed] [Google Scholar]
- Serra MJ, Turner DH. Predicting thermodynamic properties of RNA. Methods in Enzymology. 1995;259:242–261. doi: 10.1016/0076-6879(95)59047-1. [DOI] [PubMed] [Google Scholar]
- Song W, Strack RL, Jaffrey SR. Imaging bacterial protein expression using genetically encoded RNA sensors. Nature Methods. 2013;10(9):873–875. doi: 10.1038/nmeth.2568. http://dx.doi.org/10.1038/nmeth.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparano BA, Koide K. A strategy for the development of small-molecule-based sensors that strongly fluoresce when bound to a specific RNA. Journal of the American Chemical Society. 2005;127(43):14954–14955. doi: 10.1021/ja0530319. http://dx.doi.org/10.1021/Ja0530319. [DOI] [PubMed] [Google Scholar]
- Sparano BA, Koide K. Fluorescent sensors for specific RNA: A general paradigm using chemistry and combinatorial biology. Journal of the American Chemical Society. 2007;129(15):4785–4794. doi: 10.1021/ja070111z. http://dx.doi.org/10.1021/Ja070111z. [DOI] [PubMed] [Google Scholar]
- Stojanovic MN, Kolpashchikov DM. Modular aptameric sensors. Journal of the American Chemical Society. 2004;126(30):9266–9270. doi: 10.1021/ja032013t. http://dx.doi.org/10.1021/ja032013t. [DOI] [PubMed] [Google Scholar]
- Strack RL, Disney MD, Jaffrey SR. A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat-containing RNA. Nature Methods. 2013;10(12):1219–1224. doi: 10.1038/nmeth.2701. http://dx.doi.org/10.1038/Nmeth.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack RL, Song W, Jaffrey SR. Using Spinach-based sensors for fluorescence imaging of intracellular metabolites and proteins in living bacteria. Nature Protocols. 2014;9(1):146–155. doi: 10.1038/nprot.2014.001. http://dx.doi.org/10.1038/nprot.2014.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang ZW, Mallikaratchy P, Yang RH, Kim YM, Zhu Z, Wang H, et al. Aptamer switch probe based on intramolecular displacement. Journal of the American Chemical Society. 2008;130(34):11268–11269. doi: 10.1021/ja804119s. http://dx.doi.org/10.1021/Ja804119s. [DOI] [PubMed] [Google Scholar]
- Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249(4968):505–510. doi: 10.1126/science.2200121. http://dx.doi.org/10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Tyagi S, Kramer FR. Molecular beacons: Probes that fluoresce upon hybridization. Nature Biotechnology. 1996;14(3):303–308. doi: 10.1038/nbt0396-303. http://dx.doi.org/10.1038/Nbt0396-303. [DOI] [PubMed] [Google Scholar]
- Whitcombe D, Theaker J, Guy SP, Brown T, Little S. Detection of PCR products using self-probing amplicons and fluorescence. Nature Biotechnology. 1999;17(8):804–807. doi: 10.1038/11751. http://dx.doi.org/10.1038/11751. [DOI] [PubMed] [Google Scholar]
- Xayaphoummine A, Bucher T, Isambert H. Kinefold web server for RNA/DNA folding path and structure prediction including pseudoknots and knots. Nucleic Acids Research. 2005;33:W605–W610. doi: 10.1093/nar/gki447. Web server issue. http://dx.doi.org/10.1093/nar/gki447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Zhang W, Zhang DY. Molecular zipper: A fluorescent probe for realtime isothermal DNA amplification. Nucleic Acids Research. 2006;34(11):e81. doi: 10.1093/nar/gkl261. http://dx.doi.org/10.1093/nar/gkl261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin P, Choi HM, Calvert CR, Pierce NA. Programming biomolecular self-assembly pathways. Nature. 2008;451(7176):318–322. doi: 10.1038/nature06451. http://dx.doi.org/10.1038/nature06451. [DOI] [PubMed] [Google Scholar]
- Yurke B, Mills A., Jr. Using DNA to power nanostructures. Genetic Programming and Evolvable Machines. 2003;4(2):111–122. http://dx.doi.org/10.1023/a:1023928811651. [Google Scholar]
- Yurke B, Turberfield AJ, Mills AP, Simmel FC, Neumann JL. A DNA-fuelled molecular machine made of DNA. Nature. 2000;406(6796):605–608. doi: 10.1038/35020524. [DOI] [PubMed] [Google Scholar]
- Zadeh JN, Steenberg CD, Bois JS, Wolfe BR, Pierce MB, Khan AR, et al. NUPACK: Analysis and design of nucleic acid systems. Journal of Computational Chemistry. 2011;32(1):170–173. doi: 10.1002/jcc.21596. http://dx.doi.org/10.1002/jcc.21596. [DOI] [PubMed] [Google Scholar]
- Zhang DY. Dynamic DNA strand displacement circuits. California Institute of Technology; Pasadena, CA: 2010. [Google Scholar]
- Zhang DY, Chen SX, Yin P. Optimizing the specificity of nucleic acid hybridization. Nature Chemistry. 2012;4(3):208–214. doi: 10.1038/nchem.1246. http://dx.doi.org/10.1038/Nchem.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DY, Seelig G. Dynamic DNA nanotechnology using strand-displacement reactions. Nature Chemistry. 2011;3(2):103–113. doi: 10.1038/nchem.957. http://dx.doi.org/10.1038/nchem.957. [DOI] [PubMed] [Google Scholar]
- Zhang DY, Winfree E. Control of DNA strand displacement kinetics using toehold exchange. Journal of the American Chemical Society. 2009;131(47):17303–17314. doi: 10.1021/ja906987s. http://dx.doi.org/10.1021/ja906987s. [DOI] [PubMed] [Google Scholar]