

Abstract
In this paper, we describe the design of a microfluidic sample preparation chip for human stool samples infected with Clostridium difficile. We established a polymerase chain reaction able to distinguish C. difficile in the presence of several other organisms found in the normal intestinal flora. A protocol for on-chip extraction of nucleic acids from clinical samples is described that can detect target DNA down to 5.0×10−3 ng of template. The assay and sample preparation chip were then validated using known positive and known negative clinical samples. The work presented has potential applications in both the developed and developing world.
Keywords: Sample preparation, Microfluidics, Clostridium difficile, PCR
1. Introduction
Infectious diarrhea is a major global health problem, causing more than 1.4 million preventable deaths of children under the age of 5 each year worldwide (Prüss-Üstün et al., 2008). Diarrheal illnesses often lead to chronic malnutrition, thus amplifying the impact on affected populations. Infectious diarrhea is most often spread via contaminated water supplies and, as such, identifying the sources of outbreaks has the potential to save many lives (Morgan et al.,1998). Detection can involve culturing the organism, microscopy and/or a variety of molecular assays but is most often not performed at all. Simplifying the laboratory operations required to identify the offending pathogen with a view toward treatment is an important component of the effort to deliver diagnostic and surveillance tests to the developing world. In the developed world, faster diagnostics for specific food-borne and nosocomial diarrheas also have the potential to save lives by distinguishing between illnesses likely to be self-limiting versus those that require treatment.
Stool is a challenging diagnostic sample for several reasons. First, stool contains myriad of microorganisms that comprise the normal intestinal flora. Second, stool samples can range widely in consistency from a clay-like semi-solid to a turbid liquid. Third, stool is known to contain many inhibitors of the polymerase chain reaction (Clarke and Dufour, 2006). As a result, isolating and purifying nucleic acids from stool is challenging, and only a few reports have proposed methods for on-chip sample preparation using this kind of sample (Bjerketorp et al., 2008, Liu and Zhu, 2005, Weigl et al., 2006).
Here we describe a stool sample preparation chip designed to take in a sample of stool ultrafiltrate mix it with lysis buffer, extract bacterial DNA from the sample, and then clean, concentrate and elute it for downstream molecular diagnosis. The entire device is made from polymeric materials with the exception of the silica particles embedded in the solid phase extraction monolith. Thermoplastic sample preparation chips may be more rugged for application in the developing world due to their superior toughness (less likely to fail by brittle fracture than glass or silicon) and dimensional stability (as compared to PDMS and PDMS hybrid chips.) The range of material properties (e.g. glass transition temperatures and optical properties) makes this class of materials particularly attractive for mass-market applications.
To demonstrate the feasibility of extracting and detecting DNA from infectious microorganisms in human stool samples, we performed the following study on Clostridium difficile infected and non-infected samples obtained from patients at Boston University Medical Center. C. difficile is the most common cause of nosocomial, or institution acquired, diarrhea. The spectrum of disease caused by C. difficile infection is broad, ranging from acute watery diarrhea with abdominal pain, low-grade fever, and leukocytosis to the major complications of dehydration, hypotension, toxic megacolon, septicemia perforation, and death. Typically, C. difficile-associated diarrhea occurs in hospitalized patients following antibiotic treatment; it is debilitating, and prolongs hospitalization (McFarland et al., 1990). Recently, cases of the infection have been documented in patients outside of the usual affected groups: younger people and people not in a hospital or institutional environment (Warny et al., 2005). This development has been a great cause of concern in the medical community and in the popular press (Brodkin, 2006, Brody, 2006), as the new strains appear to cause a more severe disease. Distinguishing C. difficile from other less serious infections with similar symptoms at onset is critical to effective patient care.
Unlike the management of other infectious diarrheas, stool cultures have limited clinical utility in C. difficile-associated disease largely because C. difficile is difficult to culture (thus the moniker difficile). C. difficile infection is established by (1) a stool bioassay for cytotoxins that cause rounding of cultured fibroblasts or (2) immunoassays for the stool toxins themselves (Bartlett, 2002). The cytotoxicity bioassay is considered the gold standard against which other cytotoxin assays are compared, given its high sensitivity (94–100%) and specificity (99%) (George et al., 1982). In this bioassay, stool is diluted with a buffer, filtered to remove bacteria and solids, and then placed in a cultured monolayer of fibroblasts. Both C. difficile toxins A and B disrupt the cytoskeleton and, when present at levels as low as a few molecules per cell, will cause rounding of fibroblasts. Drawbacks of the cytotoxicity assay are its labor-intensive nature, attendant high cost, and the 48–72 h it typically takes to complete. The work presented here is a first step toward the development of a fully integrated plastic microfluidic diagnostic for C. difficile in stool samples at the point of care.
2. Materials and methods
2.1. Materials
Cyclic polyolefin (Zeonor 690R) was obtained as a gift from Zeon Chemicals Inc. (Louisville, KY). Butyl methacrylate (99%, BuMA), ethylene glycol dimethacrylate (98%, EGDMA), methyl methacrylate (99%, MMA), 1-dodecanol (98%), cyclohexanol (99%), benzophenone (99%), and 2,2-dimethoxy-2-phenylacetophenone (99%, DMPAP), were purchased from Sigma-Aldrich (St. Louis, MO). A Qiagen QIAamp DNA Stool Mini Kit was purchased from Qiagen Inc. (Valencia, CA). Reagents from this kit were also used for the microchip extractions. Luria Broth and antibiotic Medium 3 was purchased from DIFCO (Franklin Lakes, NJ). Illustra PureTaq Ready-To-Go PCR Beads made by GE Healthcare (Buckinghamshire, HP79NA UK) were used for all PCR reactions. Genomic DNA from C. difficile, Escherichia coli, Clostridium perfringens, and Shigella flexneri was obtained from ATCC (Manassas, VA). PCR primers were synthesized by Invitrogen (Grand Island, NY). Nalgene 4 mm 0.22 μm syringe filters and Excel 1 cm3 disposable syringes were purchased from Fisher Scientific (Fairlawn, NJ). Silica microspheres (0.7 μm) were purchased from Polysciences, Inc. (Warrington, PA). Polyetheretherketone (PEEK) capillaries of 360 μm-i.d. and NanoPort assemblies for device-based fluidic connections were purchased from Upchurch Scientific (Oak Harbor, WA).
2.2. Microchip fabrication
The microchannels were formed by hot embossing a Zeonor plaque with a nickel–cobalt electroplated mold (NiCoForm, Inc., Rochester, NY) made from a silicon master. The channels were 4 cm in length, 400 μm wide and 100 μm deep. The silicon master was fabricated by spinning a negative resist (NR5-8000, Futurrex, Franklin, NJ) to a thickness of 12 μm onto the wafers. After pre-baking the wafers for 1 min at 150 °C, the pattern was transferred through a transparency mask by proximity contact lithography. This step was followed by post-exposure baking, developing with RD6 resist developer (Futurrex, Franklin, NJ) and hard-baking the wafers for 2 min at 100 °C. The exposed areas of the wafer were then etched on an STS DRIE (STS, Newport, UK). Hot embossing was performed with a hot press (Heated Press 4386, Carver, Wabash, IN) at 176 °C (40° above the glass transition temperature (Tg) of Zeonex 690R) at a pressure of 500 psi (hot press gauge reading) for 4 min. After embossing, the master and the substrate were removed from the hot press and allowed to cool at room temperature for 30 s on an aluminum plate and were manually separated. 1.5 mm diameter wells were drilled at the ends of the hot-embossed channels. The embossed channels were sealed with another Zeonor plaque of the same dimensions by thermally bonding at the Tg for 4 min at 500 psi. Nanoports (Upchurch Scientific, Oak Harbor, WA) were epoxied to the chip at the location of the wells to provide secure attachment of PEEK tubing attached to a syringe on a syringe pump (Fig. 1).
Fig. 1.
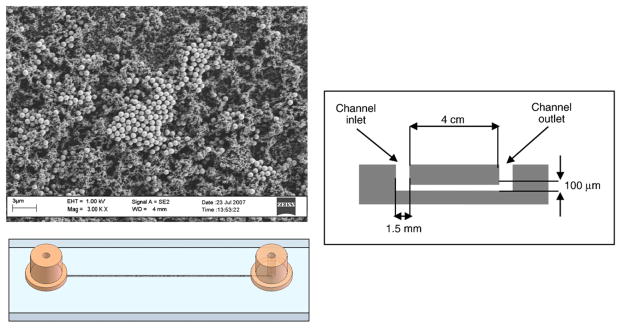
Scanning electron micrograph of a typical solid phase extraction column and schematic drawings of the thermoplastic test chip.
2.3. Extraction column fabrication
The open pore polymer monoliths were made inside of the channels after heat sealing as described previously (Bhattacharyya and Klapperich, 2006, Rohr et al., 2001). Briefly, the hot-embossed and sealed channels were surface modified with MMA and 3% benzophe-none to allow for covalent attachment of the polymer monolith. The channels were filled with the monomer solution and photopolymerized for 10 min at 365 nm with 200 mJ/cm2 energy in a UV crosslinker (CL-1000 UV Crosslinker, UPV, Inc., Upland, CA). Excess monomer solution was washed out with 50 μL of methanol pushed through using a hand syringe with vacuum assist on the other side of the channel. After surface-modification of the channel walls, the monolith containing silica particles was formed. The silica particles were purchased in aqueous solution and a 100 μL aliquot of the silica–water suspension was used to make 100 μL of monolith solution. The silica–water suspension was centrifuged and the water was decanted. The particles were resuspended in 100 μL of the monolith pre-polymer solution (15% BuMA, 10% EGDMA, 52.5% 1-dodecanol, 22.5% cyclohexanol, with 1.13% w/v DMPAP added to the solution) (Bhattacharyya and Klapperich, 2006). The mixture was ultrasonicated to ensure good distribution of the particles (Sonifier S-250D, Branson Ultrasonic, Danbury, CT). The pre-polymer solution was pipetted into the channels, and the channels were placed in the UV crosslinker at 200 mJ/cm2 for 1.8 min and then washed with 50 μL of methanol. The washing times here are greatly reduced from our previous reports without any noticeable impact on channel performance.
2.4. PCR primer selection for identification of toxin A or toxin B producing C. difficile
The primer sequences selected were described by Bélanger et al. for toxin A and Giulbault et al. for toxin B (Belanger et al., 2003, Guilbault et al., 2002). The forward primer for C. difficile toxin A was TCT ACC ACT GAA GCA TTA C and the reverse primer was TAG GTA CTG TAG GTT TAT TG. The forward primer for C. difficile toxin B was GTG GCC CTG AAG CAT ATG and the reverse primer was TCC TCT CTC TGA ACT TCT TGC. The specificity of the primers was confirmed by PCR amplification of genomic DNA from C. difficile, E. coli, C. perfringens, and S. flexneri. The PCR protocol included an initial denaturation step at 94 °C for 3 min, followed by 30 cycles of 94 °C for 45 s, 56 °C for 45 s, and 72 °C for 75 s, followed by a final extension step at 72 °C for 10 min completing the protocol at 4 °C. To determine the least amount of DNA detectable (sensitivity) using these primers at 30 cycles, the amount of genomic DNA added to the reaction was varied by serial dilution from 5.0 to 5.0×10−5 ng. To confirm that the DNA was extracted from bacteria, the presence of 16srRNA gene was detected by PCR using the same PCR conditions listed above. The primers for the 16srRNA gene used were not pathogen specific and amplify 16srRNA in all bacterial samples containing DNA (Ott et al., 2004). The 16srRNA forward primer was ACT GAG ACA CGG TCCA and the reverse primer was AAG GAG GTG ATC CAN CCR CA.
After the PCR was complete, agarose loading buffer (Boston Bioproducts, Boston, MA) was added to each sample and 6% poly-acryl amide or 1% agarose gels were run at 112 V for 1 h. The gels were incubated in ethidium bromide and TBE for 20 min at room temperature, washed with water, viewed under UV light and photographed.
2.5. Human stool samples
Previously confirmed positive (n=9) and negative (n=5) stool samples were acquired from the microbiology lab at Boston Medical Center. The presence or absence of C. difficile in these samples was determined using a cytotoxicity assay (Toxi-Titre™ Plate-HF Cells (B1029-70D), Trinity Biotech, Bray, Ireland) waiting 48 h to observe the cytopathic effect.
2.6. DNA extraction using microfluidic channels
DNA was extracted from stool samples using the Qiagen kit per manufacturer’s instructions as a positive control. Both known positive and known negative samples were extracted using the microfluidic channels. 200 mg of each negative and positive C. difficile infected stool was suspended in 1 mL PBS, vortexed until it was homogenized, spun at 500 rpm for 12.5 min, and supernatant collected. The supernatant was then frozen once at −20°for 1 h and thawed on ice. After thawing was complete, an equal amount of guanidine isothiocyanate was added to the stool supernatant (stool mixture). 200 μL of the stool mixture was passed though each channel using a syringe pump to allow a steady delivery of the stool mixture through the channels; a flow rate of 200 μL/h was used. The stool mixture was passed through the channels using a start–stop method, passing it through for 10 min and stopping it for 1 min to allow the mixture to incubate in the channels. Once the 200 μL stool mixture completely passed through the channel, the mixture was discarded. The channel was washed by passing 40 μL of the chaotropic buffer through followed by 70% cold ethanol for 20 min. Finally 200 μL RNAase free water was passed through the channel, again using the start–stop method. The eluent that was produced contained DNA from the stool. PCR was then performed using the eluent as the template.
3. Results
3.1. Specificity and sensitivity of C. difficile primers
First, we confirmed that the sequence for the primers for C. difficile toxins A and B was specific to C. difficile by running PCR reactions with these primers in the presence of genomic DNAs from other bacterial pathogens common in stool. These experiments showed that there was only amplification of an appropriate sized product with C. difficile and no amplification was observed in the other pathogenic genomic DNAs including E. coli, C. perfringens, and S. flexneri (Fig. 2). The next step was to establish the sensitivity of the primers which amplified as little as 5.0×10−3 ng of C. difficile (Fig. 3). Below this dilution, no amplification was seen.
Fig. 2.

PCR primers are specific for amplifying genomic DNA from toxin A and toxin B containing C. difficile. Lanes 1 and 2 are DNA ladders. Lanes 3–12 are genomic DNA from various diarrhea causing bacteria. Two primer sets were used for amplification; primers for toxin A were used in lanes 3–7 and primers for toxin B were used in lanes 8–12. Only PCR products are generated in the lanes containing toxin producing C. difficile genomic DNA (arrows).
Fig. 3.
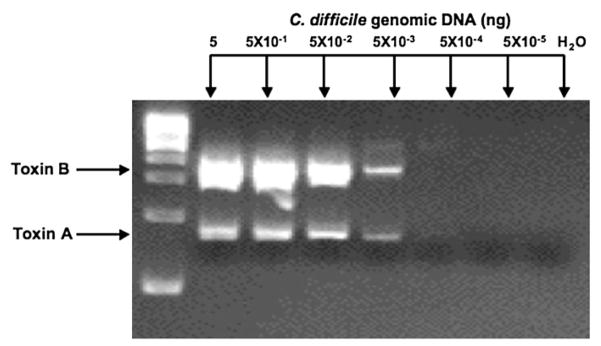
Primers for toxin A or toxin B can identify as little as 5×10−3 ng of C. difficile genomic DNA. Varying amounts of genomic DNA were subjected to PCR amplification with primer sets for toxin A and toxin B.
3.2. DNA extraction using Qiagen stool DNA kit
Following the manufacturer’s protocol, DNA was extracted from stool with the kit and PCR was performed with the C. difficile and 16srRNA as the primers and using the Qiagen extracts as the templates. The 16srRNA primers (Fig. 4A) amplified appropriate products from C. difficile negative stool, positive stool and commercial C. difficile genomic DNA. This indicates that there was DNA present in the extraction. RNase-free water was run as a negative control. In all circumstances this control did not amplify. The C. difficile toxin B primers (Fig. 4A) amplified a sequence in the C. difficile positive stool and C. difficile genomic DNA samples. Therefore, these primers are appropriate for use with DNA isolated from human stool samples.
Fig. 4.
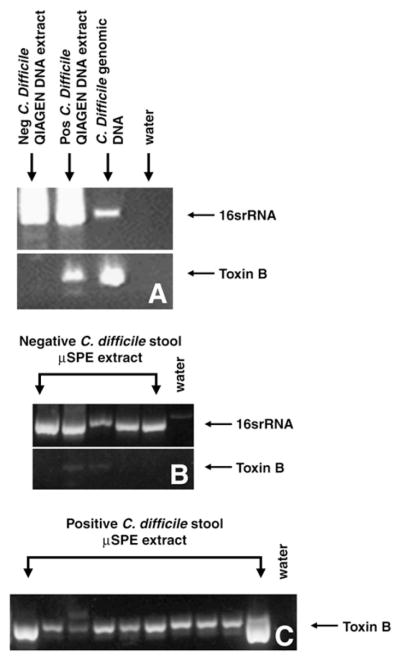
DNA was extracted from C. difficile negative (neg) and positive (pos) stool samples using a Qiagen DNA Stool Mini Kit and PCR was run with both sets of primers (A). All samples were positive for 16srRNA confirming that lysis occurred and that only toxin containing samples were positive for C. difficile toxin B. For extraction using the microfluidic columns (μSPE), no prominent toxin B amplification was observed in negative stool (B) although the microfluidic columns were able to extract DNA (16srRNA). Faint amplification of toxin B was present in 2 of 5 negative stool samples. C. difficile toxin B was detected in DNA extracts from all 9 positive samples using SPE channels (C). Each lane represents separate samples.
3.3. Microfluidic channel DNA extraction
We next determined if DNA extraction from raw stool was possible using the microfluidic channel. The stool from a total of 14 different patients (5 cell culture negative, 9 cell culture positive) was passed through the microfluidic channel as described, washed and eluted in water, and the eluent was used as the PCR template. PCR was then run with both 16srRNA and C. difficile toxin B primer pairs. The 16srRNA primers (Fig. 4A) confirmed that DNA extraction using the microfluidic channels was successful by amplifying the appropriate sized product from stool and genomic DNA, but not from the water sample. The reaction with the C. difficile toxin B primers (Fig. 4B) did not amplify product from 3 of the 5 C. difficile negative stool samples; faint amplification was observed from DNA isolated from 2 stool samples classified as negative by the cytotoxicity assay. The primers did amplify product from all 9 of the C. difficile positive stool as well as from the genomic DNA (Fig. 4C).
4. Discussion
Our long-term goal is to develop a simplified handheld molecular assay that can specifically detect several infectious organisms in stool. Taking C. difficile as our target to demonstrate clinical feasibility, we established that the toxin B primer set distinguishes pathogenic, toxin producing C. difficile from other strains of bacteria. Next, we developed a method to extract bacterial DNA from crude stool samples using a chip-based device that is miniaturized, less labor intensive and keeps the infectious agents more contained than the available gold standard isolation kits. We have demonstrated that microfluidic channels can be used to harvest DNA from crude stool suspensions with a degree of purity suitable for performing PCR. The next step is to couple our chip-based DNA extraction with a chip-based nucleic acid amplification step to make C. difficile detection possible in a completely handheld device.
In all nine positive samples (determined by cytotoxicity assay) obtained from Boston Medical Center, we were able to amplify C. difficile toxin B from DNA isolated by our microfluidic columns (Fig. 4). In 2 of the 5 negative stool samples we detected faint amplification of C. difficile toxin B. This amplification was not observed in the remaining negative samples or in water lane indicating that this is not due to contamination of our PCR reagents. Furthermore, amplification from these 2 samples was also observed using a separate C. difficile primer pair (not shown). Therefore, this result is due to the presence of C. difficile toxin B in the stool. Considering the sensitivity of PCR, it is certainly conceivable that our new diagnostic method is able to detect C. difficile that would be missed by current methods. Larger studies are necessary to accurately determine the sensitivity and specificity of our new diagnostic tool.
The total running time for the DNA extraction as presented here was roughly 3 h. The Qiagen Stool Mini Kit preps took about the same time to perform. It is important to note, however, that the running time of the microfluidic prep can be significantly decreased. We used 200 μL of the initial sample in order to ensure that we would obtain enough template DNA to observe amplicons on an agarose gel after 30 cycles of PCR amplification. Elution was also performed in a 200 μL aliquot. Each of these steps required almost 1 h to get the entire sample through the chip as designed. Gels were the preferred method of detection for this demonstration in order to confirm that the DNA amplified in the PCR reactions was of the right size and in order to detect the presence of undesired amplification products. A fully developed molecular assay would not use agarose gels as a readout. The sensitivity of the PCR assay can be greatly improved by redesigning the primer set as a Taqman assay (Applied Biosystems, Foster City, CA) or other primer–probe designs. This redesign would reduce the amount of initial sample needed and the amount of eluent required to perform the downstream amplification resulting in a much shorter running time overall. Furthermore, probe-based assays detect DNA amplification via fluorescence eliminating the need for gel electrophoresis.
The “gold” standard method for diagnosis for C. difficile has been a cytotoxicity assay for C. difficile toxin A or B identification. It is a tissue culture process that is highly sensitive and specific for C. difficile but can take 48 to 72 h to complete. Due to the need for rapid diagnosis, an ELISA based diagnostic test that takes only 2–4 h to complete has been developed. While highly specific for C. difficile, the ELISA is not very sensitive requiring 100 to 1000 pg of toxin for detection. Considering the necessity for fast, accurate diagnosis in patient care, it would be of clinical importance to develop a more rapid, highly sensitive and specific C. difficile detection assay that could be modified to require a less complex methodology. The work presented here is a major step towards such a diagnostic. Further, the results presented here could be extended to other infectious diarrheas, including those that cause massive devastation each year in the developing world.
Contributor Information
Catherine Klapperich, Email: catherin@bu.edu.
Satish K. Singh, Email: singhsk@bu.edu.
References
- Bartlett JG. Clinical practice. Antibiotic-associated diarrhea. N Engl J Med. 2002;346:334–339. doi: 10.1056/NEJMcp011603. [DOI] [PubMed] [Google Scholar]
- Belanger SD, Boissinot M, Clairoux N, Picard FJ, Bergeron MG. Rapid detection of Clostridium difficile in feces by real-time PCR. J Clin Microbiol. 2003;41:730–734. doi: 10.1128/JCM.41.2.730-734.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya A, Klapperich CM. Thermoplastic microfluidic device for on-chip purification of nucleic acids for disposable diagnostics. Anal Chem. 2006;78:788–792. doi: 10.1021/ac051449j. [DOI] [PubMed] [Google Scholar]
- Bjerketorp J, Ng Tze Chiang A, Hjort K, Rosenquist M, Liu WT, Jansson JK. Rapid lab-on-a-chip profiling of human gut bacteria. J Microbiol Methods. 2008;72:82–90. doi: 10.1016/j.mimet.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Brodkin J. Hospitals Eye Deadly Strain of Bacteria. The Boston Herald; Boston, MA: 2006. p. 6. [Google Scholar]
- Brody JE. Trouble in the Gut, When Antibiotics Work Too Well. InThe New York Times; New York: 2006. p. 7. [Google Scholar]
- Clarke WC, Dufour DR. Contemporary Practice in Clinical Chemistry. AACC Press; Washington, DC: 2006. [Google Scholar]
- George WL, Rolfe RD, Finegold SM. Clostridium difficile and its cytotoxin in feces of patients with antimicrobial agent-associated diarrhea and miscellaneous conditions. J Clin Microbiol. 1982;15:1049–1053. doi: 10.1128/jcm.15.6.1049-1053.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbault C, Labbe AC, Poirier L, Busque L, Beliveau C, Laverdiere M. Development and evaluation of a PCR method for detection of the Clostridium difficile toxin B gene in stool specimens. J Clin Microbiol. 2002;40:2288–2290. doi: 10.1128/JCM.40.6.2288-2290.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WT, Zhu L. Environmental microbiology-on-a-chip and its future impacts. Trends Biotechnol. 2005;23:174–179. doi: 10.1016/j.tibtech.2005.02.004. [DOI] [PubMed] [Google Scholar]
- McFarland LV, Surawicz CM, Stamm WE. Risk factors for Clostridium difficile carriage and C. difficile-associated diarrhea in a cohort of hospitalized patients. J Infect Dis. 1990;162:678–684. doi: 10.1093/infdis/162.3.678. [DOI] [PubMed] [Google Scholar]
- Morgan UM, Pallant L, Dwyer BW, Forbes DA, Rich G, Thompson RC. Comparison of PCR and microscopy for detection of Cryptosporidium parvum in human fecal specimens: clinical trial. J Clin Microbiol. 1998;36:995–998. doi: 10.1128/jcm.36.4.995-998.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott SJ, Musfeldt M, Ullmann U, Hampe J, Schreiber S. Quantification of intestinal bacterial populations by real-time PCR with a universal primer set and minor groove binder probes: a global approach to the enteric flora. J Clin Microbiol. 2004;42:2566–2572. doi: 10.1128/JCM.42.6.2566-2572.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prüss-Üstün A, Bos R, Gore F, Bartram J. Safer Water, Better Health: Costs, Benefits and Sustainability of Interventions to Protect and Promote Health. World Health Organization; Geneva, Switzerland: 2008. [Google Scholar]
- Rohr T, Yu C, Davey MH, Svec F, Frechet JMJ. Electrophoresis. 2001;22:3959–3967. doi: 10.1002/1522-2683(200110)22:18<3959::AID-ELPS3959>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Warny M, Pepin J, Fang A, Killgore G, Thompson A, Brazier J, Frost E, McDonald LC. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet. 2005;366:1079–1084. doi: 10.1016/S0140-6736(05)67420-X. [DOI] [PubMed] [Google Scholar]
- Weigl BH, Gerdes J, Tarr P. Fully integrated multiplexed lab-on-a-card assay for enteric pathogens. Proceedings of SPIE International Society for Optical Engineering; 2006. p. 6112. [Google Scholar]