

Abstract
A key question in the field of autoimmunity concerns the fact that experimental disease is generally induced more easily with closely related, but not completely identical, tissue restricted antigens. Here, the possibility that naturally occurring regulatory T cells (Tregs) for self antigens are more potent than those for related antigens was investigated. The self-antigen specificity of naturally occurring Tregs was tested in experimental autoimmune encephalomyelitis (EAE) induced with mouse (self) or closely related (rat) myelin oligodendrocyte glycoproteins (MOGs). Surprisingly, Treg depletion increased EAE severity in mice immunized with mouse, but not rat MOG. This increase was associated with increased T-cell activation and infiltration of the central nervous system, as well as increased IL-17 production and a higher ratio of IFNγ to IL-10 producing cells. These data suggest that Tregs are specific for self-antigen and do not “cross-protect” against autoimmunity even when disease is induced with closely related foreign antigens.
Keywords: EAE, myelin oligodendrocyte glycoprotein, T cells, cytokines, autoimmunity
1. Introduction
Multiple sclerosis (MS), a paralyzing disease of the central nervous system (CNS), is mediated in part by autoreactive T and B lymphocytes (Iglesias et al., 2001). Experimental autoimmune encephalomyelitis (EAE), an animal model of MS, can be induced by injection of myelin oligodendrocyte glycoprotein (MOG), a minor component of the myelin sheath. C57BL/6 mice immunized with the immunodominant rodent MOG peptide (aa 35-55) or recombinant E.coli-derived non-glycosylated extracellular domain of MOG (aa 1-125) develop acute EAE (Lyons et al., 1999; Oliver et al., 2003). Despite similar clinical signs, we have previously shown that MOG of different origins induces EAE via different mechanisms. That is, EAE induced with MOG of rat origin is B cell independent, whereas that induced with MOG of human origin is B cell dependent and antibody mediated (Marta et al., 2005; Oliver et al., 2003).
Naturally occurring CD4+CD25+ regulatory T cells (Tregs) have been identified as regulators of T-cell mediated autoimmunity in both humans and mice (Bach, 2003). In the mouse, Tregs can be distinguished from naïve or activated T cells by their expression of the transcription factor Foxp3; more than 90% of CD4+CD25+ T cells and fewer than 4% of CD4+CD25− T cells stain with antibody to Foxp3 (Fontenot et al., 2005). Naturally occurring Tregs are selected in the thymus by self-antigens (Jordan et al., 2001); thus, suggesting that higher affinity to tissue-restricted antigen during selection may be important in controlling the ability of Tregs to recognize self-antigens as opposed to foreign antigens.
Tregs can suppress MOG35-55 induced EAE, as illustrated by the fact that Treg supplementation decreases (Kohm et al., 2002; Zhang et al., 2004), while Treg depletion increases (McGeachy et al., 2005) EAE severity. It appears that Tregs mediate their suppression by dampening the activation of autoreactive T cells (Kohm et al., 2002; Zhang et al., 2004). Currently, it is not known how efficiently Tregs can suppress EAE induced with the extracellular domain of MOG, and whether Tregs can control EAE induced with MOG of self or foreign origin. In addition, it is not known whether the Treg suppressive effect occurs early on during EAE induction or later in the effector phase by acting directly on autoreactive T cells in the CNS.
Here, we show that early depletion of naturally occurring CD4+CD25+ Tregs results in increased EAE severity following immunization with self (mouse MOG) but not foreign (rat MOG) antigens. The reappearance of Tregs in the CNS following depletion does not reduce disease severity. These data suggest that Tregs are highly specific for mouse MOG and that Tregs do not “cross-protect” against highly similar, yet foreign, rat MOG.
2. Materials and methods
2.1. Animals, Treg depletion, and immunization
8–12 week old C57BL/6 female mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Each mouse received a total of three i.p. injections (100 μg/injection) of either anti-CD25 antibody (Clone PC61 (Onizuka et al., 1999) a gift from Mark Shlomchik (Yale University School of Medicine) or control rat IgG (Zymed Laboratories, South San Francisco, CA) at 48hr intervals.
EAE was induced as previously described (Suen et al., 1997). Seven days after completion of treatment with either PC61 or rat IgG, mice were immunized by s.c. flank injections of either 300 μg of MOG35-55 peptide or 100 μg of recombinant extracellular domain MOG of mouse, or rat origin in CFA (Difco, Detroit, MI) with 300 μg of Mycobacterium tuberculosis on day 0. In some experiments mice received an additional boost injection of MOG in CFA on day 7. Mice were injected i.p. with 500 ng of pertussis toxin (List Biological Laboratories, Campbell, CA) on days 0 and 2. All animal use and husbandry protocols were approved by the Yale University Institutional Animal Care and Use Committee.
2.2. MOG proteins and MOG peptides
Constructs consisting of mouse or rat MOG extracellular domain were expressed in E.coli DH5α bacteria (Oliver et al., 2003). Residue 47 of mouse MOG was corrected from alanine to valine by site directed mutagenesis as previously described (Oliver et al., 2003). Rodent MOG35-55 and a series of mouse MOG peptides were synthesized and sequenced by the W.M. Keck Biotechnology Resource Center at Yale University.
2.3. Clinical disease scoring
Mice were monitored daily for clinical signs of EAE. Clinical scores were based on a scale of 0–5, with a score of 0 indicating no disease, 1 - limp tail, 2 - paresis or partial paralysis of the hind limbs, 3 - total hind limb paralysis, 4 - hind and front limb paralysis, and 5 indicating death. Disease onset was determined as the first day of clinical signs. Mean maximum clinical score was calculated as the average of the maximal clinical score of all mice per group. Disease indices for each group were calculated as: ((sum of the mean clinical scores)/(day of disease onset)) × 100.
2.4. FACS analysis
The following antibodies were obtained from BD Biosciences - Pharmingen: anti-CD4, anti-CD25 (7D4), anti-CD8, anti-CD45RB/B220, anti-CD62L, anti-CD69, and anti-TCRVβ8. Intracellular Foxp3 staining was done according to the manufacturer’s instruction (eBioscience, CA). Data were collected on a Becton Dickinson FACScan™ flow cytometer and analyzed using FlowJo software.
2.5. Immunofluorescence
Animals were perfused intracardially with cold PBS and spinal cords were collected. 7 μm frozen sections were applied onto poly-L-lysine–coated glass slides (Sigma-Aldrich), fixed with acetone, air-dried and blocked with 5% mouse serum/3% BSA in PBS. Slides were then incubated with anti-CD4 (PE) antibody (BD Biosciences - Pharmingen) and analyzed on an Axioskop fluorescence microscope (Carl Zeiss Microimaging, Inc).
2.6. Isolation of CNS Mononuclear Cells
Mice were deeply anesthetized and perfused intracardially with RPMI 1640 medium (Life Technologies). Brain and spinal cord cell suspensions were incubated with 1 mg/ml collagenase II (Sigma) at 37°C for 20 minutes, and mononuclear cells were isolated by discontinuous Percoll (Pharmacia, Piscataway, NJ) gradient.
2.7. Enzyme-linked immunospot (ELISPOT) and ELISA analysis
ELISPOT assays for IFNγ, IL-4, and IL-10 were performed as previously described (Juedes et al., 2000). Antibody pairs used for capture and detection of IFNγ, IL-4, and IL-10 were R4-6A2 and XMG1.2-biotin; 11B11 and JES5-2A5-biotin; BVD6-2462 and J3S5-16E3-biotin (BD Pharmingen), respectively. Spot-forming cells were enumerated with the aid of a dissecting microscope. ELISA for the detection of IL-17A was done using the IL-17 ELISA Ready-Set-Go kit according to the manufacturer’s instructions (eBioscience, CA).
2.8. Proliferation assay
LN or spleen cells (2×105) were cultured in 96-well plates in triplicate with either medium only, or medium supplemented with 50 μg/ml of various MOG peptides or 100 μg/ml of mouse MOG. After 48hr [3H]Thymidine (1 μCi/well) was added, and cultures were harvested 18hr later and analyzed using a Wallac Microbeta 1450 liquid scintillation counter (Gaithersburg, MD). Calculations were done as previously described (Suen et al., 1997).
3. Results
3.1. Treg depletion enhances disease severity in mice immunized with mouse but not rat MOG
Previous reports have shown efficient Treg depletion using various reagents, such as cyclophosphamide and anti-CD25 depleting antibodies (Matsushita et al., 2008) as well as the use of diphteria toxin in Foxp3 DTR transgenic mice (Lahl et al., 2007). Since in the current study, naturally occurring Treg were depleted in naïve animals prior to immunization with antigen (where more than 90% of CD4+CD25+ cells are Foxp3+ (Fontenot et al., 2005)), we chose to use the anti-CD25 antibody (clone PC61) for depletion of Tregs. We tested the efficacy of Treg depletion by examining the frequency of CD4+CD25+ Tregs in the spleen and LNs of mice treated with anti-CD25 (PC61) or IgG control. PC61 treatment did not alter the frequencies of total CD4+, CD8+, or B220+ cells (Fig. 1A). In contrast, PC61 significantly reduced CD4+CD25+ T cells in both LNs and spleen of naïve mice (Fig. 1B). Treg depletion, as defined by reduction of CD4+CD25+ cells was observed in the spleens and LNs of animals treated with PC61 (Fig 1B), and was appreciably sustained in the naïve mice for at least 10 days after completion of treatment (data not shown). Since mouse MOG shares a high degree of sequence homology with rat MOG (Fig. 2), we examined the effect of CD25 depletion on mice immunized with mouse or rat MOG. First, we examined the effects of Treg depletion on EAE induced with self (mouse) MOG antigen following Treg depletion. Control animals exhibited a mild but significant EAE disease following immunization with mouse MOG, while treatment with anti-CD25 further augmented EAE severity (Fig. 3A and Table I). Surprisingly Treg depletion did not result in a statistically significant increase in disease severity in mice immunized with rat MOG (Fig. 3B and Table I), although Treg depletion did lead to an earlier mean day of onset following immunization with rat MOG.
Fig. 1.
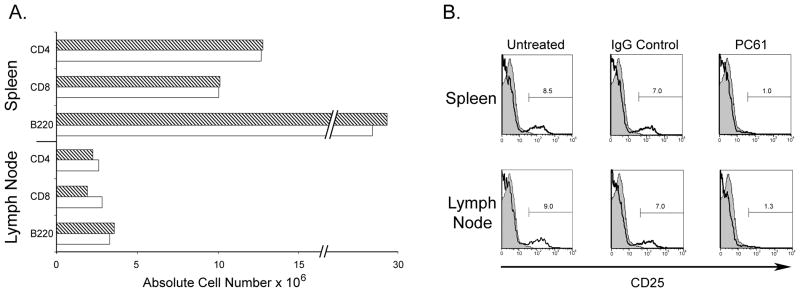
PC61 treatment results in an efficient depletion of CD4+CD25+ T cells. Mice received three i.p. injections of 100 μg/injection of either PC61 or rat IgG every 48hr and were sacrificed 7d post third injection. (A) Absolute cell numbers in pooled spleens and LNs of control IgG (dashed bars) or PC61-treated groups (open bars). (B) Expression levels of CD25 in either spleen or LN of control IgG or PC61-treated mice. Cells were stained with anti-CD4 and anti-CD25 (7D4). All plots are gated on CD4+ cells; shaded histograms show isotype control staining.
Fig. 2.

Comparison of amino acid sequence of mouse (M) and rat (R) MOG extracellular domain. Amino acid residues which differ from mouse MOG are underlined.
Fig. 3.

Treg depletion results in increased EAE severity in mice immunized with mouse MOG but not rat MOG. Mice receiving three i.p. injections of either rat IgG (□) or PC61 (◆) were immunized by standard protocol with mouse MOG (A), rat MOG (B), or with rat MOG omitting boost (C). Animals were monitored and clinical scores were collected daily. ┼, indicates death. Dead animals were noted and omitted from subsequent graphs but were included in all other calculations (see Table I).
Table I.
Increased Incidence and Disease Severity in Treg-Depleted Mice Immunized with Mouse, but not Rat, MOG
| Immunogen | Treatmenta | Incidence | Mean Day of Onset | Mean Maximum Disease Score | p Value | Day 40 Disease indexb |
|---|---|---|---|---|---|---|
| Mouse rMOG | Rat IgG | 11/15 | 14 | 1.8 | 0.035 | 172 |
| PC 61 | 14/15 | 14 | 2.8 | 397 | ||
| Rat rMOG | Rat IgG | 7/7 | 16 | 3.4 | 424 | |
| PC61 | 9/9 | 14 | 3.4 | 476 | ||
| Rat rMOG | Rat IgG | 3/5 | 19 | 1.4 | 135 | |
| No Boost | PC61 | 3/5 | 22 | 1.6 | 133 |
C57BL/6 mice that had received three i.p. injections of either rat IgG or PC61 were immunized by standard protocol with mouse MOG, rat MOG, or with rat MOG omitting boost, and observed daily for signs of clinical disease. Onset and scores were calculated for mice that developed clinical signs while maximum disease scores were calculated for all mice.
Disease indices for each group were calculated as: ((sum of the mean clinical scores)/(day of disease onset)) × 100.
The fact that rat MOG induced a more severe EAE than mouse MOG in control IgG-treated mice, might suggest that increased disease severity could mask the effects of Treg depletion in rat MOG immunized mice. To test this possibility, we immunized mice with only one injection of rat MOG without the additional boost injection on day 7. As expected, immunized mice developed a milder disease and a delayed mean day of onset when compared with mice receiving a boost (compare Fig 3B and 3C and Table I). Even though disease severity was reduced by this immunization protocol, Treg depletion still did not induce a higher disease index or earlier mean day of onset in rat MOG immunized mice. In summary, Treg depletion increases EAE severity in mice immunized with mouse, but not rat MOG under both optimal and suboptimal EAE induction conditions.
3.2. Treg depletion increases T cell activation in the periphery and the CNS
To evaluate the effects of Treg depletion on T cell activation in the periphery, mice immunized with mouse MOG were sacrificed on day 20, at the time of peak clinical score. Spleen and LNs were harvested and cells were purified for FACS analysis. CD4+ T cells were stained for CD69 or CD62L expression in order to differentiate activated from resting T cells, respectively. Similar proportions of CD4+CD62L+ cells were detected in anti-CD25 and control-Ig treated mice (Fig 4A); however, the percentage of CD4+CD69+ T cells was 1.5 times higher in the spleens of anti-CD25 treated mice when compared with controls, while no difference was detected in the LN (Fig. 4B). The effects of PC61 treatment were observed even as late as day 20 post immunization as the proportion of CD4+CD25+Foxp+ was still somewhat reduced when compared with IgG treated controls (Fig 4C). Moreover, anti-CD25 treated mice showed increased frequencies of activated CD4+CD25+Foxp3− cells when compared with control-Ig treated mice (Fig. 4C). Next, we compared the proliferative response of LN T cells in Treg depleted or Ig-control mice immunized with either mouse or rat MOG. The in vitro response to mouse MOG of LN cells from anti-CD25 treated mouse MOG immunized mice was 1.5 times higher than that obtained with cells from IgG-treated mice (Fig. 4D). In contrast, LN cells from PC61-treated rat MOG immunized mice showed no increase in response to mouse MOG (Fig. 4D). Similar results were obtained when LN cells from rat MOG immunized mice were stimulated in vitro with rat MOG (data not shown). CD4+IL-17+ LN T cells have been implicated as important mediators of EAE pathogenesis (Komiyama et al., 2006). We measured the in vitro production of IL-17A by LN cells isolated from either Treg-depleted or Treg-sufficient mice immunized with mouse or rat MOG. Anti-CD25 treatment resulted in a significance increase in IL-17 production in LN cells from mouse MOG immunized mice when compared with cells from control mice (Fig. 4E). No increase in IL-17 production was observed following Treg depletion of rat MOG immunized mice (Fig. 4E).
Fig. 4.


T cells from spleen and LNs of Treg-depleted mice immunized with mouse MOG are hyper-responsive when stimulated with mouse MOG. Mice receiving three i.p. injections of either rat IgG (n=3) or PC61 (n=3) were immunized with mouse MOG and sacrificed on day 20. Spleens or LNs isolated from each group were pooled and CD4+ gated cells were stained for expression of (A) CD62L and (B) CD69. (C) Spleen cells were also tested for anti-CD4, anti-CD25, and anti-Foxp3. (D) Cells isolated from mice immunized with mouse or rat MOG were stimulated with mouse MOG and proliferation was measured as described in Materials and Methods. For (D) data are expressed as stimulation indices and represent one of 2 independent experiments with three mice per group. * p<0.0002, § p<0.003 (E) LN cells from mouse or rat MOG immunized mice were stimulated with mouse MOG protein and supernatants were collected for IL-17A detection by ELISA. Dashed bars- control IgG treated; opened bars- PC61-treated animals
To test the effects of Treg depletion on T cell infiltration in the CNS, we analyzed lymphocytes isolated from the CNS of animals treated with anti-CD25 or IgG control. Treg-depleted animals showed elevated numbers of infiltrating lymphocytes in the CNS 20 days post immunization with mouse MOG protein, as demonstrated by both FACS analysis (Table II) and immunofluorescence (Fig. 5A). Despite this increase in cell number, similar frequencies of CD69+ or CD62L+CD4+ T cells were observed in both groups (Fig. 5B). Next, we examined the frequency of Tregs in the CNS of these mice. The frequency of CD4+CD25+Foxp3+ cells was similar in PC61 and control IgG-treated mice and more than 90% of CD4+CD25+ cells stained positive for Foxp3 (Fig. 5C). A small fraction of CD4+CD25− cells was also positive for Foxp3 in the CNS. Interestingly, fewer CD4+CD25−Foxp3+ cells were detected in the CNS of PC61-treated mice when compared with IgG-treated controls (Fig. 5C). Since T cells isolated from the CNS do not proliferate in vitro (Juedes et al., 2000), we analyzed their ability to produce cytokines. Treg depleted mice showed a higher number of mouse MOG specific T cells when compared with control mice, as shown by increased numbers of IFNγ producing cells stimulated with MOG in vitro (Fig. 5D). No differences were detected in the numbers of IL-4 and IL-10 producing cells.
Table II.
Increased Lymphocytic Infiltration of the CNS of Treg-Depleted Mice
| Treatment | Cell Numbers in the CNS × 103
|
||
|---|---|---|---|
| CD4+ T Cells | CD8+ T Cells | B220+ B Cells | |
| Rat IgG | 13 | 3 | 9 |
| PC61 | 49 | 14 | 24 |
C57BL/6 mice that had received three i.p. injections of either rat IgG orPC61 were immunized by standard protocol with mouse MOG, then sacrificed on day 20. Mononuclear cells were isolated from the CNS and stained with anti-CD4, anti-CD8 and anti-B220 antibodies as described in Methods. Cell frequencies were used to calculate the absolute cell numbers of each population out of the total number of isolated cells.
Fig. 5.

Treg depletion results in increased numbers of infiltrating CD4+ cells and increased numbers of IFNγ producing cells in the CNS. Mice treated with either rat IgG (n=3) or PC61 (n=3) were immunized with mouse MOG and sacrificed on day 20. (A) Isolated SC were sectioned and stained for the presence of CD4+ T cells. (B) Mononuclear cells from the CNS were gated on CD4+ cells and stained for the expression of CD62L or CD69. (C) Cells isolated as in (B) were stained with anti-CD4, anti-CD25, and anti-Foxp3. (D) ELISPOT analysis. CNS tissues from each group were pooled together and 105 cells were plated and stimulated with either mouse MOG or MOG35-55. Dashed bars- control IgG treated; open bars- PC61-treated animals. Data are expressed as stimulation indices and represent one of 2 independent experiments with three mice per group.
In summary, Treg depletion resulted in an increased frequency of MOG-specific IFNγ-producing T cells in the periphery and augmented total T cell infiltration of the CNS, despite similar precentages of infiltrating Tregs at the peak of clinical disease. These data suggest that CNS infiltrating Tregs are unable to diminish EAE severity at the site of inflammation.
3.3. The effect of Treg depletion on autoreactive T cell specificity to MOG
In order to test the possibility that depletion of Tregs could lead to the emergence of subdominant clones of T cells capable of responding to epitopes in addition to MOG35-55, thus, further contributing to increased disease severity, we analyzed the ability of LN or spleen cells from Treg-depleted mouse MOG immunized mice to respond to a panel of peptides that encompassed the entire mouse MOG extracellular domain (Oliver et al., 2003). Cell proliferation with a stimulation index greater than 1 was detected in the spleen (Fig. 6A) and LN (Fig. 6B) of mice day 10 post immunization. Interestingly, cells isolated from spleen (Fig. 6A) or LN (Fig. 6B) of PC61-treated mice proliferated more vigorously in response to the immunodominant peptide MOG35-55 than did cells isolated from IgG-treated mice (Fig. 6A and 6B) as indicated by increased stimulation indices.
Fig. 6.
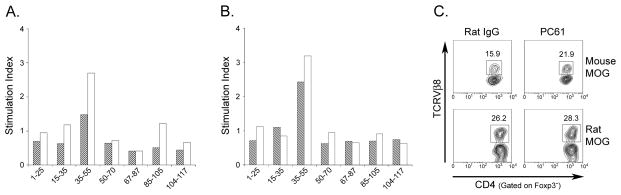
Depletion of Tregs does not result in the emergence of T cells reactive to new encephalitogenic epitopes but does lead to increased proliferation to MOG35-55 in the periphery as well as an enrichment of CD4+TCRVβ8+Foxp3− T cells in the CNS. (A) Spleens or (B) LNs from individual mice treated with either rat IgG (hatched bars, n=3) or PC61 (open bars, n=3) and immunized with mouse MOG were isolated and pooled on day 10 post immunization. Cells were stimulated with different peptides of MOG extracellular domain. Data are expressed as stimulation indices and represent one of two independent experiments with three mice per group. * p<0.0002. (C) Mice treated with either rat IgG or PC61 and immunized with mouse or rat MOG were sacrificed on day 20 post immunization and cells from the CNS were collected. Cells were stained with anti-CD4, anti-TCRVβ8 and anti-Foxp3 antibodies.
T cell lines from C57BL/6 mice immunized with MOG35-55 show an increase in the proportion of CD4+ T cells expressing the TCRVβ8 chain ((Mendel et al., 1999) and data not shown). Concurrently, we also found a ~40% increase in CD4+TCRVβ8+Foxp3− cells in the CNS of PC61-treated mouse MOG immunized mice relative to controls (Fig. 6C). In contrast, Treg depletion of mice immunized with rat MOG resulted in a minor increase of already high levels of CD4+ TCRVβ8+Foxp3− cells in the CNS (~10%) (Fig. 6C). Taken together, these data indicate that Treg depletion does not alter T cell reactivity to MOG epitopes other than MOG35-55, but rather results in increased activation and accumulation of MOG35-55 specific T cells in the CNS.
4. Discussion
This report examines the role of Tregs in EAE induced with the extracellular Ig-like domain of MOG of mouse or rat origin. Taken together, the data presented here demonstrate that Tregs are highly protective against autoimmunity induced with self-antigen (mouse MOG) but do not protect against autoimmunity induced with a closely related foreign antigen (rat MOG). This protection likely occurs during priming of autoreactive T cells in the periphery and results in reduced infiltration and altered cytokine production in the CNS.
The efficient induction of EAE in some models of the disease requires the use of foreign rather than self antigens. In our hands, mouse MOG does not induce a severe form of EAE. Similarly, C3H mice respond vigorously to immunization with recombinant guinea pig myelin basic protein, but not mouse myelin basic protein (Targoni and Lehmann, 1998). Here, we examined whether Tregs could suppress disease development in EAE induced with mouse or rat myelin oligodendrocyte glycoproteins. Treg depletion resulted in increased EAE severity in mice immunized with mouse MOG, but did not significantly increase EAE severity in mice immunized with rat MOG. The fact that rat MOG shares a high degree of homology with mouse MOG raises the question of why Treg depletion did not increase EAE severity in rat MOG immunized mice. The selection of Tregs in the thymus during their development requires high affinity self-antigens (Jordan et al., 2001), suggesting the possibility that naturally occurring Tregs preferentially recognize self over foreign antigens. Moreover, existing evidence suggests that Tregs have a TCR repertoire that is distinct from that of effector T cells (Hsieh et al., 2004; Masteller et al., 2005). The detection of mouse MOG mRNA transcript in the thymus (Derbinski et al., 2001) raises the possibility that natural Tregs are generated exclusively by self (mouse) MOG determinants which are absent in non-self (rat) MOG. A recent report showing that only a minority of Tregs isolated from MOG35-55 immunized mice were capable of recognizing this peptide (O’Connor et al., 2007), suggests that determinants other than peptide 35-55 in the MOG protein may stimulate Tregs. An examination of mouse and rat MOG protein sequences reveals several differences in amino-acids outside the immunodominant peptide 35-55. Our observation of an effect of Treg depletion only in mouse MOG immunized mice supports the hypothesis that self (mouse) MOG may preferentially stimulate Tregs in this model of murine EAE and promote a more profound suppression of the disease when compared with foreign (rat) MOG induced EAE. Alternatively, the inability of Tregs to suppress EAE induced with rat MOG may be due to a different repertoire of effector T cells. These autoreactive T cells may be more strongly stimulated and are thus less susceptible to Treg suppression. A recent report by Sweenie et al. showing that T cells generated after immunization with the extracellular domain of mouse MOG protein are inherently different from those generated with MOG35-55 peptide supports this hypothesis (Sweenie et al., 2007).
Tregs’ ability to control EAE may depend on the degree of disease severity. The fact that mouse MOG induced EAE is considerably milder than rat MOG induced EAE prompted us to test whether a milder rat MOG induced EAE may reveal Tregs’ action in this model. In support of this hypothesis, Stephens et al. demonstrated that following treatment with anti-CD25, only mice which were immunized with suboptimally encephalitogenic peptides of the acetylated myelin basic protein 1–9 exhibited increased EAE severity (Stephens et al., 2005). The data presented here following a single immunization with rat MOG demonstrate that Tregs do not protect against EAE induced with a foreign antigen, even under conditions of reduced disease severity. The fact that EAE induced with rat MOG cannot be suppressed by Tregs demonstrates, at least in our model, that antigen source is key in facilitating Tregs’ suppression of autoimmunity.
In EAE induced with MOG35-55, Treg supplementation results in a skewing of the T cell cytokine profile (Th1 vs. Th2) and is highly dependent on IL-10 production in the periphery (Kohm et al., 2002; Zhang et al., 2004). Recently, the production of IL-17 by Th17 cells has been identified in the development of EAE (Guo et al., 2008; Hofstetter et al., 2007; Komiyama et al., 2006; Sutton et al., 2006). The increased proliferation and production of IL-17 by T cells from mice immunized with mouse MOG following Treg depletion suggests that Tregs play a role in reducing T cell activation to mouse MOG protein. The fact that no increase in T cell proliferation or IL-17 production was observed in anti-CD25 -treated rat MOG immunized mice further support the hypothesis that Tregs are highly specific in their ability to suppress T cell responses to mouse MOG. IFNγ is a key cytokine produced by cells infiltrating the CNS (Juedes et al., 2000). In our hands, mice that were immunized with mouse MOG showed a dramatic increase in the numbers of IFNγ producing cells in the CNS when compared with controls, while similar numbers of IL-10 producing cells were detected in both groups. These observations are particularly interesting, since they suggest that the ratio of IFNγ to IL-10 producing cells, rather than the number of IL-10 producing cells alone, determines disease severity. This ratio was nearly doubled when Tregs were depleted prior to induction of disease. Altered IFNγ to IL-10 ratios have been shown to be important in other autoimmune disease such as type 1 diabetes and in pregnant women with MS (Gilmore et al., 2004; Schloot et al., 2002)
Although cells from the CNS of Treg depleted and control mice immunized with mouse MOG showed different cytokine profiles, we did not detect any difference in the frequencies of Tregs in the CNS of these mice. These similar frequencies may be explained by two non-mutually exclusive possibilities. First, the absence of Tregs during priming may allow for a stronger stimulation of autoreactive T cells in the periphery, thus, contributing to an increase in autoreactive T cells in the CNS as evident by increased IFNγ levels without increased IL-10 in Treg depleted mouse MOG immunized mice. This hypothesis is supported by a report by Korn et al., demonstrating that MOG35-55 specific Foxp3+ T cells from CNS of MOG35-55 immunized mice were incapable of suppressing activated effector T cell function in vitro (Korn et al., 2007). Second, although CD4+CD25+Foxp3+ cells were present in the CNS of anti-CD25 treated animals, these cells, which reappear 10 days following depletion, may not possess the “correct” TCR repertoire and fail to recognize mouse MOG and protect against EAE at the site of inflammation.
In addition to CD4+CD25+Foxp3+, we have also detected CD4+CD25−Foxp3+ cells in the CNS of mouse MOG immunized mice. These cells were also detected in the CNS of mice immunized with acetylated MBP1-9 (Stephens et al., 2005). In our hands CD4+CD25−Foxp3+ cells were reduced by as much as 30% in the CNS of anti-CD25 treated mice when compared with IgG-treated controls. Although our results suggest a link between CD4+CD25+Foxp3+ and CD4+CD25−Foxp3+ cells, as anti-CD25 reduces the number of the latter in the CNS, the origin and regulatory function of CD4+CD25−Foxp3+ cells remain to be determined.
Treg depletion may enhance EAE severity by a retraction of suppression of subdominant T cell clones capable of responding to epitopes in addition to MOG35-55. Our data show that T cells from anti-CD25 treated mice exhibited an increase in their response to MOG35-55. Although less pronounced, a slight increase in stimulation index to MOG85-105 and MOG15-35 in spleen cells from PC61 treated mice was also noted. These activated clones could further contribute to increased disease severity following Treg depletion. In addition, we cannot exclude the possibility that autoreactive T cell clones capable of recognizing MOG35-55 with medium to low affinity may also be allowed to proliferate in the absence of Tregs. An additional hallmark of T cell response to MOG, appears as an increase in the proportion of CD4+TCRVβ8+ T cells in T cell lines isolated from MOG35-55 immunized mice ((Mendel et al., 1999; O’Connor et al., 2007) and data not shown). Our results show an accumulation of CD4+TCRVβ8+Foxp3− cells in the CNS of mice immunized with mouse MOG. Interestingly, treatment with anti-CD25 led to an increase in the proportion of these cells in the CNS. Based on these findings, we suggest that Tregs control the activation of autoreactive T cells that are specific for what is usually the dominant epitope in mouse MOG; and that Treg depletion does not promote the activation of T cell clones capable of recognizing cryptic epitopes of MOG.
In summary, this study suggests that Tregs are exquisitely specific for mouse-MOG and may not “cross-protect” against even highly homologous, yet not identical, rat MOG. Tregs’ ability to suppress EAE is independent of disease severity and appears to occur early on during T cell priming in the periphery. These observations shed a new light on the role of molecular mimicry in autoimmunity, where foreign antigens bearing a high similarity to self, are capable of stimulating cross reactive pathogenic T cells, but fail to stimulate self-antigen specific Tregs. It will be intriguing to determine whether the high specificity of Tregs to self-antigen in MOG EAE is apparent in other autoimmune diseases.
Acknowledgments
Supported by National Multiple Sclerosis Society Grants NMSS RG2394, NMSS RG 4126, and NIH CA16885.
We thank Minetta Gardinier for murine MOG-expressing bacteria, Christopher Linington for rat and human MOG-expressing bacteria and for advice on protein expression and purification, Mark Shlomchik for the PC61 hybridoma, and Fred Oliver for helpful discussions.
Reference List
- Bach JF. Regulatory T cells under scrutiny. Nat Rev Immunol. 2003;3:189–198. doi: 10.1038/nri1026. [DOI] [PubMed] [Google Scholar]
- Derbinski J, Schulte A, Kyewski B, Klein L. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol. 2001;2:1032–1039. doi: 10.1038/ni723. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- Gilmore W, Arias M, Stroud N, Stek A, McCarthy KA, Correale J. Preliminary studies of cytokine secretion patterns associated with pregnancy in MS patients. J Neurol Sci. 2004;224:69–76. doi: 10.1016/j.jns.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Guo B, Chang EY, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest. 2008;118:1680–1690. doi: 10.1172/JCI33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofstetter HH, Toyka KV, Tary-Lehmann M, Lehmann PV. Kinetics and organ distribution of IL-17-producing CD4 cells in proteolipid protein 139-151 peptide-induced experimental autoimmune encephalomyelitis of SJL mice. J Immunol. 2007;178:1372–1378. doi: 10.4049/jimmunol.178.3.1372. [DOI] [PubMed] [Google Scholar]
- Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity. 2004;21:267–277. doi: 10.1016/j.immuni.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Iglesias A, Bauer J, Litzenburger T, Schubart A, Linington C. T- and B-cell responses to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis and multiple sclerosis. Glia. 2001;36:220–234. doi: 10.1002/glia.1111. [DOI] [PubMed] [Google Scholar]
- Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- Juedes AE, Hjelmstrom P, Bergman CM, Neild AL, Ruddle NH. Kinetics and cellular origin of cytokines in the central nervous system: insight into mechanisms of myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. J Immunol. 2000;164:419–426. doi: 10.4049/jimmunol.164.1.419. [DOI] [PubMed] [Google Scholar]
- Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol. 2002;169:4712–4716. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, Backstrom BT, Sobel RA, Wucherpfennig KW, Strom TB, Oukka M, Kuchroo VK. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007 doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, Sparwasser T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med. 2007;204:57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons JA, San M, Happ MP, Cross AH. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur J Immunol. 1999;29:3432–3439. doi: 10.1002/(SICI)1521-4141(199911)29:11<3432::AID-IMMU3432>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Marta CB, Oliver AR, Sweet RA, Pfeiffer SE, Ruddle NH. Pathogenic myelin oligodendrocyte glycoprotein antibodies recognize glycosylated epitopes and perturb oligodendrocyte physiology. Proc Natl Acad Sci U S A. 2005;102:13992–13997. doi: 10.1073/pnas.0504979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masteller EL, Warner MR, Tang Q, Tarbell KV, McDevitt H, Bluestone JA. Expansion of functional endogenous antigen-specific CD4+CD25+ regulatory T cells from nonobese diabetic mice. J Immunol. 2005;175:3053–3059. doi: 10.4049/jimmunol.175.5.3053. [DOI] [PubMed] [Google Scholar]
- Matsushita N, Pilon-Thomas SA, Martin LM, Riker AI. Comparative methodologies of regulatory T cell depletion in a murine melanoma model. J Immunol Methods. 2008;333:167–179. doi: 10.1016/j.jim.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J Immunol. 2005;175:3025–3032. doi: 10.4049/jimmunol.175.5.3025. [DOI] [PubMed] [Google Scholar]
- Mendel I, Gur H, Kerlero dR, Ben Nun A. Experimental autoimmune encephalomyelitis induced in B6. C-H-2bm12 mice by myelin oligodendrocyte glycoprotein: effect of MHC class II mutation on immunodominant epitope selection and fine epitope specificity of encephalitogenic T cells. J Neuroimmunol. 1999;96:9–20. doi: 10.1016/s0165-5728(99)00009-0. [DOI] [PubMed] [Google Scholar]
- O’Connor RA, Malpass KH, Anderton SM. The inflamed central nervous system drives the activation and rapid proliferation of Foxp3+ regulatory T cells. J Immunol. 2007;179:958–966. doi: 10.4049/jimmunol.179.2.958. [DOI] [PubMed] [Google Scholar]
- Oliver AR, Lyon GM, Ruddle NH. Rat and human myelin oligodendrocyte glycoproteins induce experimental autoimmune encephalomyelitis by different mechanisms in C57BL/6 mice. J Immunol. 2003;171:462–468. doi: 10.4049/jimmunol.171.1.462. [DOI] [PubMed] [Google Scholar]
- Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 1999;59:3128–3133. [PubMed] [Google Scholar]
- Schloot NC, Hanifi-Moghaddam P, Goebel C, Shatavi SV, Flohe S, Kolb H, Rothe H. Serum IFN-gamma and IL-10 levels are associated with disease progression in non-obese diabetic mice. Diabetes Metab Res Rev. 2002;18:64–70. doi: 10.1002/dmrr.256. [DOI] [PubMed] [Google Scholar]
- Stephens LA, Gray D, Anderton SM. CD4+CD25+ regulatory T cells limit the risk of autoimmune disease arising from T cell receptor crossreactivity. Proc Natl Acad Sci U S A. 2005;102:17418–17423. doi: 10.1073/pnas.0507454102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen WE, Bergman CM, Hjelmstrom P, Ruddle NH. A critical role for lymphotoxin in experimental allergic encephalomyelitis. J Exp Med. 1997;186:1233–1240. doi: 10.1084/jem.186.8.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweenie CH, Mackenzie KJ, Rone-Orugboh A, Liu M, Anderton SM. Distinct T cell recognition of naturally processed and cryptic epitopes within the immunodominant 35-55 region of myelin oligodendrocyte glycoprotein. J Neuroimmunol. 2007;183:7–16. doi: 10.1016/j.jneuroim.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Targoni OS, Lehmann PV. Endogenous myelin basic protein inactivates the high avidity T cell repertoire. J Exp Med. 1998;187:2055–2063. doi: 10.1084/jem.187.12.2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Koldzic DN, Izikson L, Reddy J, Nazareno RF, Sakaguchi S, Kuchroo VK, Weiner HL. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol. 2004;16:249–256. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]