

Introduction
Mitochondria are central to cellular metabolism. The mitochondria’s metabolic pathways include fatty acid oxidation, glucose oxidation and glutaminolysis. The initial step in glucose metabolism occurs in the cytosol, where glycolysis converts glucose to pyruvate1 (Figure 1).
Figure 1.

Mechanism of impaired glucose oxidation and enhanced aerobic glycolysis in PAH. Changes in redox signaling, such as downregulation of SOD2 and the resultant decrease in H2O2 signaling, can activate transcription factors (i.e. HIF-1α) which in turn upregulate PDK. PDK inhibits PDH, which impairs oxidative glucose metabolism, causing the cell to rely on other forms of metabolism, such as aerobic glycolysis. The small molecular inhibitor of PDK, dichloroacetate, can reactivate PDH and restore oxidative glucose metabolism. Abbreviations: ETC = electron transport chain, FOXO1 = Forkhead box protein O1, HK = hexokinase, HIF-1α= Hypoxia inducible factor 1α, H2O2 = hydrogen peroxide, LDHA = lactate dehydrogenase A, PDH = Pyruvate dehydrogenase, PDK = Pyruvate dehydrogenase kinase, PFK = phosphofructokinase. Reprinted with permission from 2.
Normally, glycolysis is coupled to glucose oxidation, meaning that the pyruvate is transported into the mitochondria where it serves as a substrate for pyruvate dehydrogenase (PDH)3. Under pathologic conditions, such as inhibition of PDH, glycolysis may be uncoupled from glucose oxidation and remain a wholly cytosolic reaction that terminates in the generation of lactate.
Metabolism is quite plastic and the relative importance of each pathway can change in response to environmental stimuli, such as substrate availability, the organism’s developmental stage, and pathologic stimuli, such as hypoxia, shear stress, pressure overload, ischemia and hypertrophy. In addition, the activity of one metabolic pathway alters the activity of competing pathways. Examples of this metabolic crosstalk include the reciprocal relationship between fatty acid and glucose oxidation. Fatty acid oxidation suppresses glucose oxidation, through a mechanism called the Randle cycle (Figure 2), named after Phillip Randle who first described the phenomenon3. Another example of metabolic plasticity is the uncoupling of glycolysis from glucose oxidation, so called aerobic glycolysis. Aerobic glycolysis is also called the Warburg effect, in honor of Otto Warburg who first described the phenomenon in cancer cells5. Warburg noted that this shift to glycolysis contributed to the growth and survival advantage of cancer cells5. He also observed, but could not explain, accumulation of ammonia in his cancer tissue culture. Ultimately this proved to relate to a concomitant upregulation of glutaminolysis in cancer cells. Aerobic glycolysis results in a reliance on glycolysis to produce ATP despite the presence of sufficient oxygen to have allowed pyruvate generation and mitochondrial glucose oxidation. Aerobic glycolysis usually reflects active inhibition of one or more mitochondrial enzymes, notably inhibition of PDH by pyruvate dehydrogenase kinases (PDK). These acquired changes in metabolism alter the cell’s bioenergetics status, susceptibility to hypertrophy and fibrosis, rates of proliferation and apoptosis, angiogenesis and contractility. Importantly, the cell’s metabolic choices can be pharmacologically manipulated, offering the potential for metabolic therapies.
Figure 2.

Manipulating fatty acid and glucose oxidation in PAH: The Randle’s cycle. Randle’s cycle is the reciprocal relationship between glucose oxidation and fatty acid oxidation. Note how the acetyl CoA and citrate produced by β-oxidation of fatty acids inhibits PDH (in the mitochondria) and phosphofructokinase (in the cytosol). This feedback (and other indicated feedback mechanisms) slow glucose oxidation under conditions where there is substantial fatty acid oxidation. The pharmacologic inhibitors of fatty acid oxidation, trimetazidine and ranolazine, can restore glucose oxidation by partially inhibiting fatty acid oxidation and activating Randle’s cycle. Abbreviations: CPT1/2 = Carnitine palmitoyltransferase 1/2, FA-CoA = fatty acyl-CoA, FATP1/6 = Fatty acid transport protein 1/6, Glut1/4 = Glucose transporter ¼, HK = hexokinase, HIF-1α= Hypoxia inducible factor 1α, LDHA = lactate dehydrogenase A, PDH = Pyruvate dehydrogenase, PDK = Pyruvate dehydrogenase kinase, PFK = phosphofructokinase, OMM=outer mitochondrial membrane, IMM=inner mitochondrial membrane, TMZ=trimetazidine, RAN=ranolazine. Reprinted with permission from 4.
In addition to generating adenosine triphosphate (ATP), mitochondria are constantly dividing and joining together6. These highly conserved and regulated processes are called fission and fusion, respectively7. These non-canonical mitochondrial functions (fission, fusion), as well as migration, are called mitochondrial dynamics.8 Mitochondrial dynamics are important in physiology, participating in oxygen sensing9 and the distribution of mitochondria to daughter cells during mitosis10. Mitochondrial dynamics are also involved in cellular quality control, notably participating in mitophagy and apoptosis. Acquired and inherited disorders of mitochondrial dynamics are involved in diseases, including pulmonary arterial hypertension (PAH), cancer, and cardiac ischemia reperfusion injury7. Both metabolic plasticity and mitochondrial dynamics are relevant to the pathogenesis of PAH and offer new therapeutic targets in the pulmonary vasculature and the right ventricle.
Mitochondria and metabolism in PAH
Vascular cells and right ventricular cardiomyocytes in PAH have a mitochondrial-metabolic phenotype similar to that seen in cancer. The cancer-like metabolic phenotype in PAH includes increased energetic reliance on aerobic glycolysis, inhibition of mitochondrial respiration, due to pathologic activation of transcription factors such as cMyc, Forkhead transcription factor (FOXO1) and hypoxia inducible factor (HIF-1α), and PDK-induced PDH inhibition. In the hypertrophied right ventricle, cancer-like metabolic changes, aerobic glycolysis and glutaminolysis, reduce energy production and contractility1. PAH and cancer cells also share a mitochondrial morphologic phenotype (increased mitochondrial fragmentation) that is due to a fission/fusion imbalance.11 Mitochondrial fragmentation contributes to the proliferative, apoptosis-resistant phenotype of both diseases.
Although the analogy between PAH and cancer is imperfect, both syndromes share a propensity for cell enlargement, proliferation and apoptosis resistance that is attributable in part to acquired disorders of mitochondrial metabolism and mitochondrial dynamics. Preclinical studies in rodent models of PAH have identified the therapeutic benefits of targeting these mitochondrial abnormalities in the lung, to regress vascular obstruction and improve hemodynamics, and in the right ventricle, to improve contractility, increase cardiac output and reduce hypertrophy.10,12 In this review, we will summarize the mechanism of several mitochondrial abnormalities in PAH and discuss potential therapeutic targets in the pulmonary vasculature and right ventricle.13 Readers are referred to several recent reviews on the subject of metabolism13–16 and mitochondrial dynamics7 in PAH.
A brief review of metabolism
In the fetus, glucose oxidation and glycolysis are the major sources of cardiac ATP and circulating levels of free fatty acids are low17. In the adult heart, the predominant energy source is fatty acid oxidation (60–90%); however, glucose metabolism continues to contribute to ATP production18. Although classically considered a secondary source of ATP production in the adult heart19, direct measurement shows that glucose oxidation remains an important source of ATP in the normal right ventricle, accounting for 48% of total ATP produced20.
Despite the complexities of the metabolic pathways, glucose oxidation, fatty acid oxidation and glutaminolysis have some common features. First, each pathway imports its substrate through a transporter into the cytosol. The transporters for glucose, fatty acids and glutamine are, the glucose transporters (Glut 1–4), the fatty-acid transport proteins (FATP 1 and 6) and the solute carrier proteins (SLC 1A5 and 7A5), respectively (Fig 1–3). When substrate utilization is increased, transporter expression rises, as is the case for Glut and SLC1A5 in the hypertrophied right ventricle in PAH20, 21. Second, each of the pathways drives Krebs’ cycle and promotes ATP production. The glucose and fatty acid oxidation pathways ultimately increase Acetyl CoA levels and provide the electron donors that fuel the majority of cellular ATP generation. Third, it appears that most of the pathways display cross-talk, such that when one is increased another is depressed. This reciprocal relationship is well established for glucose and fatty acid oxidation (the Randle cycle); however, it also appears to be the case for glutaminolysis and glucose oxidation21, although independent corroboration is required. Fourth, it appears that most of the metabolic changes seen in the right ventricle and pulmonary artery in PAH are maladaptive, in that metabolic modulators that restore depressed glucose oxidation or inhibit upregulated fatty acid oxidation and glutaminolysis are beneficial to the animal’s hemodynamic and functional state.
Figure 3.
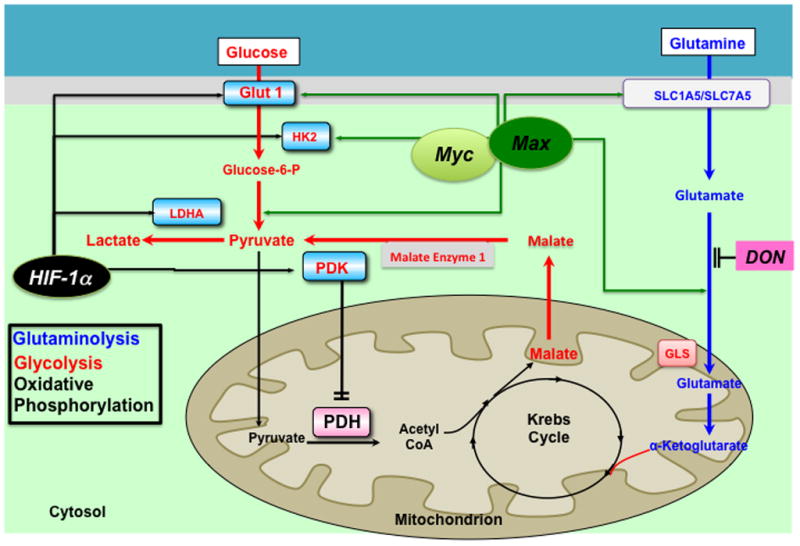
Proposed mechanism of glutaminolysis in the hypertrophied right ventricle. RV ischemia and capillary rarefaction activate cMyc and its binding partner Max, which increases the expression of the glutamine transporters (SLC 1A5 and 1A7) and augments glutamine uptake. This drives the production of α-ketoglutarate (α-KG). α-KG enters Krebs’ cycle leading to production of malate. Krebs’ cycle-derived malate generates cytosolic pyruvate, which is converted by lactate dehydrogenase A (LDHA) to lactate. In conditions of high glutaminolysis, glucose oxidation is inhibited. DON can inhibit glutaminolysis and restore glucose oxidation. HIF-1α increases the transcription of the some of the same glycolytic mediators as cMyc and Max, notably Glut1 and HK2. Abbreviations: Glut1 = Glucose transporter 1, HK = hexokinase, HIF-1α= Hypoxia inducible factor 1α, LDHA = lactate dehydrogenase A, PDH = Pyruvate dehydrogenase, PDK = Pyruvate dehydrogenase kinase, PFK = phosphofructokinase. DON= 6-diazo-5-oxo-l-norleucine. Reprinted with permission from 21.
Despite these commonalities the pathways vary greatly in their bioenergetic yield. A fatty acid containing 6 carbons subjected to beta-oxidation in the mitochondria can yield 48 ATP. However, fatty acid oxidation comes at a price in that it uses approximately 10% more oxygen than glucose oxidation to generate the same amount of ATP22 (Figure 2). The energetic premium associated with fatty acid oxidation reflects the inhibition of glucose oxidation via the Randle cycle, which leads to aerobic glycolysis and lactate accumulation. The resulting acidosis must be corrected by transporters and pumps at an energetic cost. In contrast, although oxidation of 6 carbon-containing glucose has lower ATP yield it is more efficient in that it does not elicit aerobic glycolysis and does not engender a excess production of lactate. In aerobic glycolysis only 2 ATP are generated per mole of glucose and lactate is produced2.
Metabolism of the amino acid glutamine via glutaminolysis, also occurs in PAH and cancer. In glutaminolysis, glutamine is hydrolysed to glutamate and converted to α-ketoglutarate in the mitochondria. α-ketoglutarate then enters Krebs’ cycle and replenishes metabolic intermediates, thereby supporting rapid cell growth. In addition, glutaminolysis can increase nitrogen anabolism which further supports cell growth23. Glutaminolysis was originally identified in cancer cells23, but has recently been found to be induced in the heart during RVH (Figure 3)21. Glutaminolysis in the RV generates modest energy but rewards cells with amino acid intermediates that permit rapid cell growth21.
A Brief Review of Mitochondrial Dynamics
Mitochondrial fusion is mediated by large GTPases, mitofusin-1 and mitofusin-212, which reside in the outer mitochondrial membrane, and a GTPase called optic atrophy-1, in the inner mitochondria membrane7. Fission is mediated by the GTPase, dynamin-related protein 1 (DRP1)10, which upon activation moves from the cytosol to the outer mitochondrial membrane. There it interacts with non-GTPase binding partners, such as mitochondrial fragmentation factor (MFF) and fission factor 1 (Fis1), resulting in multimerization and division of the mitochondria (reviewed in7).
Many of the abnormalities that occur in PAH promote fission, notably, increased intracellular calcium, increased activity of the mitosis promoter, cyclin B1/CDK1, and normoxic activation of HIF-1α10. Inhibition of mitochondrial fission, achieved by administration of inhibitors of DRP1, such as mdivi-1, regress PAH in rodent models by arresting pulmonary artery smooth muscle cells in the G2/M phase of the cell cycle and promoting apoptosis10. Mitochondrial fusion is also decreased in PAH. The decrease in mitochondrial fusion reflects, in part, a reduced expression of both mitofusin-2 and its transcriptional co-activator, peroxisome proliferator-activated receptor γ coactivator 1-α12. Augmentation of mitofusin-2 expression is antiproliferative, proapoptotic and improves hemodynamics in rodent PAH models12. Although the linkage between mitochondrial dynamics and metabolism is poorly understood in PAH, there are examples where form and function clearly intersect. For example, in skeletal muscle cells decreases in mitofusin-2 reduce glucose oxidation and oxygen consumption24. In myotubes decreases in mitofusin-2 similarly reduce the oxidation of pyruvate, palmitate and glucose25. Thus, mitofusin-2 deficiency may contribute to the glycolytic shift observed in pulmonary artery smooth muscle cells in PAH (Figure 4)10, 12, 26.
Figure 4.
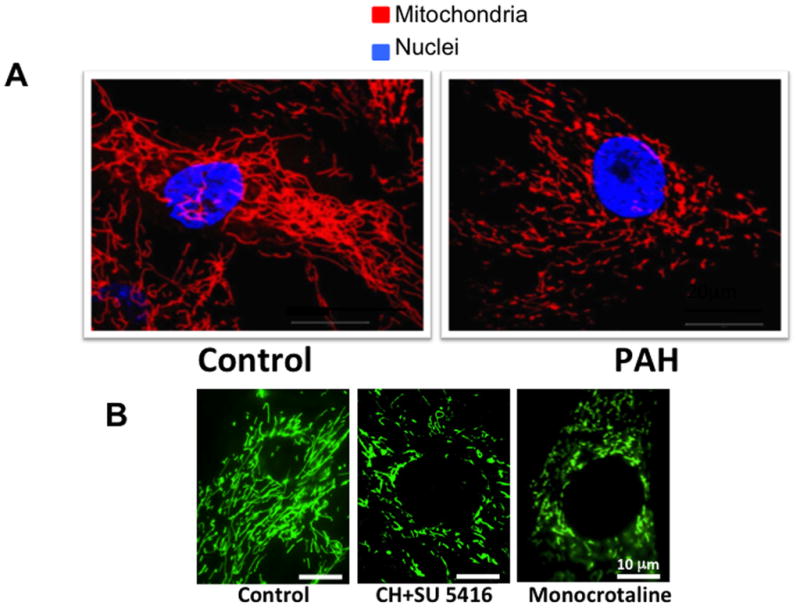
Mitochondrial fragmentation in Pulmonary Arterial Hypertension (PAH). A. Mitochondria are more fragmented in PAH versus control pulmonary artery smooth muscle cells (PASMCs). Quantification of the mitochondrial fragmentation count reveals a doubling of the number of individual mitochondria in PAH versus control PASMCs. Scale bar = 20 μm. Reprinted with permission from 10. B. Increased mitochondrial fragmentation observed in PASMC of rats with PAH induced by exposure to chronic hypoxia plus the VEGF receptor antagonist, SU5416 (CH+SU 5416) or monocrotaline. Mitochondria were imaged by infection of cells with BacMam virus carrying a mitochondrial-targeted green fluorescent protein transgene. Reprinted with permission from 12.
Impaired oxygen sensing and normoxic HIF-1α activation in the pulmonary vasculature in PAH
The mitochondria in pulmonary artery smooth muscle cells normally serve as oxygen sensors10. However in PAH there is normoxic activation of HIF-1α, which creates a “pseudo-hypoxic” environment despite normal oxygen availability26. The term pseudo-hypoxia conveys the concept that changes in mitochondrial metabolism and redox signaling normally seen in response to environmental hypoxia are occurring despite adequate oxygen supply. In the lung the pseudo-hypoxic state is associated with impairment of a well-established mitochondria-ROS-HIF-1α-Kv1.5 oxygen-sensing pathway26. In PAH, downregulation of the mitochondrial hydrogen peroxide generating enzyme superoxide dismutase 2 (SOD2) decreases production of the redox signaling molecule, hydrogen peroxide, thus creating an hypoxia-like redox milieu that activates HIF-1α27. HIF-1α in turn transcriptionally upregulates PDK, which inhibits PDH and further reduces production of reactive oxygen species (ROS) by the mitochondrial electron transport chain. Loss of physiologic levels of ROS inhibit and downregulate expression of the oxygen- and voltage-sensitive potassium channel, Kv1.5, resulting in depolarization and calcium overloading of the smooth muscle cells. Thus, the mitochondria-ROS-HIF-1α-Kv1.5 oxygen-sensing pathway is subverted in PAH, contributing to downstream changes in mitochondrial metabolism and dynamics.
Pseudohypoxia can be modeled in cell culture by activating hypoxic transcriptional pathways in a PO2 independent manner. For example, adenoviral overexpression of constitutively activated HIF-1α in normoxic human arterial endothelial cells leads to overexpression of hundreds of hypoxia responsive genes28. This concept of pseudohypoxia in PAH suggests the feasibility of treatments directed either at restoring oxygen sensing (targeting the mitochondrial electron transport chain and SOD2) or correcting the downstream mitochondrial-metabolic abnormalities that result from impaired oxygen sensing14.
The interrelatedness of disorders in mitochondrial oxygen sensing and metabolism is shown in Figure 1. One example of an acquired but heritable mechanism by which the mitochondrial metabolic changes of PAH can occur is the epigenetic inhibition of the expression and activity of SOD227, 29. SOD2 is a nuclear-encoded, mitochondrial enzyme responsible for the production of the diffusible, redox signaling molecule hydrogen peroxide (H2O2)26, 27, 30. Loss of SOD2-mediated production of hydrogen peroxide activates HIF-1α10, 27. The relationship between SOD2 and HIF-1α is robust. Simply downregulating SOD2 in normal pulmonary artery smooth muscle cells using small inhibitory RNA, activates HIF-1α despite normal PO2. Likewise, once HIF-1α is activated there is a clear phenotypic shift in pulmonary artery smooth muscle cells, which become hyperproliferative31 and display increased mitochondrial fragmentation10, 12. The importance of epigenetic inhibition of SOD2 and activation of HIF-1α was first identified in the Fawn-hooded rat, a strain that spontaneously develops PAH26. HIF-1α activation in smooth muscle cells from Fawn-hooded rats and PAH patients persist in culture, despite abundant O227.
Transcriptional repression of SOD2 expression occurs through methylation of two key CpG islands in the SOD2 gene. Methylation in the promoter and enhancer regions of the gene halves SOD2 expression and the resulting reduction of hydrogen peroxide production initiates HIF-1α activation. Prolonged activation of HIF-1α and the associated mitochondrial fragmentation promotes a glycolytic shift in metabolism and hyperproliferation of pulmonary artery smooth muscle cells. In Fawn-hooded rats, demethylation of the SOD2 gene, by the DNA methyltransferase inhibitor 5-azacytidine, restores SOD2 expression and inhibits pulmonary artery smooth muscle cell proliferation. It remains unclear why the dysregulation of DNA methyltransferases in the Fawn-hooded rat is confined to the lung, although there is selective upregulation of DNA methyltransferases in the lung that is not seen in the systemic vasculature. Interestingly, epigenetic inhibition of SOD2 also contributes to the hyperproliferative phenotype of some cancer cells32, 33.
Additional evidence implicating HIF-1α in the pathogenesis of PAH comes from Chuvash pulmonary hypertension. This syndrome affects individuals in the mid Volga river region of Russia. They develop a hypoxic phenotype that is characterized by polycythemia and pulmonary hypertension resulting from inappropriate normoxic activation of HIF-1α and increased expression of genes for erythropoietin, Glut1 and vascular endothelial growth factor (VEGF)34. They also have PDK activation and elevated plasma lactate levels. This pseudohypoxic pathophysiology is similar to the Fawn-hooded rat but results from a homozygous mutation in the von Hippel-Lindau gene (VHL 598C to T) that removes the ubiquitination signal for HIF-1α degradation35 and thus impairs proteosomal degradation of HIF-1α. Further implicating a role for HIF-1α activation in PAH is the observation that normoxic cobalt-induced HIF-1α activation causes mitochondrial fragmentation within 2–3 hours. Interestingly, cobalt does not cause cell proliferation in culture, likely due to nonspecific toxicity. However, chronic, in vivo administration of cobalt engenders pulmonary hypertension and right ventricular hypertrophy with evidence of HIF-1α activation and increased mitochondrial fission in the pulmonary artery smooth muscle cells10. Furthermore, inhibitors of HIF-1α, for example chemotin, can reverse the hyperproliferative metabolic effects of this transcription factor in PAH31. Finally, the role of HIF-1α in PAH is also evident from the observation that HIF-1α haploinsufficiency disrupts oxygen sensing and reduces hypoxic pulmonary hypertension in mice36, 37.
Metabolic remodeling in the hypertrophied right ventricle in PAH
Altered right ventricular metabolism in PAH is transcriptionally mediated. However, in the right ventricle the likely precipitant is ischemia, rather than impaired oxygen sensing. In RVH there is ischemia38 and decreased coronary flow reserve39; however, it is unclear whether this reflects impaired epicardial perfusion pressure, capillary rarefaction or both. The right coronary artery normally fills during both systole and diastole because the right ventricle systolic pressure is low relative to the driving pressure in the aorta. In right ventricle pressure overload, the systolic perfusion gradient between the aortic and right ventricle systolic pressures may disappear limiting right coronary artery flow to diastole. This essentially halves the amount of blood being supplied to the hypertrophied right ventricle, which has increased metabolic demands40. At extremes of pulmonary hypertension, when right coronary artery perfusion pressure falls below 50 mmHg, right ventricle contractile function declines41. Right ventricular ischemia in PAH may also result from impairment in angiogenesis, also referred to as capillary rarefaction (Figure 5A). The impairment in angiogenesis may result from decreased expression of genes, such as insulin-like growth factor 1, VEGF, apelin and angiopoeitin-1 (Figure 5B)21. Occlusive microvascular disease and capillary rarefaction has been seen in the right ventricle in animal models of maladaptive PAH20, 42 and in patients with scleroderma-associated PAH (Figure 5C)21.
Figure 5.

Adaptive versus Maladaptive Forms of RVH. A. Catheterization data shows simultaneous RV pressure and aortic pressure (AoP) in various rat RVH models. Despite similar coronary perfusion pressure (defined as the Δ Pressure Ao-RV), there is worse RV function and exercise capacity in monocrotaline (MCT) versus pulmonary artery banding (PAB) (not shown). B. Representative images and mean data showing greater RV capillary rarefaction (loss of small arteries) in a maladaptive form of RVH (Monocrotaline-RVH) than in an adaptive form of RVH (created by pulmonary artery banding, PAB). Reprinted with permission from 21. C. Representative images and mean data showing greater right ventricular capillary rarefaction in scleroderma-PAH patients, known for the greater propensity to develop RV failure, than in normal subjects or patients with idiopathic PAH. Reprinted with permission from 21. Red stain: CD31 (an endothelial cell marker); Green stain (smooth muscle actin).
The role played by HIF-1α in the metabolic remodeling of the right ventricle in PAH is less clear than its role in the pulmonary vasculature. Several investigators find HIF-1α is increased in rodent RVH models43, 44. However, there are differences in the role of HIF-1α in the ventricle versus the lung in terms of the predominant downstream PDK isoform expression profile that is elicited and in HIF-1α’s temporal profile (reviewed in the section on controversies).
A central role for inhibition of pyruvate dehydrogenase in PAH
In PAH and cancer, PDH is phosphorylated and inhibited by PDK. PDK expression is increased in these syndromes. Phosphorylation of the α-subunit of the E1 (pyruvate decarboxylase) component of the PDH complex by any PDK isoform rapidly inhibits PDH. When PDH is inhibited by PDK, the supply of electron donors to Krebs’ cycle is limited and energy production is reduced45 (Figure 1).
The four PDK isoforms differ in their transcriptional regulation and tissue distribution46. PDK2 appears to be the predominant human isoform in many tissues46, however the magnitude and consequences of regional isoform heterogeneity amongst tissues is not adequately studied. For example, PDK1 expression is transcriptionally upregulated by HIF-1α28, 47, whereas PDK4, which lacks a hypoxia recognition element in its promoter and is thus not directly regulated by HIF-1α is induced by FOXO120, 48. However, HIF-1α can transcriptionally upregulate estrogen-related receptor γ (ERRγ), which is capable of transcriptionally increasing PDK4 expression49.
The dominant PDK isoforms in a specific tissue can change with disease and this pathologic isoform variation is largely unstudied. For example in rodent PAH the dominant PDK isoforms upregulated in the right ventricle are PDK 2 and 420. Anecdotal evidence suggests PDK4 is also upregulated in the human right ventricle50. In the lung the predominant isoforms upregulated in PAH are PDK1 and PDK22, 51–53.
Tissue heterogeneity in PDK expression and disease specific regulation of PDK and PDH in PAH merit further study. In some tissues there appears to be minimal basal PDK activity whilst in others PDK is active under physiologic condition. In skeletal muscle, tonic activation of PDK2 contributes to regulation of carbohydrate oxidation and production of reducing equivalents for the electron transport chain47. In contrast, there appears to be little tonic PDK activity in the normal right ventricle and pulmonary artery, as indicated by the absence of effect of a pan-PDK inhibitor, dichloroacetate (Figure 1), on metabolism and the cellular electrophysiology of normal right ventricular and pulmonary vascular cells2, 51–53.
When the mitochondrial PDH complex is active, pyruvate is converted to acetyl coenzyme A, which fuels Krebs’ cycle, generating electron donors for the electron transport chain and fueling generation of ATP19. However, in PAH PDH inhibition inhibits the electron transport chain and increases reliance on aerobic glycolysis13, 44. This metabolic shift contributes to the hyperproliferative, apoptosis-resistant state of pulmonary artery smooth muscle cells53. The PDK-mediated metabolic switch to aerobic glycolysis is associated with decreased cardiac output and reduced right ventricle contractility20, 2. Increased lactate production further impairs right ventricle function secondary to acidosis. Suppression of glucose oxidation reflects the cell’s acceptance of reduced efficiency of ATP generation in exchange for reduced risk of mitochondrial-mediated apoptosis and an increased ability to hypertrophy1.
Increased glycolysis is associated with increased glucose flux, allowing right ventricular glycolysis to be detected on cardiac 18F-fluorodeoxyglucose-positron emission tomography (18FDG-PET) scans in patients and in animal models (Figure 6)31, 54. There is also preliminary evidence that reduction of right ventricle afterload by initiation of pulmonary vasodilators reduces right ventricular uptake of 18FDG in patients (Figure 7)55. In a case report comparing a PAH patient who died from rapidly decompensating right ventricular failure with a long-term survivor, expression of both PDK4 and the Glut-1 transporter were more elevated in the patient with the rapidly fatal RVH.
Figure 6.
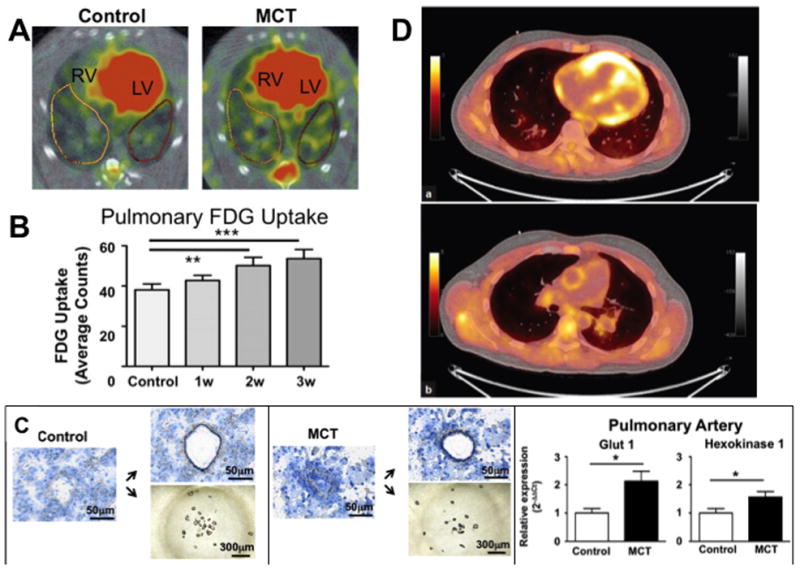
Detection of enhanced aerobic glycolysis in the right ventricle and small pulmonary arteries. A. Increased 18F-fluorodeoxyglucose (FDG) uptake in the right ventricle (RV) and the lung parenchyma of MCT animals. LV=left ventricle. B. Quantification of pulmonary 18F-FDG uptake measured with PET. By week 2 after monocrotaline injection, lung 18F-FDG uptake is significantly greater than in control lungs. C. Laser capture microdissection (LCM) confirms the vascular origin of the glycolytic signal in the lungs of monocrotaline (MCT) rats. Small pulmonary precapillary resistance vessels (<100 μm in diameter) or pieces of airway tissue were collected by LCM. Reprinted with permission from 31. D. (a) PET and (b) fused PET/CT of the right ventricle and pulmonary trunk of an idiopathic PAH subject showing increased 18F-FDG uptake. Reprinted with permission from 54.
Figure 7.
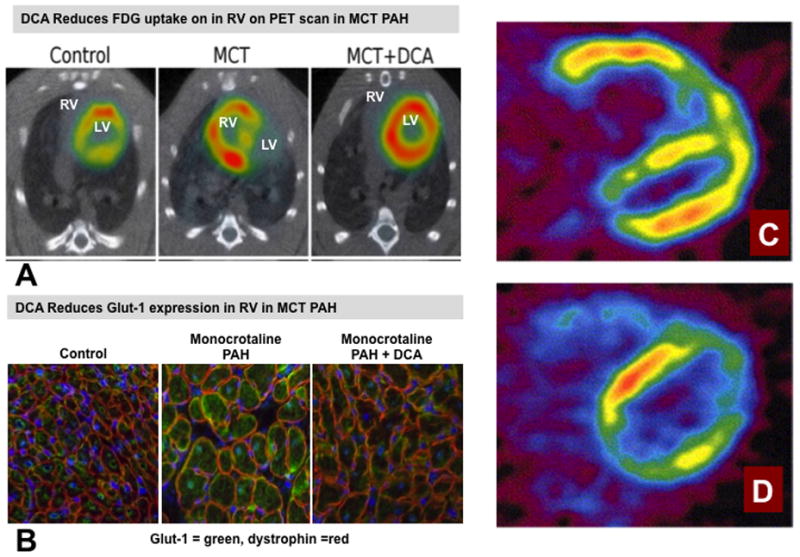
Reversibility of metabolic changes in the RV in PAH. A. Representative images and mean data showing that the increased 18F-fluorodeoxyglucose uptake in the right ventricle (RV) on PET scans in monocrotaline (MCT) rats which is reduced by dichloroacetate (DCA) (See Figure 1). Reprinted with permission from 2. B. Immunostaining showing that the increased Glut1 expression in RV myocytes in monocrotaline-induced RVH is reduced by chronic oral consumption of the PDK inhibitor, dichloroacetate (DCA). The merge of the staining of Glut1 (green) and dystrophin (red) in RV shows that more Glut1 is expressed at the myocyte membrane in RVH. Reprinted with permission from 2. C & D. Representative midventricular transaxial 18F-fluorodeoxyglucose PET images of a patient with idiopathic pulmonary hypertension before and after the pulmonary vasodilator therapy with epoprostenol for three months. Before the pulmonary vasodilator therapy, the RV FDG accumulation was highly increased (C). After the therapy, the RV FDG accumulation was markedly decreased (D). Reprinted with permission from 55.
Dichloroacetate is a small molecule pyruvate analog. Dichloroacetate inhibits all 4 PDK isoforms by binding a conserved, allosteric site in the N-terminal domain55. The dichloroacetate binding pocket is relatively small (volume = 211 Å3)57 and it is buried within the PDK structure, making it relatively inaccessible to molecules larger than pyruvate26. These allosteric constraints make it difficult to modify dichloroacetate to enhance its potency. Consequently, recent studies have attempted to inhibit PDK by targeting its larger (volume = 865 Å3), more accessible ATP-binding pocket57. Dichloroacetate-induced metabolic changes depolarize mitochondria and induce apoptosis while inhibiting pulmonary artery smooth muscle cell proliferation26. Dichloroacetate is relatively specific for abnormal tissues and has little effect on normal cardiac or vascular cells, in which PDK isoforms are inactive51, 52. Oral dichloroacetate is effective in regressing pulmonary vascular disease and improving functions in preclinical models of pulmonary hypertension, including chronic hypoxic pulmonary hypertension52, 53, monocrotaline-PAH52, spontaneous PAH in Fawn-hooded rats20 and PAH induced by transgenic overexpression of the serotonin transporter in mice51. The same dose of dichloroacetate that is effective in the lung vasculature also decreases PDH phosphorylation, activates PDH, enhances glucose oxidation and increases contractility in a variety of other rodent PAH models2, 58. Dichloroacetate has been used as chronic experimental therapy in adults with glioblastoma multiforme59 and in children with inherited mitochondrial diseases and lactic acidosis60. Dichloroacetate is currently the subject of a clinical trial to determine if it is a safe and tolerated therapy in patients with moderate PAH (NCT 01083524). To date dichloroacetate’s main toxicity appears to be a dose dependent, reversible peripheral neuropathy61, 62, although it is well tolerated in patients at appropriate doses.
Fatty Acid Oxidation in PAH
Fatty Acid Oxidation in the Pulmonary Vasculature
Fatty acid oxidation plays a role in the pulmonary vasculature in PAH. Mice deficient in malonyl-coenzyme A decarboxylase have little fatty acid oxidation and are protected from developing hypoxia-induced pulmonary hypertension63. Malonyl-coenzyme A decarboxylase deficiency exerts its beneficial effects by activating the Randle cycle and promoting glucose oxidation.
Fatty Acid Oxidation in the Right Ventricle
In normal rats, the contribution ratio of glucose oxidation, fatty acid oxidation and glycolysis to cardiac ATP production is 48 %/37%/15%, respectively20. This is evidence of the importance of glucose oxidation in the normal heart. In fawn hooded rats with RVH and PAH the ratios change reflecting an increased reliance on glycolysis (37%/39%/24%)20. Dichloroacetate increases the contribution of glucose oxidation to ATP production at the expense of fatty acid oxidation (70%/15%/15%), an illustration of the Randle cycle mechanism20.
There appear to be differences in fatty acid oxidation amongst different models of RVH, with increases being reported in the pulmonary artery banding model4 versus decreases in Fawn-hooded rats20. There are limited data available regarding oxidative metabolism of fatty acids in human PAH. Acetate is rapidly metabolized into acetyl-CoA and enters into Krebs’ cycle. Consequently, 11C-acetate uptake on PET scans can measure net oxidative metabolism in vivo. 11C-acetate PET was performed in 27 patients with WHO functional class II/III PAH and 9 healthy individuals64. The RV oxidative metabolic rate was increased in PAH patients relative to controls, although no intervention was performed to assess whether this change was beneficial or maladaptive nor was the relative contribute of fatty acid versus glucose oxidation determined.
Partially inhibiting fatty acid oxidation appears beneficial in RVH models in which fatty acid oxidation is increased. This can be achieved using ranolazine and trimetazidine (Figure 2). These partial inhibitors of fatty acid oxidation are approved for use in patients with angina (USA) and left heart failure (Europe), respectively. Inhibition of fatty acid oxidation in RVH increases glucose oxidation and right ventricular ATP levels4. In rats with pulmonary artery banding-induced RVH, inhibition of fatty acid oxidation increases exercise tolerance, cardiac output and improves cardiac repolarization, evident clinically by normalization of the QT interval on the surface electrocardiogram4. The potential therapeutic benefit of trimetazidine has also been observed in monocrotaline-induced RVH65. Trimetazidine reduces the creation of free oxygen radicals, increases oxygen consumption, and improves mitochondrial function in cardiac myocytes66. 18FDG-PET studies have demonstrated that inhibition of fatty acid oxidation with trimetazidine in dilated cardiomyopathy results in a corresponding increase in glucose oxidation67. Ranolazine is now being studied in PAH patients in Phase 1 clinical trials (NCT01757808 and NCT01174173).
Glutaminolysis in PAH
There is little if any glutaminolysis in the normal heart. However in RVH, glutaminolysis is selectively induced in the right ventricle21. Increased glutaminolysis in monocrotaline-RVH is accompanied by increased right ventricle expression of mitochondrial malic enzyme and the glutamine transporters, SLC1A5 and SLC7A521. Glutaminolysis appears to be induced by ischemic activation of the cMyc transcriptional pathway21. Preliminary evidence suggests that glutaminolysis may provide a therapeutic target. In vivo, chronic glutamine antagonism with 6-Diazo-5-oxo-L-norleucine (DON) (Figure 3), increases cardiac output, reduces RVH, restores PDH activity and increases glucose oxidation21. However, there is limited preclinical data supporting this strategy in RVH and PAH and the attempt to exploit this pathway as a cancer therapy was confounded by toxicity68. It has yet to be assessed whether glutaminolysis is also induced in the hypertensive pulmonary vasculature.
Adaptive versus maladaptive RVH
There is increasing recognition of heterogeneity in RVH with some forms being well-tolerated (adaptive RVH) and other forms rapidly resulting in right ventricle failure (maladaptive RVH). PAH patients with adaptive RVH remain stable for many years, whereas others, with maladaptive RVH, rapidly decompensate despite similar right ventricular mass and similar increases in right ventricle pressure50. In adaptive RVH, cardiac output remains relatively normal, as does right ventricular ejection fraction and exercise capacity. In maladaptive RVH, cardiac output falls significantly, as does right ventricular ejection fraction and exercise capacity. Maladaptive RVH and right ventricular failure is much more common in scleroderma-associated PAH69, 70, than in PAH associated with congenital heart disease (i.e. Eisenmenger’s syndrome71), which is often adaptive. Similarly, in isolated right ventricle pressure overload due to pulmonic stenosis, adaptive RVH, characterized by concentric hypertrophy and minimal fibrosis, is common and is associated with preserved contractility72, 73.
The determinants of progression to right ventricle failure in PAH are poorly understood. In carefully controlled rodent models with identical right ventricular mass and RVH severity, there are dramatic differences in cardiac output and likelihood of progression to failure. Adaptive RVH is evident in models of pulmonary artery banding whereas maladaptive RVH occurs in PAH models induced by monocrotaline or the combination of chronic hypoxia plus the VEGF-2 receptor antagonist, SU541642. There is greater aerobic glycolysis and PDH inhibition in maladaptive monocrotaline-RVH than in adaptive pulmonary artery banding-RVH2.
There appears to be a transition point at which RVH changes from being adaptive to maladaptive44. The transition to maladaptive RVH is associated with a decrease in angiogenesis, inhibition of HIF-1α in the right ventricle, and a decrease in glucose uptake45. In maladaptive RVH, there is also chamber-specific dysregulation of the autonomic nervous system with desensitization and downregulation of α-, β- and dopaminergic receptors in the right ventricle74. In maladaptive RVH, many of these changes extend into the left ventricle74. Factors that determine whether RVH will be adaptive or maladaptive include the presence and severity of right ventricular ischemia, autonomic dysregulation, fibrosis, angiogenesis, and metabolic changes.
The combination of ischemia, metabolic abnormalities and impaired contractility suggest that the hypokinetic right ventricle may be a form of myocardial hibernation50. Supporting this argument, successful lung transplantation for PAH usually results in reversal of right ventricular dysfunction75. Similarly, in chronic thromboembolic pulmonary hypertension, the function of the right ventricle typically returns to normal within weeks after pulmonary endarterectomy76.
Controversies
There remains controversy as to whether HIF-1α prevents or promotes failure of the right ventricle in PAH. Sutendra compared the initially adaptive hypertrophy seen in rats with monocrotaline-induced PAH to rats who were later in their disease course and had developed signs of heart failure. The compensated right ventricle had low production of mitochondria-derived reactive oxygen species (mROS) and increased expression of HIF-1α with evidence of activation of its downstream pathway (increased expression of Glut1, VEGF, and stromal-derived factor 1)44. As a result of HIF-1α activation there was increased angiogenesis and increased 18F-fluorodeoxyglucose uptake on PET. The transition to decompensated RVH was marked by a sharp rise in mROS and an associated inhibition of HIF-1α, and activation of p53, both of which contributed to down-regulation of PDK and decreased glucose uptake. The authors found that decompensation was associated with a decrease in angiogenic factors and angiogenesis44. This latter finding is consistent with the work of others, who have noted capillary rarefaction in maladaptive RVH20, 42. However, the conclusion that this decrease in RV angiogenesis reflects loss of HIF-1α44 differs from that of Drake et al, who noted preserved HIF-1α expression in severe RVH and attributed impaired angiogenesis to failure of downstream angiogenic signaling, evident as reduced VEGF and Akt expression77.
The debate about whether activation of the glycolytic pathways is adaptive or maladaptive is also informed by the cancer literature. In malignancy, excessive activation of oncogenes, such as cMyc, results in a lethal oncogenic stress response that is caused by enhanced aerobic glycolysis and glutaminolysis78. Likewise in the failing right ventricle in PAH there appears to be more activation of PDK4 and greater upregulation of Glut1 than in a compensated right ventricle50. This would suggest that ongoing or excessive aerobic glycolysis might be expected to be maladaptive.
There is also debate about the predominant transcriptional pathways activated in RVH. Although some groups report that HIF-1α is the predominant transcription factor in RVH, we have observed that much of the transcriptional basis for metabolic remodeling in the right ventricle results from activation of cMyc21 and FOXO119. In contrast, HIF-1α appears to be the predominant transcription factor governing the metabolic shift to aerobic glycolysis in the lung vasculature10, 31.
Metabolism may also be abnormal in the left ventricle in PAH. Myocardial metabolism in the interventricular septum, as measured using the labeled fatty acid, β-methyl-p-123I-iodophenyl-pentadecanoic acid (BMIPP), is reduced in PAH patients79. The impairment of septal BMIPP uptake is proportional to the degree of pulmonary hypertension. The coronary flow reserve of the left ventricle is also impaired in patients with PAH39. The role of metabolic changes in the left ventricle merits further investigation.
Prediction whether a metabolic change in the heart will be adaptive or maladaptive is likely contextual, depending on the disease and species and time course of the change80. For example, a mouse model of diabetic cardiomyopathy, created by transgenic overexpression of PDK4, would have been predicted to be deleterious. However, chronic activation of PDK4 led to transcriptional and post-transcriptional changes in metabolism that adapted the heart to cope with the observed suppression of glucose oxidation and the resulting chronic high rates of fatty acid oxidation81. PDK4 overexpressing mice had increased cardiac levels of AMP-activated protein kinase and its target, peroxisome proliferator- activated receptor γ coactivator-1.
Not all metabolic derangements require a metabolic solution. For example, increased right ventricular 18F-fluorodeoxyglucose uptake in PAH patients is reduced by long term therapy using intravenous epoprostenol, a vasodilator prostaglandin55 (Figure 7C–D). Similarly, lung 18F-fluorodeoxyglucose uptake in monocrotaline rats is reduced by Imatinib, a tyrosine kinase inhibitor31. In both cases this suggests that reducing the pressure overload and shear stress in PAH may turn off an ongoing metabolic program. An additional strategy to indirectly correct metabolic abnormalities in PAH would be to reduce right ventricle ischemia using β-blockers. Indeed, capillary rarefaction in the right ventricle of rats with PAH is reversible with β-blockers82. β-blockers are currently being studied in clinical trials in PAH patients (NCT 01246037) and are reported to be well tolerated83.
Limitations
Although this review focuses on metabolism, many other factors determine the success of the right ventricle’s response to pressure and volume overload, notably the occurrence of fibrosis. Right ventricle fibrosis is an important predictor of maladaptive physiology, both in the lung and right ventricle. In PAH patients, late gadolinium enhancement at the RV insertion point into the septum is indicative of fibrosis. Late gadolinium enhancement is associated with right ventricle dilation, reduced right ventricle ejection fraction, and predicts time to clinical worsening61. The accumulation of collagen with a resulting loss of right ventricle compliance is also seen in the chronic hypoxia plus SU5416 rat model of PAH80. This maladaptive response can also present itself as a therapeutic target, and in rodents can be prevented by inhibitors of the angiotensin-converting enzyme, such as enalapril62. The importance of mitochondrial metabolic abnormalities in promoting fibrosis is an area of where research is required.
Conclusions
Metabolic abnormalities are observed in the right ventricle and pulmonary circulation in PAH, both in preclinical models and in patients. Therapies that promote glucose oxidation or inhibit fatty acid oxidation or glutaminolysis may represent new therapeutic targets in PAH. These therapies would be predicted to have benefits both on the right ventricle and pulmonary vasculature. The potential benefit of metabolic therapies for shared abnormalities in the cardiopulmonary unit suggests a new and attractive therapeutic paradigm in PAH. However, carefully designed clinical trials are required to assess the safety and therapeutic value of metabolic therapies.
Supplementary Material
Acknowledgments
The authors thank Dr. E. Kenneth Weir, University of Minnesota, for his review of this article. His suggestions improved the article’s clarity.
Funding Sources: This work is supported by NIH-RO1-HL071115 and 1RC1HL099462, a Canada Research Chair in Mitochondrial Dynamics and Translational Medicine and the Canada Foundation for Innovation (SLA).
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Paulin R, Michelakis ED. The metabolic theory of pulmonary arterial hypertension. Circ Res. 2014;115:148–164. doi: 10.1161/CIRCRESAHA.115.301130. [DOI] [PubMed] [Google Scholar]
- 2.Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, Toth PT, Marsboom G, Zhang HJ, Haber I, Rehman J, Lopaschuk GD, Archer SL. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: Resuscitating the hibernating right ventricle. J Mol Med (Berl) 2010;88:47–60. doi: 10.1007/s00109-009-0524-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 4.Fang YH, Piao L, Hong Z, Toth PT, Marsboom G, Bache-Wiig P, Rehman J, Archer SL. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: Exploiting randle’s cycle. J Mol Med (Berl) 2012;90:31–43. doi: 10.1007/s00109-011-0804-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warburg O, Wind F, Negelein E. Über den stoffwechsel von tumoren im körper. Klinische Wochenschrift. 1926;5:829–832. [Google Scholar]
- 6.Lewis MR, Lewis WH. Mitochondria in tissue culture. Science. 1914;39:330–333. doi: 10.1126/science.39.1000.330. [DOI] [PubMed] [Google Scholar]
- 7.Archer SL. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369:2236–2251. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 8.Bereiter-Hahn J, Voth M. Dynamics of mitochondria in living cells: Shape changes, dislocations, fusion, and fission of mitochondria. Microsc Res Tech. 1994;27:198–219. doi: 10.1002/jemt.1070270303. [DOI] [PubMed] [Google Scholar]
- 9.Hong Z, Kutty S, Toth PT, Marsboom G, Hammel JM, Chamberlain C, Ryan JJ, Zhang HJ, Sharp WW, Morrow E, Trivedi K, Weir EK, Archer SL. Role of dynamin-related protein 1 (drp1)-mediated mitochondrial fission in oxygen sensing and constriction of the ductus arteriosus. Circ Res. 2013;112:802–815. doi: 10.1161/CIRCRESAHA.111.300285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J, Chen Y, Morrow E, Weir EK, Rehman J, Archer SL. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res. 2012;110:1484–1497. doi: 10.1161/CIRCRESAHA.111.263848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C, Archer SL. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. Faseb J. 2012;26:2175–2186. doi: 10.1096/fj.11-196543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryan JJ, Marsboom G, Fang YH, Toth PT, Morrow E, Luo N, Piao L, Hong Z, Ericson K, Zhang HJ, Han M, Haney CR, Chen CT, Sharp WW, Archer SL. Pgc1alpha-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2013;187:865–878. doi: 10.1164/rccm.201209-1687OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Archer SL, Fang YH, Ryan JJ, Piao L. Metabolism and bioenergetics in the right ventricle and pulmonary vasculature in pulmonary hypertension. Pulm Circ. 2013;3:144–152. doi: 10.4103/2045-8932.109960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: A mitochondria-ros-hif-1alpha-kv1.5 o2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol. 2008;294:H570–578. doi: 10.1152/ajpheart.01324.2007. [DOI] [PubMed] [Google Scholar]
- 15.Sutendra G, Michelakis ED. The metabolic basis of pulmonary arterial hypertension. Cell Metabol. 2014;19:558–573. doi: 10.1016/j.cmet.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Tuder RM, Davis LA, Graham BB. Targeting energetic metabolism: A new frontier in the pathogenesis and treatment of pulmonary hypertension. Am J Respir Crit Care Med. 2012;185:260–266. doi: 10.1164/rccm.201108-1536PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. Return to the fetal gene program protects the stressed heart: A strong hypothesis. Heart Fail Rev. 2007;12:331–343. doi: 10.1007/s10741-007-9034-1. [DOI] [PubMed] [Google Scholar]
- 18.Neely JR, Morgan HE. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu Rev Physiol. 1974;36:413–459. doi: 10.1146/annurev.ph.36.030174.002213. [DOI] [PubMed] [Google Scholar]
- 19.Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res. 1997;33:243–257. doi: 10.1016/s0008-6363(96)00245-3. [DOI] [PubMed] [Google Scholar]
- 20.Piao L, Sidhu VK, Fang YH, Ryan JJ, Parikh KS, Hong Z, Toth PT, Morrow E, Kutty S, Lopaschuk GD, Archer SL. Foxo1-mediated upregulation of pyruvate dehydrogenase kinase-4 (pdk4) decreases glucose oxidation and impairs right ventricular function in pulmonary hypertension: Therapeutic benefits of dichloroacetate. J Mol Med (Berl) 2013;91:333–346. doi: 10.1007/s00109-012-0982-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piao L, Fang YH, Parikh K, Ryan JJ, Toth PT, Archer SL. Cardiac glutaminolysis: A maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J Mol Med (Berl) 2013;91:1185–1197. doi: 10.1007/s00109-013-1064-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abozguia K, Clarke K, Lee L, Frenneaux M. Modification of myocardial substrate use as a therapy for heart failure. Nat Clin Pract Cardiovasc Med. 2006;3:490–498. doi: 10.1038/ncpcardio0583. [DOI] [PubMed] [Google Scholar]
- 23.Dang CV. Glutaminolysis: Supplying carbon or nitrogen or both for cancer cells? Cell Cycle. 2010;9:3884–3886. doi: 10.4161/cc.9.19.13302. [DOI] [PubMed] [Google Scholar]
- 24.Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR, Rabasa-Lhoret R, Wallberg-Henriksson H, Laville M, Palacin M, Vidal H, Rivera F, Brand M, Zorzano A. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem. 2003;278:17190–17197. doi: 10.1074/jbc.M212754200. [DOI] [PubMed] [Google Scholar]
- 25.Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, Palacin M, Zorzano A. The charcot-marie-tooth type 2a gene product, mfn2, up-regulates fuel oxidation through expression of oxphos system. Hum Mol Genet. 2005;14:1405–1415. doi: 10.1093/hmg/ddi149. [DOI] [PubMed] [Google Scholar]
- 26.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1alpha-kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation. 2006;113:2630–2641. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 27.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JR, Gomberg-Maitland M, Thebaud B, Husain AN, Cipriani N, Rehman J. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation. 2010;121:2661–2671. doi: 10.1161/CIRCULATIONAHA.109.916098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by hif-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 29.Xu W, Erzurum SC. Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. Compr Physiol. 2011;1:357–372. doi: 10.1002/cphy.c090005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weir EK, Hong Z, Chen Y. Superoxide dismutase: Master and commander? Eur Respir J. 2010;36:234–236. doi: 10.1183/09031936.00062510. [DOI] [PubMed] [Google Scholar]
- 31.Marsboom G, Wietholt C, Haney CR, Toth PT, Ryan JJ, Morrow E, Thenappan T, Bache-Wiig P, Piao L, Paul J, Chen CT, Archer SL. Lung (1)(8)f-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;185:670–679. doi: 10.1164/rccm.201108-1562OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hitchler MJ, Oberley LW, Domann FE. Epigenetic silencing of sod2 by histone modifications in human breast cancer cells. Free Radic Biol Med. 2008;45:1573–1580. doi: 10.1016/j.freeradbiomed.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hitchler MJ, Wikainapakul K, Yu L, Powers K, Attatippaholkun W, Domann FE. Epigenetic regulation of manganese superoxide dismutase expression in human breast cancer cells. Epigenetics. 2006;1:163–171. doi: 10.4161/epi.1.4.3401. [DOI] [PubMed] [Google Scholar]
- 34.Ang SO, Chen H, Hirota K, Gordeuk VR, Jelinek J, Guan Y, Liu E, Sergueeva AI, Miasnikova GY, Mole D, Maxwell PH, Stockton DW, Semenza GL, Prchal JT. Disruption of oxygen homeostasis underlies congenital chuvash polycythemia. Nature genetics. 2002;32:614–621. doi: 10.1038/ng1019. [DOI] [PubMed] [Google Scholar]
- 35.Gladwin MT. Polycythemia, hif-1alpha and pulmonary hypertension in chuvash. Haematologica. 2006;91:722. [PubMed] [Google Scholar]
- 36.Shimoda LA, Manalo DJ, Sham JS, Semenza GL, Sylvester JT. Partial hif-1alpha deficiency impairs pulmonary arterial myocyte electrophysiological responses to hypoxia. Am J Physiol Lung Cell Mol Physiol. 2001;281:L202–208. doi: 10.1152/ajplung.2001.281.1.L202. [DOI] [PubMed] [Google Scholar]
- 37.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest. 1999;103:691–696. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gomez A, Bialostozky D, Zajarias A, Santos E, Palomar A, Martinez ML, Sandoval J. Right ventricular ischemia in patients with primary pulmonary hypertension. J Am Coll Cardiol. 2001;38:1137–1142. doi: 10.1016/s0735-1097(01)01496-6. [DOI] [PubMed] [Google Scholar]
- 39.Vogel-Claussen J, Skrok J, Shehata ML, Singh S, Sibley CT, Boyce DM, Lechtzin N, Girgis RE, Mathai SC, Goldstein TA, Zheng J, Lima JA, Bluemke DA, Hassoun PM. Right and left ventricular myocardial perfusion reserves correlate with right ventricular function and pulmonary hemodynamics in patients with pulmonary arterial hypertension. Radiology. 2011;258:119–127. doi: 10.1148/radiol.10100725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Wolferen SA, Marcus JT, Westerhof N, Spreeuwenberg MD, Marques KM, Bronzwaer JG, Henkens IR, Gan CT, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur Heart J. 2008;29:120–127. doi: 10.1093/eurheartj/ehm567. [DOI] [PubMed] [Google Scholar]
- 41.Bian X, Williams AG, Jr, Gwirtz PA, Downey HF. Right coronary autoregulation in conscious, chronically instrumented dogs. Am J Physiol. 1998;275:H169–175. doi: 10.1152/ajpheart.1998.275.1.H169. [DOI] [PubMed] [Google Scholar]
- 42.Bogaard HJ, Natarajan R, Henderson SC, Long CS, Kraskauskas D, Smithson L, Ockaili R, McCord JM, Voelkel NF. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation. 2009;120:1951–1960. doi: 10.1161/CIRCULATIONAHA.109.883843. [DOI] [PubMed] [Google Scholar]
- 43.Bohuslavova R, Kolar F, Kuthanova L, Neckar J, Tichopad A, Pavlinkova G. Gene expression profiling of sex differences in hif1-dependent adaptive cardiac responses to chronic hypoxia. J Appl Physiol (1985) 2010;109:1195–1202. doi: 10.1152/japplphysiol.00366.2010. [DOI] [PubMed] [Google Scholar]
- 44.Sutendra G, Dromparis P, Paulin R, Zervopoulos S, Haromy A, Nagendran J, Michelakis ED. A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J Mol Med (Berl) 2013;91:1315–1327. doi: 10.1007/s00109-013-1059-4. [DOI] [PubMed] [Google Scholar]
- 45.Sugden MC, Langdown ML, Harris RA, Holness MJ. Expression and regulation of pyruvate dehydrogenase kinase isoforms in the developing rat heart and in adulthood: Role of thyroid hormone status and lipid supply. Biochem J. 2000;352(Pt 3):731–738. [PMC free article] [PubMed] [Google Scholar]
- 46.Gudi R, Bowker-Kinley MM, Kedishvili NY, Zhao Y, Popov KM. Diversity of the pyruvate dehydrogenase kinase gene family in humans. J Biol Chem. 1995;270:28989–28994. doi: 10.1074/jbc.270.48.28989. [DOI] [PubMed] [Google Scholar]
- 47.Dunford EC, Herbst EA, Jeoung NH, Gittings W, Inglis JG, Vandenboom R, LeBlanc PJ, Harris RA, Peters SJ. Pdh activation during in vitro muscle contractions in pdh kinase 2 knockout mice: Effect of pdh kinase 1 compensation. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1487–1493. doi: 10.1152/ajpregu.00498.2010. [DOI] [PubMed] [Google Scholar]
- 48.Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, Shelton JM, Gerard RD, Rothermel BA, Gillette TG, Lavandero S, Hill JA. Metabolic stress-induced activation of foxo1 triggers diabetic cardiomyopathy in mice. J Clin Invest. 2012;122:1109–1118. doi: 10.1172/JCI60329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee JH, Kim EJ, Kim DK, Lee JM, Park SB, Lee IK, Harris RA, Lee MO, Choi HS. Hypoxia induces pdk4 gene expression through induction of the orphan nuclear receptor errgamma. PLoS One. 2012;7:e46324. doi: 10.1371/journal.pone.0046324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rich S, Pogoriler J, Husain AN, Toth PT, Gomberg-Maitland M, Archer SL. Long-term effects of epoprostenol on the pulmonary vasculature in idiopathic pulmonary arterial hypertension. Chest. 2010;138:1234–1239. doi: 10.1378/chest.09-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guignabert C, Tu L, Izikki M, Dewachter L, Zadigue P, Humbert M, Adnot S, Fadel E, Eddahibi S. Dichloroacetate treatment partially regresses established pulmonary hypertension in mice with sm22alpha-targeted overexpression of the serotonin transporter. Faseb J. 2009;23:4135–4147. doi: 10.1096/fj.09-131664. [DOI] [PubMed] [Google Scholar]
- 52.McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, Michelakis ED. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004;95:830–840. doi: 10.1161/01.RES.0000145360.16770.9f. [DOI] [PubMed] [Google Scholar]
- 53.Michelakis ED, McMurtry MS, Wu XC, Dyck JR, Moudgil R, Hopkins TA, Lopaschuk GD, Puttagunta L, Waite R, Archer SL. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: Role of increased expression and activity of voltage-gated potassium channels. Circulation. 2002;105:244–250. doi: 10.1161/hc0202.101974. [DOI] [PubMed] [Google Scholar]
- 54.Hagan G, Southwood M, Treacy C, Ross RM, Soon E, Coulson J, Sheares K, Screaton N, Pepke-Zaba J, Morrell NW, Rudd JH. (18)fdg pet imaging can quantify increased cellular metabolism in pulmonary arterial hypertension: A proof-of-principle study. Pulm Circ. 2011;1:448–455. doi: 10.4103/2045-8932.93543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oikawa M, Kagaya Y, Otani H, Sakuma M, Demachi J, Suzuki J, Takahashi T, Nawata J, Ido T, Watanabe J, Shirato K. Increased [18f]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J Am Coll Cardiol. 2005;45:1849–1855. doi: 10.1016/j.jacc.2005.02.065. [DOI] [PubMed] [Google Scholar]
- 56.Klyuyeva A, Tuganova A, Popov KM. Amino acid residues responsible for the recognition of dichloroacetate by pyruvate dehydrogenase kinase 2. FEBS Lett. 2007;581:2988–2992. doi: 10.1016/j.febslet.2007.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tso SC, Qi X, Gui WJ, Wu CY, Chuang JL, Wernstedt-Asterholm I, Morlock LK, Owens KR, Scherer PE, Williams NS, Tambar UK, Wynn RM, Chuang DT. Structure-guided development of specific pyruvate dehydrogenase kinase inhibitors targeting the atp-binding pocket. J Biol Chem. 2014;289:4432–4443. doi: 10.1074/jbc.M113.533885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nagendran J, Gurtu V, Fu DZ, Dyck JR, Haromy A, Ross DB, Rebeyka IM, Michelakis ED. A dynamic and chamber-specific mitochondrial remodeling in right ventricular hypertrophy can be therapeutically targeted. J Thorac Cardiovasc Surg. 2008;136:168–178. 178 e161–163. doi: 10.1016/j.jtcvs.2008.01.040. [DOI] [PubMed] [Google Scholar]
- 59.Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra34. doi: 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]
- 60.Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN, Hutson AD, Neiberger RE, O’Brien RG, Perkins LA, Quisling RG, Shroads AL, Shuster JJ, Silverstein JH, Theriaque DW, Valenstein E. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006;117:1519–1531. doi: 10.1542/peds.2005-1226. [DOI] [PubMed] [Google Scholar]
- 61.Abdelmalak M, Lew A, Ramezani R, Shroads AL, Coats BS, Langaee T, Shankar MN, Neiberger RE, Subramony SH, Stacpoole PW. Long-term safety of dichloroacetate in congenital lactic acidosis. Mol Genet Metab. 2013;109:139–143. doi: 10.1016/j.ymgme.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Calcutt NA, Lopez VL, Bautista AD, Mizisin LM, Torres BR, Shroads AL, Mizisin AP, Stacpoole PW. Peripheral neuropathy in rats exposed to dichloroacetate. J Neuropathol Exp Neurol. 2009;68:985–993. doi: 10.1097/NEN.0b013e3181b40217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, Lopaschuk GD, Dyck JR, Michelakis ED. Fatty acid oxidation and malonyl-coa decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med. 2010;2:44ra58. doi: 10.1126/scitranslmed.3001327. [DOI] [PubMed] [Google Scholar]
- 64.Yoshinaga K, Ohira H, Tsujino I, Oyama-Manabe N, Mielniczuk L, Beanlands RS, Katoh C, Kasai K, Manabe O, Sato T, Fujii S, Ito YM, Tomiyama Y, Nishimura M, Tamaki N. Attenuated right ventricular energetics evaluated using (1)(1)c-acetate pet in patients with pulmonary hypertension. Eur J Nucl Med Mol Imaging. 2014;41:1240–1250. doi: 10.1007/s00259-014-2736-4. [DOI] [PubMed] [Google Scholar]
- 65.Guarnieri C, Muscari C. Beneficial effects of trimetazidine on mitochondrial function and superoxide production in the cardiac muscle of monocrotaline-treated rats. Biochem Pharmacol. 1988;37:4685–4688. doi: 10.1016/0006-2952(88)90338-3. [DOI] [PubMed] [Google Scholar]
- 66.Guarnieri C, Muscari C. Beneficial effects of trimetazidine on mitochondrial function and superoxide production in the cardiac muscle. Cardiovasc Drugs Ther. 1990;4 (Suppl 4):814–815. doi: 10.1007/BF00051282. [DOI] [PubMed] [Google Scholar]
- 67.Tuunanen H, Engblom E, Naum A, Nagren K, Scheinin M, Hesse B, Juhani Airaksinen KE, Nuutila P, Iozzo P, Ukkonen H, Opie LH, Knuuti J. Trimetazidine, a metabolic modulator, has cardiac and extracardiac benefits in idiopathic dilated cardiomyopathy. Circulation. 2008;118:1250–1258. doi: 10.1161/CIRCULATIONAHA.108.778019. [DOI] [PubMed] [Google Scholar]
- 68.Tennant DA, Duran RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10:267–277. doi: 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- 69.Kawut SM, Taichman DB, Archer-Chicko CL, Palevsky HI, Kimmel SE. Hemodynamics and survival in patients with pulmonary arterial hypertension related to systemic sclerosis. Chest. 2003;123:344–350. doi: 10.1378/chest.123.2.344. [DOI] [PubMed] [Google Scholar]
- 70.Kuhn KP, Byrne DW, Arbogast PG, Doyle TP, Loyd JE, Robbins IM. Outcome in 91 consecutive patients with pulmonary arterial hypertension receiving epoprostenol. Am J Respir Crit Care Med. 2003;167:580–586. doi: 10.1164/rccm.200204-333OC. [DOI] [PubMed] [Google Scholar]
- 71.Hopkins WE, Ochoa LL, Richardson GW, Trulock EP. Comparison of the hemodynamics and survival of adults with severe primary pulmonary hypertension or eisenmenger syndrome. J Heart Lung Transplant. 1996;15:100–105. [PubMed] [Google Scholar]
- 72.Oosterhof T, Tulevski II, Vliegen HW, Spijkerboer AM, Mulder BJ. Effects of volume and/or pressure overload secondary to congenital heart disease (tetralogy of fallot or pulmonary stenosis) on right ventricular function using cardiovascular magnetic resonance and b-type natriuretic peptide levels. Am J Cardiol. 2006;97:1051–1055. doi: 10.1016/j.amjcard.2005.10.047. [DOI] [PubMed] [Google Scholar]
- 73.Haddad F, Doyle R, Murphy DJ, Hunt SA. Right ventricular function in cardiovascular disease, part ii: Pathophysiology, clinical importance, and management of right ventricular failure. Circulation. 2008;117:1717–1731. doi: 10.1161/CIRCULATIONAHA.107.653584. [DOI] [PubMed] [Google Scholar]
- 74.Piao L, Fang YH, Parikh KS, Ryan JJ, D’Souza KM, Theccanat T, Toth PT, Pogoriler J, Paul J, Blaxall BC, Akhter SA, Archer SL. Grk2-mediated inhibition of adrenergic and dopaminergic signaling in right ventricular hypertrophy: Therapeutic implications in pulmonary hypertension. Circulation. 2012;126:2859–2869. doi: 10.1161/CIRCULATIONAHA.112.109868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weir EK, Archer SL. Counterpoint: Hypoxic pulmonary vasoconstriction is not mediated by increased production of reactive oxygen species. J Appl Physiol (1985) 2006;101:995–998. doi: 10.1152/japplphysiol.00480a.2006. discussion 998. [DOI] [PubMed] [Google Scholar]
- 76.Ward JP. Point: Hypoxic pulmonary vasoconstriction is mediated by increased production of reactive oxygen species. J Appl Physiol (1985) 2006;101:993–995. doi: 10.1152/japplphysiol.00480.2006. discussion 999. [DOI] [PubMed] [Google Scholar]
- 77.Drake JI, Bogaard HJ, Mizuno S, Clifton B, Xie B, Gao Y, Dumur CI, Fawcett P, Voelkel NF, Natarajan R. Molecular signature of a right heart failure program in chronic severe pulmonary hypertension. American journal of respiratory cell and molecular biology. 2011;45:1239–1247. doi: 10.1165/rcmb.2010-0412OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dengler MA, Staiger AM, Gutekunst M, Hofmann U, Doszczak M, Scheurich P, Schwab M, Aulitzky WE, van der Kuip H. Oncogenic stress induced by acute hyper-activation of bcr-abl leads to cell death upon induction of excessive aerobic glycolysis. PLoS One. 2011;6:e25139. doi: 10.1371/journal.pone.0025139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nagaya N, Satoh T, Ishida Y, Uematsu M, Hirose Y, Okano Y, Kyotani S, Nakanishi N, Katafuchi T, Kunieda T. Impaired left ventricular myocardial metabolism in patients with pulmonary hypertension detected by radionuclide imaging. Nucl Med Commun. 1997;18:1171–1177. doi: 10.1097/00006231-199712000-00009. [DOI] [PubMed] [Google Scholar]
- 80.Sutendra G, Michelakis ED. Pulmonary arterial hypertension: Challenges in translational research and a vision for change. Sci Transl Med. 2013;5:208sr205. doi: 10.1126/scitranslmed.3005428. [DOI] [PubMed] [Google Scholar]
- 81.Chambers KT, Leone TC, Sambandam N, Kovacs A, Wagg CS, Lopaschuk GD, Finck BN, Kelly DP. Chronic inhibition of pyruvate dehydrogenase in heart triggers an adaptive metabolic response. J Biol Chem. 2011;286:11155–11162. doi: 10.1074/jbc.M110.217349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bogaard HJ, Natarajan R, Mizuno S, Abbate A, Chang PJ, Chau VQ, Hoke NN, Kraskauskas D, Kasper M, Salloum FN, Voelkel NF. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med. 2010;182:652–660. doi: 10.1164/rccm.201003-0335OC. [DOI] [PubMed] [Google Scholar]
- 83.Grinnan D, Bogaard HJ, Grizzard J, Van Tassell B, Abbate A, DeWilde C, Priday A, Voelkel NF. Treatment of group i pulmonary arterial hypertension with carvedilol is safe. Am J Respir Crit Care Med. 2014;189:1562–1564. doi: 10.1164/rccm.201311-2025LE. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.