

Abstract
Cell-Penetrating Peptides (CPPs) are known as efficient transporters of molecular cargo across cellular membranes. Their properties make them ideal candidates for in vivo applications. However, challenges in development of effective CPPs still exist: CPPs are often fast degraded by proteases and large concentration of CPPs required for cargo transporting can cause cytotoxicity. It was previously shown that restricting peptide flexibility can improve peptide stability against enzymatic degradation and limiting length of CPP peptide can lower cytotoxic effects. Here we present peptides (30-mers) that efficiently penetrate cellular membranes by combining very short CPP sequences and collagen-like folding domains. The CPP domains are hexa-arginine (R6) or arginine/glycine (RRGRRG). Folding is achieved through multiple proline-hydroxyproline-glycine (POG)n repeats that form a collagen-like triple helical conformation. The folded peptides with CPP domains are efficiently internalized, show stability against enzymatic degradation in human serum, and have minimal toxicity. Peptides lacking correct folding (random coil) or CPP domains are unable to cross cellular membranes. These features make triple helical cell penetrating peptides promising candidates for efficient transporters of molecular cargo across cellular membranes.
Keywords: cell-penetrating peptides, collagen peptides, triple helix, intracellular delivery, internalization, enzymatic degradation
1. Introduction
Molecular transporters across the plasma membrane have been long sought after to improve cellular uptake efficiency of therapeutic drugs and imaging agents [1–3]. One of the most popular classes of molecular transporters is cell-penetrating peptides (CPPs) [4,5]. CPPs proved to be capable of transporting a variety of cargos: small molecules, proteins, nucleic acids, quantum dots and contrast agents [6,7]. General classification of CPPs divides them into three major groups: (1) cationic peptides where more than eight positive charges are needed for peptide cellular uptake, (2) hydrophobic peptides, and (3) amphipathic peptides with alternating charged/hydrophobic residues [5].
Although cationic CPPs are one of the best molecular transporters, their use have limitations [8]. The multitude of positive charges located within cationic peptides can lead to side effects including diminished cell viability and disrupted membrane integrity upon uptake [5,9,10]. It was shown that decreasing the length of CPP effectively diminished peptide toxicity [12]. Yet, limiting the number of charges (below eight) prevents cationic peptides from entering the cell [13]. The number of charges associated with CPPs can be decreased in some amphipathic peptides, where hydrophobic and charged domains within the sequence allow the peptide to fold into secondary structure like α-helixes, β-sheets or polyproline helixes (PPII) [13–16]. In these cases, cooperation of positive charges, hydrophobic domains and, in some cases receptor specific sequence, allows for translocation of peptide with molecular cargo across the membrane.
The mechanism of CPP translocation is still a matter of discussion [17]. The similarity in sequence, size, and chemical character within the CPP family would suggest a single mechanism of cellular entry. This is contrary to experimental observations where several different mechanisms of entry have been identified [18–21]. In view of this, the presence of the receptor binding sequence or particular secondary structure (or lack of it) is not necessary for peptide translocation across the membrane.
The main interest in developing CPPs comes from their potential application in transporting therapeutic and imaging agents, but many peptides are susceptible to enzymatic degradation [22]. Peptide stability against enzymatic degradation can be often improved by introduction of unnatural amino-acids. Another approach is to restrict their conformational flexibility by peptide cyclization, sequence-induced folding into well-defined secondary structure or introduction of covalent modifications i.e. cysteine knots in collagen-like structures [23–25].
Here we present characterization of synthetic cell-penetrating peptides folded into a collagen triple helix. The rationale behind designing new synthetic CPP was to limit the length of a CPP sequence to diminish possible peptide toxicity [12]. The number of arginines in the designed polyarginine CPP sequence is too small for the peptide to be able to penetrate the cell membrane in coil conformation [26–29], but when folded into higher order structure, will be able to cross the cell membrane. Therefore peptide folding was used as a strategy to accumulate enough charges for peptide internalization. Additional reason to employ peptide folding was to decrease peptide flexibility and thus protect it against enzymatic degradation.
To accomplish this goal designed peptides have two domains: “CPP domain” and “folding domain”. Two different CPP domain sequences were studied in this work: R6 and RRGRRG. Both CPP domains within designed peptides contain insufficient number of arginines to penetrate cell membrane when peptide is in coil conformation (unfolded). To introduce secondary structure into the peptides the folding domain was used. The folding domain is based on collagen peptide sequence: multiple repeats of Proline-Hydroxyproline-Glycine (POG)n that has a high propensity to form triple helix, and thus allows the peptide to fold into a helical conformation.
In this work, synthesized peptides were characterized with circular dichroism spectroscopy and confocal microscopy. Cytotoxicity and enzymatic degradation studies were performed in view of potential application of peptides as molecular transporters.
2. Materials and Methods
2.1. Materials
All reagents were used as received unless noted otherwise. All peptides (Table 1) were purchased from Tufts University Core Facility (solid support synthesis and HPLC purification). All but T5 were modified with FITC (fluorescein isothiocyanate, EX= 494 nm, EM= 521 nm) at the N-terminus via a β–alanine-glycine-glycine linker. Distilled water was purified by a Milli-Q (Millipore) deionizing system.
Table 1.
Peptide sequences. “O” represents hydroxyproline.
| Peptide | Sequence |
|---|---|
| V1 | FITC-AβGG-POGPOGPOGPOGPOGPOGPOGPOG-RRGRRG |
| V2 | FITC-AβGG-POGPOGPOGPOGPOGPOGPOGPOG-RRRRRR |
| V2R | FITC-AβGG-GPPOOGPGGGPOOPGOOPGGOOPP-RRRRRR |
| FL8 | FITC-AβGG-POGPOGPOGPOGPOGPOGPOGPOG |
| FL4 | FITC-AβGG-POGPOGPOGPOG |
| T5 | TTNYT |
2.2. Circular Dichroism
CD measurements were performed with a Jasco J-810 spectropolarimeter equipped with a Peltier temperature control system, using quartz cells with a path length of 0.2 cm. Prior to measurements, the peptides were thermally annealed: peptide solutions in water (2×10−7 M) were pre-heated to 80 °C for 5 min and slowly cooled down to 4°C, then incubated for 24 h. The peptide solutions were transferred to a CD cell and equilibrated for 30 min at 37 °C. A scan speed of 50 nm/min was used, and four scans per sample were acquired. A reference spectrum containing deionized water was subtracted from the final peptide spectrum.
Thermal unfolding curves were obtained by monitoring the decrease in ellipticity in a 25 to 80 °C temperature range at a wavelength where the CD spectra show a positive maximum (224 nm) at a heating rate of 10 °C/h. The derivative of the plotted unfolding curve was calculated using Jasco Spectra Manager II software. The minimum of the derivative indicates the steepest slope of the unfolding process and determines the helix to coil transition temperature (Tm) under described conditions. All experiments were performed in duplicates or triplicates.
2.3. Cell Cultures and Cellular Uptake
NIH-3T3 Swiss mice fibroblasts and and Jurkat E6, human leukemia cells were purchased from ATCC and cared for according to provided protocols. In short, the cells were grown in complete media: DMEM, Mediatech Inc. (3T3) or RPMI, Mediatech Inc. (Jurkats); 10% FBS (Cellgrow), 100 U / mL penicillin (Fisher Scientific), and 0.1 mg / mL streptomycin (Fisher Scientific). Passages 3–10 were used for all experiments. Peptide uptake studies were performed by dissolving given peptide in phosphate buffered saline (PBS, pH=7.4, Fisher Scientific) and incubating cells with the peptide for variable times (5 min to 30 min) at 37°C. Subsequently cells were washed 3 times and examined with microscopy or flow cytometry.
2.4. Confocal Microscopy
Inverted fluorescent microscope (Nikon TE2000) and confocal microscope (Olympus Fluoview) were used for cell imaging. 3T3 Swiss mice fibroblasts were seeded at 50,000 cells in 1000 μL of media per dish in DMEM complete media and incubated overnight in 35mm petri dishes with No. 1.5 coverslip as a bottom (MatTek Corporation). The following day cells were incubated with studied peptides (PBS, 0.1 μM to 100 μM) for up to 30 min. After incubation, cells were washed three times with PBS and 1 ml of PBS was added to the plate. The cells were imaged immediately with an Olympus Fluview confocal microscope at 20X, 40X and 100X oil immersion objectives. If DAPI (Molecular Probes) staining was performed, the cells were fixed with glutaraldehyde (Aldrich), washed with PBS and stained.
2.5. Flow Cytometry
Jurkat E6 human leukemia cells were used for all flow cytometry assays. Cells (30,000 cells in 300 μl RPMI media) were placed in conical tube and 300 μl of studied peptide stock solution (in PBS) was added. Peptide concentration in final solution was varied (0.1 to 100 μM) and cells were incubated for different time intervals from 5 to 30 min. After incubation with peptide carriers, cells were spun at 3000 rpm for 2 min., washed three times with cold PBS and then resuspended in PBS containing 0.1 % BSA. Cells (10,000/sample) were analyzed using flow cytometry (Cell Lab Quanta SC-MPL) to quantify the cellular uptake of the peptides.
2.6. Cell Viability assay
Cellular toxicity of the peptides was determined using Trypan Blue assay (BioRad). 3T3 Swiss mice fibroblasts were cultured into 24 well plates at a density of 185,000 cells in 1 ml of complete media and incubated for 24h. The cells were then incubated with 15 μM concentration of peptides in PBS for 30 min. Following incubation, the cells were washed thrice with PBS and treated with 100 μl of trypsin (Mediatech Inc.) for 2 min. The cells were transferred to culture tube, incubated with Trypan Blue for 1 minute and counted on a hemocytometer (Bright Line, Hausser Sci.).
2.6. Enzymatic digestion
Enzymatic digestion of peptides was performed according to a previously published protocol [30]. In short, stock solutions of each peptide FL4, FL8, T5, V2R, V1 and V2 (10 mg/ml) were prepared in distill water . 95 μl of 25% human serum/RPMI (v/v) was equilibrated at 37ºC for 15 min before adding 5 μl of the given peptide stock solution. The digestion was performed for different time intervals at 37ºC. After incubation, 200 μl of 95% ethanol was added to stop the reaction and the cloudy sample was incubated at 4ºC for 15 min. Following the cooling procedure, the samples were spun at 14,000 rpm for 2 min. to obtain the supernatant, free of precipitated serum proteins. Peptides were analyzed by reverse-phase HPLC (Varian) using a (Vydac, Agilent) C-18 column and a 10 – 100 % gradient (0.08% trifluoro-acetic acid in water, 0.08% trifluoro-acetic acid in acetonitrile) over 10 min. (flow rate = 2 ml/min, detection at 214 nm) at room temperature. Kinetic analysis was performed on the bases of peak area and pseudo first order reaction rate was assumed according to reference [30].
3. Results
3.1 Peptides
Peptides were synthesized, purified and characterized (HPLC, MS) by the Tufts University Core Facility. Table 1 lists all the sequences studied in this work. With the exception of T5 all peptides were modified with fluorescein isothiocyanate (FITC) at the N-terminus (via a β–Alanine-Glycine-Glycine linker) to visualize cellular uptake. The FITC linker sequence was designed to prevent possible quenching of the fluorescence when three peptides fold into helical conformation [31]. The C-terminus of all peptides was blocked by amidation to avoid unintended reactions.
3.2 Conformational studies of peptides
The secondary structure of all peptides was studied with circular dichroism (CD) spectroscopy. The CD spectra of polyarginine-based peptides show only one transition (amide ππ* transition at 197 nm), since most of them exhibit coiled conformation without well-defined secondary structure. On the other hand, CD spectra of collagen and collagen peptides are dominated by two transitions: a negative amide peak at 197 nm (ππ* transition) and a positive peak at 224 nm (nπ* transition) [30]. In Figure 1, the CD spectra of peptide V1, V2, V2R, FL8, FL4 and T5 peptides recorded at 37°C are presented. The spectra of V1, V2 and FL8 peptides show well developed positive peaks at 37°C, confirming their helical conformation. The maximum at 224 nm for peptides V2R and FL4 is missing proving their random coil conformation.
Figure 1.

Circular Dichroism spectra of peptides listed in table 1 at 37°C. Arrows indicate peak characteristic for triple helical conformation.
The good representation of peptide propensity to form triple helical conformation is transition temperature (Tm) between helical and coil conformation. Tm also indicates peptide’s thermal stability. Higher Tm indicates stronger propensity to form triple helix and increased thermal stability. In Figure 2 thermal unfolding of peptides that are helical at 37°C (V1, V2, FL8) and their first derivatives are shown. Thermal unfolding is monitored at maximum molar ellipticity (224 nm). Peptides V1 and V2 have the same folding domain {(POG)n , n=8}, but different CPP domain (Table 1). V1 contains RRGRRG while V2 contains R6. Tm for V1 = 48.8 (± 0.6) °C, while V2 = 45.6 (± 0.6) °C (Table 2). Peptides FL8 and FL4 differ only by number of (POG)n repeats: in FL8 n=8 and in FL4 n=4. Tm for FL8 is observed at 63.7 (± 0.6) °C while FL4 exist only in coil conformation for measured temperatures (Tm is below 15°C) (Table 2).
Figure 2.

Circular Dichroism for triple helical peptides: thermal unfolding shows that the peak (224 nm) is eliminated in a cooperative transition which indicates the presence of a triple helix. The first derivative of molar elipticity is used to indicate the temperature of helix to coil transition (Tm).
Table 2.
Peptide properties
| Peptide | Conformation @ 37°C | Tm (°C) | FF @ 37°C | Label |
|---|---|---|---|---|
| V1 | triple helix | 48.8 | 0.90 | FITC |
| V2 | triple helix | 45.6 | 0.82 | FITC |
| V2R | random coil | --- | --- | FITC |
| FL8 | triple helix | 63.7 | 0.96 | FITC |
| FL4 | random coil | >15 | --- | FITC |
| T5 | random coil | --- | --- | --- |
Peptide T5 belongs to a different family of peptides and was used in this work as a standard positive control for enzymatic decomposition [30]. The CD spectrum of T5 peptide shows two minima at 221 and 204 nm, possibly suggesting α-helical conformation; T5 contains only 5 residues and on average a single turn of α-helix contains 3.6 residues, thus the minima observed as signature of α-helix with CD may be due to higher order organization (stacking) of multiple peptides. However this consideration is beyond the scope of this report.
3.3 Cellular Internalization of Peptides
The cellular uptake of studied peptides was performed with two cell lines. To limit the potential problems with data analysis adherent cells were used for microscopy and suspension cells were used for flow cytometry studies. NIH-3T3 Swiss mice fibroblasts were used as a model to visualize peptide uptake with confocal microscopy. Jurkat E6 human leukemia cells were used to study cellular uptake in population of cells using flow cytometry.
Figure 3 shows confocal images of fibroblasts incubated with 15 μM solution of different peptides in PBS for 30 min. The images were collected with bright field microscope or illuminated with 488nm Argon-ion laser (to excite FITC), or illuminated with 405nm diode laser (to excite DAPI) and digitally combined together. In panel A (Figure 3) cells incubated with peptide V1 (triple helix conformation, RRGRRG CPP domain) or V2 (triple helix conformation, R6 CPP domain) show well visible fluorescence from peptides modified with FITC. This proves that peptides V1 and V2 are able to effectively penetrate the cellular membrane. Additionally DAPI stain was used to localize nuclei of the cells. The colocalization of DAPI and FITC within the nucleus suggests that peptide carriers have tendency to accumulate within nucleus. However, the two-color (FITC/DAPI) cell images do not clearly show possible differences in V1 and V2 peptide localization due to artifacts of DAPI staining protocol. Thus 3T3 fibroblasts were incubated with V1 or V2 peptide and imaged without DAPI (Figure 3, panel C). Evidently, V1 accumulates to larger extend in cytoplasm than V2, with only small amount reaching the nucleus. V2 has a stronger preference to accumulate within the nuclear bodies that contain high concentration of DNA. Taking into account the size and number of nuclear bodies, as well as their deep staining with DAPI they are likely nucleosomes. The behavior of V2 peptide is very similar to the previously observed cationic CPPs containing more than nine arginines in the sequence or specific nuclear localization factor [33,34]. Panel B in Figure 3 shows the images of 3T3 cells incubated with control peptides: FL8 (triple helix conformation, lack of CPP domain), FL4 (coil conformation, lack of CPP domain) or V2R (coil conformation, CPP domain: R6) that are either lacking CPP domain or correct folding domain, or both. In all of the above cases the lack of fluorescence emitted from the cells indicates lack of cellular uptake. Clearly these peptides are unable to penetrate the cellular membrane.
Figure 3.

Cellular uptake of peptides listed in table 1 by 3T3 Swiss mice fibroblasts. Cells were incubated with 15 μM peptide in PBS for 30 min. The images were taken by confocal microscope in bright field, with FITC filter, DAPI filter (for coloclization studies), and all images were digitally combined. Panel A: peptides containg folding and CPP domains with DAPI: V1 and V2, panel B: control peptides FL8, FL4 and V2R. Panel C: peptides containg folding and CPP domains without DAPI: V1 and V2
Flow cytometry was used to measure the effectiveness of V1 and V2 cellular uptake with Jurkat E6 human leukemia cells. The cells were incubated with variable concentration of different peptides (in PBS) at 37ºC for 10 min, washed three times with cold PBS and analyzed by flow cytometry. In Figure 4 the dependence of peptide concentration during incubation period on mean cell fluorescence shows that peptides V1 and V2 are readily internalized by Jurkats at concentrations below 10 μM, with noticeably higher uptake of V2 peptide. Peptide V2R has the same amino acid composition as V2 peptide, but scrambled folding domain sequence, thus is lacking the helical conformation of V2. The CPP domain in V2R is the same as in V2 (R6). There is a small fluorescence signal from cells incubated with very high concentrations of V2R (above 75 μM). The peptides lacking CPP domain (FL4 and FL8) are not uptaken by the cells.
Figure 4.

Cellurar uptake of peptides by E6 Jurkat human leukemia cells measured with flow cytomerty. Incubation time: 10 min at 37°C.
3.4 Enzymatic degradation of peptides in human serum
To measure the resistance against enzymatic degradation studied peptides were treated with human serum. According to previously publised protocol, peptides were incubated with 25% human serum (HS in RPMI) and analyzed with RP-HPLC [30] . The negative control (PBS only added to serum) was used to identify all peaks in chromatogram arising from serum proteins and peptides, and to exclude them from the analysis of peptide digestion. The positive control of the digestion assay was performed on the T5 peptide, known standard for serum digestion [30]. In Figure 5 the area of HPLC peak was measured for each peptide after digestion, normalized with respect to the initial area (0 hour incubation time in HS) and plotted vs. time. All peptides in a triple helical conformation (V1, V2, FL8) are stable and show no visible HS digestion within the tested incubation time (24h). The same conclusion is true for FL4, that has random coil conformation at 37°C, but contains the same motif of multiple POG repeats as V1, V2 and FL8. Peptides T5 (positive control) and V2R are susaptible to enzymatic digestion.
Figure 5.

Peptide degradation performed in 25% human serum/DMEM at 37ºC. Supernatant of peptide after digestion was analyzed by HPLC (integraded peak area) and ploted with respect to digestion time. Error bars represent standard deviation calculated from 3 repeats.
3.5 Peptide cytotoxicity
The cytotoxicity of all studied peptides was measured with a Trypan Blue colorimetric assay using 3T3 Swiss mice fibroblasts. The cells viability was measured after 30 minute exposure to 15 μM solution of peptide. As shown in Figure 6, peptides V1 and V2 show no significant (p>0.05) toxicity compared to the control (media and PBS), however statistically significant toxicity (p<0.05) was observed in case of FL4 peptide.
Figure 6.

Histogram of the percentage of viable cells (3T3 fibroblasts) after 30 min. measured with the Trypan Blue assay after the addition of 15 μM peptides normalized with respect to the control (no incubation). The error bars reflect standard deviation calculated from three repeats on two different plates.
4. Discussion
The peptide sequence was always the primary concern when CPP was chosen for cargo translocation. Here, the choice of simple CPP domain containing only 6 arginines (R6) was made to study the effect of the peptide folding on the cellular uptake. Previously published results showed that R6 sequence does not contain sufficient number of arginines to allow the peptide to penetrate cell membrane [11]. The addition of collagen folding domain (POG)n forces three peptides to fold into the triple helix. The net number of arginines in the folded peptide is 18 thus the peptide V2 should be able to cross the membrane, if the peptide conformation has no negative effects on cellular internalization. This hypothesis was consistent with our observations. One can argue that the R6 sequence has very low propensity to fold into triple helix, so it can be postulated that R6 located at C-terminus of peptide is not folded and exist as a coil at the end of the peptide (Figure 7). The folded fraction (FF) of peptide at physiological conditions can be calculated based on CD measurements with previously published equation [35]:
| Equ (1) |
where CDx represent molar elipticity measured at 224 nm and subscript denotes the temperature at which the measurement was acquired. At the physiological temperature (37°C) FF of V2 peptide is 0.82 (Table 2), what is clearly sufficient to translocate the peptide across the cell membrane.
Figure 7.
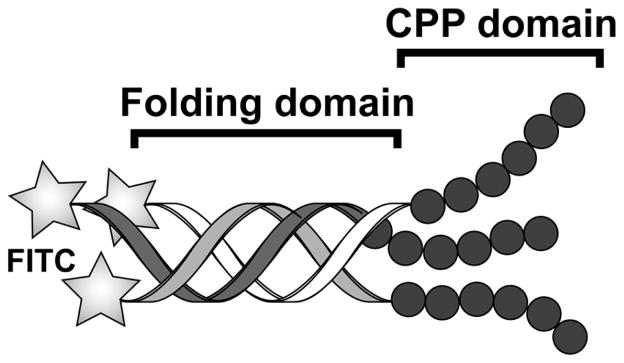
Schematic representation of V1 and V2 peptides. The peptide is folded into triple helix within the POGn sequence (folding domain). The CPP domain R6 or (RRG)2 is located at C-terminus. The N-terminus is modified with FITC.
To improve peptide folding into a triple helix, the RRGRRG sequence was designed as the CPP domain. This sequence should have higher propensity to fold into triple helix then R6 due to positioning glycine in the preferred position that decreases steric hindrance in the fold and limiting electrostatic repulsion by decreasing number of arginines. Indeed the FF for V1 peptide containing RRGRRG sequence is 0.90. Additionally, the stability of helical conformation can be defined as temperature (Tm) of helix-to-coil transition: higher Tm is observed for peptides that are more thermally stable due to tighter folding. Experimentally, Tm can be determined with CD spectroscopy by measuring the decrease of the positive peak at 224 nm with respect to temperature (Figure 2). We observe that Tm of V2 peptide is lower by 3.2°C than Tm of V1 (Table 2). Clearly in case on V2, lack of glycine in a “G” position in (POG)n peptide sequence contributes to lower Tm and smaller FF.
Jurkat human leukemia cells were used to compare the internalization of peptides V1 and V2. It is visible in Figure 4, that both peptides are able to cross the cellular membrane, however V2 is very effective, even at small concentration, while at higher concentration both peptides have similar uptake efficiency. This behavior can be attributed to larger number of arginine in folded V2 peptide (18) versus V1 peptide (12). Figure 8 presents flow cytometry results of Jurkat cells incubated with 15μM V1 or V2 for 10 min. The fluorescence intensity from cells exposed to V1 is about 20% lower, than the cells incubated with V2. However, the number of cells that uptake V1 is comparable with the number of cells that uptake V2 (71.2% vs. 72.8%). These results suggest that number of arginines present in the folded peptide is a predominant factor in enabling peptide to cross the cell membrane and folding is only necessary to accumulate sufficient number of positive charges for the translocation. This conclusion is also supported by the difference in cellular uptake between V2 and V2R. Both peptides contain the R6 CPP sequence, but only V2 has folding sequence (POG)8, while V2R has the same amino acid composition, but scrambled folding sequence. This causes V2 to be folded at physiological temperature into triple helix and effectively increases the number of arginines to 18, while unfolded V2R has only 6. Clearly based on the confocal microscopy and flow cytometry V2R is unable to cross the cell membrane in these conditions. We concluded that in case of cellular uptake efficiency of studied peptides, folding is important as a method of accumulating larger charge, but seems to have neither positive nor negative effect on the efficiency of peptide translocation. Furthermore, our observations indicate that it is not necessary for the CPP sequence to be localized within a single strand to promote effective cellular uptake.
Figure 8.

Cellurar uptake of peptides by E6 Jurkat human leukemia cells measured with flow cytomerty : histogram of mean fluorescence measured after Jurkat cells were incubated for 10 min. in 15 μM peptide solution.
The cellular uptake of peptides V1 and V2 is very efficient for both 3T3 Swiss mice fibroblasts and human leukemia Jurkat cells, but we note that the internalization of peptides takes longer time for the fibroblast than Jurkat cells, what agrees with past observations. Both peptides V1 and V2 have very similar structure, vary only by two amino acids, but this small difference seem to determine the preferential accumulation site within the cell. While V1 tends to distribute within the cytoplasm and to smaller extend in nuclear bodies, V2 has a strong preference to accumulate within nuclear bodies that contain high concentration of nucleic acids. This fact can be utilized in the potential application of the peptide in drug delivery and enable the delivery to either cytoplasm (V1) or nucleus (V2) depending on the site of drug action.
The peptide resistance to enzymatic attack is a potentially useful characteristic in view of prospective applications. The (POG)n folding sequence proved to be resistant to enzymes in human serum (Figure 5), however the resistance appears to be derived from the sequence rather than stabilization due to folding into a secondary structure. All the helical peptides (V1, V2 and FL8) are resistant to enzymatic attack as well as FL4, that has the (POG)4 sequence, however FL4 is not folded into triple helix at 37°C since it has only four POG units. Moreover, peptide V2R with the same composition as V2, but a scrambled sequence (random coil conformation) is susceptible to HS digestion. In Figure 9 the HPLC peptide peak area was correlated with digestion time of peptie suseptible to HS digestion. Pseudo-first order reaction kinetics was assumed to determine the half-life of V2R peptide digested with 25% HS. The half-life of V2R in these conditions was 832 min. The control experiment used T5 peptide as a digestion standard. We determined that the half-life of T5 is 210 min. under these conditions. This correlates very well with previously reported results by Powell et.al. [30].
Figure 9.

Peptide degradation performed in 25% human serum/DMEM at 37ºC. Plot of pseudo first order enzymatic digestion analyzed by HPLC (integraded peak area) for peptides that undergo decomposition (T5, V2R). Error bars represent standard deviation calculated from 3 repeats.
FL8 peptide has no cytotoxic effect comparing to control. FL8 has analogous sequence as FL4 except it contains more (POG)n units, n=8 vs. n=4, thus the only structural difference between them is the presence of a triple helix in FL8. Consequently the change in conformation from triple helix (FL8) to random coil (FL4) possibly causes an increase in cell death (by about 50% within 30 min.). This surprising behavior is consistent with the observed cell morphology. It can be seen in Figure 3 panel B where 3T3 fibroblasts incubated with FL4 have a different shape and size in comparison with control or with the cells incubated with the other peptides. However neither microscopy or flow cytometry shows visible binding to the cell surface or uptake of FL4. At this stage of our investigation the nature of interactions between FL4 and 3T3 fibroblast is unknown, but it may involve activation of apoptosis receptors or changes to cell membrane integrity ultimately causing cell rupture.
In summary, cell-penetrating peptides containing triple helical folding domain that determines their secondary structure have a potential to be used as molecular transporters due to ease of cellular membrane penetration, lack of toxicity and stability against enzymatic degradation. Additionally the proposed peptides are self-assembled into triple helix without need of complicated synthetic protocols. Yamazaki et. al. show effective internalization of collagen-like CPP containing cysteine-knot that keeps peptide chains covalently bound [25]. While their approach proves that collagen motif can be used as effective strategy to synthesize CPP, complex synthetic protocol would likely prevent scaling up the synthesis of the peptide for drug carrier application.
Acknowledgments
We are grateful to Dr. Zhang and Dr. Haas for assistance with FACScan; to Lisa Stutts for helpful discussions and artistic touch. This work was funded by National Institutes of Health GM099594 and National Science Foundation MRI DBI0722757 (confocal microscopy).
References
- 1.Wang B, Siahaan TJ, Soltero AR, editors. Drug Delivery: Principles and Applications. John Wiley & Sons Inc; Hoboken, NJ: 2005. [Google Scholar]
- 2.Gu Z, Biswas A, Zhaoab M, Tang Y. Tailoring nanocarriers for intracellular protein delivery. Chem Soc Rev. 2011;40:3638–3655. doi: 10.1039/c0cs00227e. [DOI] [PubMed] [Google Scholar]
- 3.Khan DR. The use of nanocarriers for drug delivery in cancer therapy. J Cancer Sci & Therapy. 2010;2:58–62. [Google Scholar]
- 4.Lindgren M, Hällbrink M, Prochiantz A, Langel U. Cell-penetrating peptides. Trends Pharmacol Sci. 2000;21:99–103. doi: 10.1016/s0165-6147(00)01447-4. [DOI] [PubMed] [Google Scholar]
- 5.Wang F, Wanga Y, Zhanga X, Zhanga W, Guo S, Jin F. Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J Controlled Release. 2014;174:126–136. doi: 10.1016/j.jconrel.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 6.Shin MC, Zhang J, Min KA, Lee K, Byun Y, David AE, He H, Yang VC. Cell-penetrating peptides: Achievements and challenges in application for cancer treatment. J Biomed Mater Res Part A. 2014;102A:575–587. doi: 10.1002/jbm.a.34859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta B, Levchenko TS, Torchilin VP. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv Drug Deliv Rev. 2005;57:637– 651. doi: 10.1016/j.addr.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Reissmann S. Cell penetration: scope and limitations by the application of cell-penetrating peptides. J Pept Sci. 2014;20:760–784. doi: 10.1002/psc.2672. [DOI] [PubMed] [Google Scholar]
- 9.Palm-Apergi C, Lorents A, Padari K, Pooga M, Hallbrink M. The membrane repair response masks membrane disturbances caused by cell-penetrating peptide uptake. FASEB J. 2009;23:214–223. doi: 10.1096/fj.08-110254. [DOI] [PubMed] [Google Scholar]
- 10.Verdurmen WP, Brock R. Biological responses towards cationic peptides and drug carriers. Trends Pharmacol Sci. 2011;32:116–124. doi: 10.1016/j.tips.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 11.El-Sayed A, Futaki S, Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. AAPS J. 2009;11:13–22. doi: 10.1208/s12248-008-9071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cardozo AK, Buchillier V, Mathieu M, Chen J, Ortis F, Ladrière L, Allaman-Pillet N, Poirot O, Kellenberger S, Beckmann JS, Eizirik DL, Bonny C, Maurer F. Cell-permeable peptides induce dose- and length-dependent cytotoxic effects. BBA Biomembranes. 2007;1768:2222–2234. doi: 10.1016/j.bbamem.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 13.Fillon YA, Anderson JP, Chmielewski J. Cell-penetrating agents based on a polyproline helix scaffolds. J Am Chem Soc. 2005;127:11798–11799. doi: 10.1021/ja052377g. [DOI] [PubMed] [Google Scholar]
- 14.Pujals S, Giralt E. Proline-rich, amphipathic cell-penetrating peptides. Adv Drug Delivery Rev. 2003;55:1679–1684. doi: 10.1016/j.addr.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 15.Geisler I, Chmielewski J. Cationic amphiphilic polyproline helices: side-chain variatrions and cell-specific internalization. Chem Biol Drugs Des. 2009;73:39–45. doi: 10.1111/j.1747-0285.2008.00759.x. [DOI] [PubMed] [Google Scholar]
- 16.Mitchell DJ, Kim DT, Steinman W, Fathman CG, Rothbard JB. Polyarginine enters cells more efficiently than other polycationic homopolymers. J Pept Res. 2000;56:318–325. doi: 10.1034/j.1399-3011.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 17.Fonseca SB, Pereira MP, Kelley SO. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv Drug Deliv Rev. 2009;61:953–964. doi: 10.1016/j.addr.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Futaki S, Nakase I, Tadokoro A, Takeuchi T, Jones A. Arginine-rich peptides and their internalization mechanisms. Biochem Soc Trans. 2007;35:784–787. doi: 10.1042/BST0350784. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt N, Mishra A, Lai GH, Wong GC. Arginine-rich cell-penetrating peptides. FEBS Lett. 2010;584:1806–1813. doi: 10.1016/j.febslet.2009.11.046. [DOI] [PubMed] [Google Scholar]
- 20.Futaki S. Arginine-rich peptides: potential for intracellular delivery of macromolecules and the mystery of the translocation mechanisms. Int J Pharm. 2002;245:1–7. doi: 10.1016/s0378-5173(02)00337-x. [DOI] [PubMed] [Google Scholar]
- 21.Nakase I, Takeuchi T, Tanaka G, Futaki S. Methodological and cellular aspects that govern the internalization mechanisms of arginine-rich cell-penetrating peptides. Adv Drug Deliv Rev. 2008;60:598–607. doi: 10.1016/j.addr.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Zhang XX, Eden HS, Chen X. Peptides in cancer nanomedicine: Drug carriers, targeting ligands and protease substrates. J Controlled Release. 2012;159:2–13. doi: 10.1016/j.jconrel.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGregor DP. Discovering and improving novel peptide therapeutics. Curr Opin Pharm. 2008;8:616–619. doi: 10.1016/j.coph.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 24.Werle M, Bernkop-Schnurch A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids. 2006;30:351–367. doi: 10.1007/s00726-005-0289-3. [DOI] [PubMed] [Google Scholar]
- 25.Yamazaki CM, Nakase I, Endo H, Kishimoto S, Mashiyama Y, Masuda R, Futaki S, Koide T. Collagen-like Cell-Penetrating Peptides. Angew Chem Int Ed. 2013;52:5497–5500. doi: 10.1002/anie.201301266. [DOI] [PubMed] [Google Scholar]
- 26.Bella J, Eaton M, Brodsky B. Crystal structure and molecular structure of a collagen-like peptide at 1. 9 Angstrom resolution. Science. 1994;266:74–81. doi: 10.1126/science.7695699. [DOI] [PubMed] [Google Scholar]
- 27.Persikov AV, Ramshaw JAM, Brodsky B. Prediction of collagen stability from amino acid sequence. J Biol Chem. 2005;280:19343–19349. doi: 10.1074/jbc.M501657200. [DOI] [PubMed] [Google Scholar]
- 28.Venugopal MG, Ramshaw JAM, Braswell E, Zhu D, Brodsky B. Electrostatic interactions in collagen-like triple-helical peptides. Biochemistry. 1994;33:7948–7956. doi: 10.1021/bi00191a023. [DOI] [PubMed] [Google Scholar]
- 29.Yu SM, Li Y, Kim D. Collagen mimetic peptides: progress towards functional applications. Soft Matter. 2011;7:7927–7938. doi: 10.1039/C1SM05329A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Powell MF, Stewart T, Otvos L, Jr, Urge L, Gaeta FCA, Sette A, Arrhenius T, Thomson D, Soda K, Colon SM. Peptide stability in drug development. 2. Effect of amino acid substitution and glycosylation on peptide reactivity in human serum. Pharm Res. 1993;10:1268–1273. doi: 10.1023/a:1018953309913. [DOI] [PubMed] [Google Scholar]
- 31.Reimer AE, Feher K, Hernandez D, Slowinska K. Self-assembly of collagen peptides into hollow microtubules. J Mater Chem. 2012;22:7701–7703. [Google Scholar]
- 32.Ky D, Liu CK, Marumoto C, Castaneda L, Slowinska K. Electrochemical Time-of-Flight in crosslinked collagen matrix solution: implications of structural changes for drug delivery systems. J Controlled Release. 2006;112:214–222. doi: 10.1016/j.jconrel.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 33.Fuchs SM, Raines RT. Polyarginine as a multifunctional fusion tag. Protein Science. 2005;14:1538–1544. doi: 10.1110/ps.051393805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakai N, Matile S. Anion-mediated transfer of polyarginine across liquid and bilayer membranes. J Am Chem Soc. 2003;125:14348–14356. doi: 10.1021/ja037601l. [DOI] [PubMed] [Google Scholar]
- 35.Hwang E, Brodsky B. Folding delay and structural perturbations caused by type IV collagen natural interruptions and nearby Gly missense mutations. J Biol Chem. 2012;287:4368–4375. doi: 10.1074/jbc.M111.269084. [DOI] [PMC free article] [PubMed] [Google Scholar]