

Abstract
Accumulating evidence suggests that nicotine, a drug that stimulates nicotinic acetylcholine receptors, may be of therapeutic value in Parkinson’s disease. Beneficial effects may be several-fold. One of these is a protective action against nigrostriatal damage. This possibility stems from the results of epidemiological studies that consistently demonstrate an inverse correlation between tobacco use and Parkinson’s disease. This reduced incidence of Parkinson’s disease has been attributed to the nicotine in tobacco products, at least in part, based on experimental work showing a protective effect of nicotine against toxic insults. Second, several studies suggest a symptomatic effect of nicotine in Parkinson’s disease, although effects are small and somewhat variable. Third, recent data in nonhuman primates show that nicotine attenuates L-dopa-induced dyskinesias, a debilitating side effect that develops in the majority of patients on L-dopa therapy. Collectively, these observations suggest that nicotine or CNS selective nicotinic receptor ligands hold promise for Parkinson’s disease therapy to reduce disease progression, improve symptoms and/or decrease L-dopa-induced dyskinesias.
Keywords: Dyskinesias, L-dopa, neuroprotection, nicotinic receptors, striatum, smoking
Although dopamine replacement therapy has proved very effective in Parkinson’s disease management, side effects develop and efficacy declines due to disease progression 1-5. There is therefore a continual search for better treatment strategies. This article will review accumulating evidence that nicotine or nicotinic receptor agonists may be useful in the therapeutic management of Parkinson’s disease. We initially describe anatomical and functional work that provides a rationale for a relationship between the nicotinic cholinergic and dopaminergic systems. This is followed by studies showing that there are several nicotinic receptor populations in the striatum that regulate dopamine release under physiological conditions and with nigrostriatal damage. These studies provide a cellular basis for behavioral studies demonstrating a functional interaction between the dopamine and nicotinic cholinergic system. Lastly, we discuss a role for nicotine and nicotinic agonists in neuroprotection against nigrostriatal damage, in the treatment of Parkinson’s disease symptoms, and in the reduction of side effects (dyskinesias) associated with L-dopa treatment.
LINK BETWEEN THE STRIATAL NICOTINIC CHOLINERGIC AND DOPAMINERGIC SYSTEMS
Anatomical studies clearly demonstrate a close association between the cholinergic and dopaminergic systems in the striatum 6. Striatal dopamine, tyrosine hydroxylase, dopamine receptors and other catecholaminergic markers 7-9 extensively colocalize with acetylcholine, choline acetyltransferase, acetylcholinesterase, and cholinergic receptors across species. All these cholinergic measures are associated with large cholinergic interneurons that comprise ~2% of the striatal neuronal population 6. Although their number is limited, cholinergic neurons have extensive axonal arbors yielding a dense local innervation in the striatum that overlaps with dopaminergic terminals from the substantia nigra. Striatal cholinergic neurons are tonically active and continually secrete acetylcholine, which interacts with acetylcholine receptors present throughout the striatum 10, 11. The two major acetylcholine receptor types in striatum include muscarinic receptors, which are activated by acetylcholine and muscarine, and nicotinic receptors, so-named because these are stimulated by acetylcholine and nicotine, but not muscarine 12-14.
Muscarinic receptors, which are G-protein linked, reside on striatal GABAergic neurons and cholinergic interneurons. Their stimulation modulates motor function. In fact, muscarinic receptor blockers were extensively used to ameliorate Parkinson’s disease symptoms before the advent of L-dopa. Their use is currently limited to the treatment of early Parkinson’s disease and/or as an adjunct therapy to L-dopa because improvement is modest and associated with significant side effects 3, 4, 15.
Nicotinic receptors are ligand gated ion channels that are also extensively present in the striatum 10, 16-19. These receptors are localized to striatal GABAergic and cholinergic neurons and, in addition, are present at presynaptic sites including nigrostriatal dopaminergic terminals and cortical glutamatergic afferents 10, 11, 20, 21. Extensive studies in experimental models show that the role of these presynaptic nicotinic receptors is to regulate dopamine release 10, 11, 22. These findings underlie the cellular basis for the functional interaction between the striatal nicotinic and dopaminergic systems, which is the current focus. Behavioral studies in rodent models further support this relationship as dopamine receptor antagonists can block nicotine-evoked changes in locomotor activity 23. These latter findings also support the idea that the nigrostriatal pathway is key in regulating motor functions. As well, administration of nicotine and nicotinic agonists influence motor function in animals with nigrostriatal damage 18, providing the basis for the idea that they may be useful in Parkinson’s disease management.
NICOTINIC RECEPTORS IN THE NIGROSTRIATAL SYSTEM
The nicotinic receptors controlling striatal dopamine function are composed of different combinations of α and β subunits, or only select α subunits 16, 19, 22. The α subunits are critical as they express the acetylcholine recognition sites, while the β subunits modulate binding of acetylcholine to the α subunit. Multiple α and β subunits have been identified including α2 through α7, and β2 through β4. This multiplicity of subunits allows for expression of numerous nicotinic receptor subtypes, as five subunits are required for formation of functional receptors. Combined evidence from in situ hybridization, receptor binding experiments and immunoprecipitation studies using receptor subunit-directed antibodies show that several major nicotinic receptor subtypes are present in rodent and nonhuman primate striatum 24-33. These include homomeric α7 nicotinic receptors, and heteromeric receptors containing the α4β2 and the α6β2 subunits (Table 1). The former two receptor populations will subsequently be designated as α4β2* and α6β2* nicotinic receptor, with the asterisks signifying the possible presence of other subunits in the receptor complex. Interestingly, the α6β2* nicotinic receptors are restricted to the striatum and only a few other brain regions 34-36. Studies to further elucidate the subunits comprising these receptors demonstrate the presence of at least two populations of α6β2* nicotinic receptor, including those composed of the α6α4β2β3 and α6β2β3 subunits 20, 37. There also appear to be multiple populations of α4β2* receptors, with the primary one composed solely of α4β2 subunits 20, 25, 26, 32. The α6α4β2β3, α6β2β3 and α4β2 nicotinic receptor populations form the major striatal subtypes, and appear fairly consistent across species, including mice, rats, monkeys (Table 1) 37. Importantly, for drug development, data suggest that these same nicotinic receptor subtypes are present in human striatum 17, 37-39.
TABLE 1.
Putative subunit composition of nicotinic receptor subtypes in rodent, monkey, and human striatum
| Species | Nicotinic receptor subtypes |
|---|---|
| Rodents, Monkeys, Humans | α4β2 |
| α6α4β2β3 | |
| α6β2β3 | |
| α7 | |
| Rodents only | α4α5β2 |
| α6β2* | |
| Monkeys only | α4α2β |
| α3β2* |
The asterisk indicates the possible presence of additional subunits in the receptor complex.
Current evidence suggests that both the α6β2* and α4β2* nicotinic receptors are present on dopaminergic terminals, and are important for controlling dopamine release in striatum 22. The magnitude of the involvement of these two subtypes appears to vary with species. In nonhuman primates, the α6β2* nicotinic receptors are responsible for about 75% of nicotinic receptor-evoked striatal dopamine release and the α4β2* the remaining 25%, with these percentages reversed in rodent striatum 20, 28, 40. Dopamine release is also indirectly mediated by a smaller population of α7 receptors present on striatal glutamatergic afferents from the cortex 21, 39.
NEURONAL NICOTINIC RECEPTORS ARE DECREASED WITH NIGROSTRIATAL DAMAGE
Since the α4β2* and α6β2* subtypes are the primary nicotinic receptors that regulate dopamine release from nigrostriatal terminals, it is important to know how nigrostriatal damage impacts their expression. Such information is critical for the design of suitable nicotinic receptor drugs for Parkinson’s disease. To address this, studies have been done in several parkinsonian animal models, including 6-hydroxydopamine-lesioned rats, MPTP-treated mice, and MPTP-lesioned nonhuman primates. The α6β2* nicotinic receptors generally appear to be the most susceptible to nigrostriatal damage across species, an observation suggesting they are primarily localized to striatal dopaminergic terminals 25, 26, 31, 35, 41. The α4β2* nicotinic receptors are decreased only with a severe nigrostriatal lesion, a finding suggesting they are localized to nigrostriatal afferents more resistant to nigrostriatal damage and other striatal neurons 25, 26, 41, 42. By contrast, α7 nicotinic receptors are unaffected by lesioning, further confirming that they are not present on dopamine terminals 25, 26, 32, 43, 44.
Since nicotinic receptors are present on neuronal elements that are lost in Parkinson’s disease 17, 45, the question arises whether there are corresponding changes in striatal nicotinic receptors in this disorder. Receptor studies done using radiolabeled nicotine, methylcarbachol and epibatidine, demonstrate 30 to 75% declines in nicotinic receptors in the caudate, putamen and substantia nigra that appear to correlate with nigrostriatal damage 17, 46-48. Subsequent studies to elucidate the receptor subtypes that are affected suggest that the more severe losses are in the α6β2* receptor population, with somewhat smaller declines in the α4β2* subtype, in agreement with results in animal models 17, 37, 38, 45, 49. α7 nicotinic receptor were generally unaffected by nigrostriatal damage, also consistent with experimental studies. The observed reductions in α6β2* nicotinic receptors generally paralleled decreases in other indices of nigrostriatal degeneration, such as dopamine and/or dopamine transporter levels 37, 38, 45, 49.
These combined findings suggest that α6β2* and α4β2* nicotinic receptors may represent promising pharmacological targets for Parkinson’s disease for neuroprotection and/or symptomatic improvement. Since α6β2* subtypes are localized primarily on dopaminergic terminals and regulate dopamine release, they may be particularly relevant for the development of Parkinson’s disease therapies, which could provide optimal therapeutic benefits with minimal side effects. However, another important consideration is that certain receptor subtypes are more vulnerable to nigrostriatal damage and preferentially lost with a mild lesion 37. These findings raise the question whether the more susceptible subtypes may be more relevant targets for drug development in the initial stages of Parkinson’s disease whereas others may be more important targets with disease progression.
NICOTINE AND PARKINSON’S DISEASE MANAGEMENT
The studies described in the preceding sections show that (1) there is extensive overlap between the striatal nicotinic and dopaminergic system, (2) that nicotinic receptors are expressed in striatum with several subtypes present on striatal dopaminergic terminals, and (3) that nicotinic receptor activation elicits dopamine release. (4) In addition, nicotine treatment enhances expression of some nicotinic receptor subtypes decreased with nigrostriatal damage 50, which may suggest that function mediated through these receptors is restored closer to control levels with nicotine treatment.
These combined data provide a rational basis for the suggestion that nicotine or nicotinic receptor agonists may enhance dopamine function. Since Parkinson’s disease is characterized by a loss of nigrostriatal dopaminergic function, stimulation of the nicotinic cholinergic system offers therapeutic potential. This is important because dopamine replacement therapy with L-dopa and dopamine agonists, the mainstay of Parkinson’s disease management, is associated with serious side effects including on-off phenomena and dyskinesias. Moreover, there is a diminished efficacy with time because of continued disease progression. There is therefore an urgent need for alternate and/or complementary therapeutic approaches. Below we describe a role for nicotine and/or nicotinic agonist in Parkinson’s disease management for (1) protection against nigrostriatal damage, (2) symptomatic relief of motor symptoms and (3) attenuation of L-dopa-induced side effects (dyskinesias).
Nicotine and neuroprotection for Parkinson’s disease
The ultimate mode of treatment for any neurological disorder is one that halts disease progression and reverses existing damage. Such a strategy for Parkinson’s disease offers the advantage that it may not only alter the development of the disease process but, in addition, delay or minimize motor and/or nonmotor symptoms, as well as complications associated with treatment. An understanding of the etiology of Parkinson’s disease would greatly facilitate the identification of suitable candidate agents. However, the origin of Parkinson’s disease is currently uncertain although it is most likely due to a complex interaction between genetic and environmental factors. These result in the activation of diverse neurodegenerative mechanisms, including oxidative stress, free radical generation, mitochondrial deficits, glutamate excitotoxicity, inflammation, neurotrophic factor deprivation, and dysfunction of the ubiquitin–proteasome system 1-5, 51. There is currently an ongoing search for drugs that delay Parkinson’s disease progression by targeting the molecular mechanisms that underlie the neurodegenerative process. These include agents that increase mitochondrial function (coenzyme Q10, creatine), enhance trophic activity (GDNF), inhibit monoamine oxidase (selegiline, rasagiline), modulate inflammatory processes (minocycline), block protein aggregation, and inhibit apoptosis 15, 52-54.
Nicotine has been proposed as a putative neuroprotective agent against nigrostriatal damage based on several lines of evidence. These include a large and compelling literature showing that (1) cigarette smoke, in which nicotine is a major component, is a robust negative risk factor for Parkinson’s disease, and (2) nicotine acts as a neuroprotectant in in vitro and in vivo experimental models, as described in greater detail in the following sections.
(1) Epidemiological studies demonstrate a reduced incidence of Parkinson’s disease with tobacco use
An extensive and compelling epidemiological literature shows that smoking is inversely associated with Parkinson’s disease. This concept initially appears counterintuitive based on the generally detrimental health-related consequences of smoking. However, over 50 prospective cohort and case-control studies over the last half century indicate that smoking and other forms of tobacco use appear protective against Parkinson’s disease in both males and females 55-61. There was a dose-response relationship between the number of pack-years smoked, such that the incidence of Parkinson’s disease was lower with increased length of time of smoking and the number of cigarettes smoked per day. Moreover, the decline in risk, or apparent protective effect, was diminished in former smokers. These inverse trends between smoking incidence and Parkinson’s disease were observed in every age group except the very elderly (>75 years), suggesting that smoking delays the onset of Parkinson’s disease 55, 62. An inverse association has also been identified with the use of cigars, pipes, chewing tobacco and snuff 55, 63. Alternative interpretations for this association have been proposed. These include a higher mortality in smokers with Parkinson’s disease 64, a genetically conferred decreased propensity to smoke, a premorbid personality, and a reduced dopaminergic tone that results in a lower reward of smoking and thus nonsmoking behavior in people fated to develop Parkinson’s disease 65. Evidence against these theories derive from a study in twin pairs discordant for Parkinson’s disease, in which twins without Parkinson’s disease had smoked more than their brothers 66. This difference was most evident in the monozygotic pairs, known to be remarkably similar in personality. Other studies of twins 67 and siblings 68 discordant for Parkinson’s disease replicate this finding, suggesting that the inverse trend between smoking and Parkinson’s disease is unlikely to result from a genetic factor determining both smoking and Parkinson’s disease risk.
In summary, the inverse relationship between Parkinson’s disease and smoking appears consistent with a true biological protective effect of tobacco use, based on findings that: (1) it is observed in large prospective cohort studies; (2) is stronger in current compared to former smokers; (3) is correlated with smoking duration and intensity; (4) is seen with other forms of tobacco use; and (5) is observed in monozygotic twins discordant for Parkinson’s disease. However, because tobacco products contain thousands of compounds, the question arises as to which of these confers the apparent protection against Parkinson’s disease. A large body of evidence derived from experimental animal models suggests that nicotine, the primary psychoactive component in tobacco, plays a key role in the observed protection from nigrostriatal damage as discussed below.
(2) Nicotine treatment protects against nigrostriatal damage
Numerous studies using culture models have shown that nicotine reduces toxicity in striatal, nigral, cortical, hippocampal, cerebellar, and spinal cord neurons, as well as in neuroblastoma and phaeochromocytoma (PC12) cell lines. Protection was observed against glutamate excitotoxicity, ischemic damage, ethanol-induced toxicity, nerve growth factor deprivation, MPTP exposure and other forms of insult 18, 69-71.
Importantly, in vivo studies also show that nicotine pretreatment attenuates damage induced by dopaminergic neurotoxins such as MPTP, 6-hydroxydopamine, methamphetamine and paraquat (Table 2). Nicotine administered prior to lesioning consistently reduced nigrostriatal degeneration in 6-hydroxydopamine lesioned rats with the extent of protection dependent on the dose of nicotine and severity of nigrostriatal damage 72-77. Nicotine pretreatment also protected against MPTP-induced dopaminergic loss in the mouse nigrostriatal system 76, 78-85. However, results have proved somewhat inconsistent in this model possibly because of the large, acute lesions that develop in mice administered MPTP or because nicotine is very rapidly metabolized in this species 77, 86-90. By contrast, chronic nicotine treatment did protect against a slow MPTP-induced neurodegenerative lesion in monkeys. There were enhanced levels of tyrosine hydroxylase, the dopamine transporter, vesicular monoamine transporter and dopamine in striatum of lesioned animals receiving nicotine compared to non-treated lesioned monkeys 91, 92. Nicotine exposure not only improved morphological measures related to the dopaminergic system but also normalized lesion-induced overactivity of the nigrostriatal pathway in monkeys and preserved synaptic plasticity lost with nigrostriatal damage 93. Continued studies in the rat and nonhuman primate model should help identify the nicotinic receptor subtypes through which this effect occurs, as well as the mechanisms of nicotine-mediated neuroprotection. It will also be important to test whether nicotine protects against nigrostriatal damage when administered after the lesion, as this would more closely resemble the clinical situation.
TABLE 2.
Nicotine treatment protects against nigrostriatal damage in parkinsonian animal models
| Animal model | Protection | Type of Lesion | Treatment | References |
|---|---|---|---|---|
| Rat | Striatum and substantia nigra | Hemitransection | Nicotine injection and minipump | (Janson et al., 1988) |
| Striatum | 6-OHDA | Nicotine minipump | (Ryan et al., 2001) | |
| Nicotine injection | (Costa et al., 2001; Abin-Carriquiry et al., 2002; Soto-Otero et al., 2002; Visanji et al., 2006) | |||
| Monkey | Striatum | MPTP | Nicotine in drinking water | (Quik et al., 2006a; Quik et al., 2006b) |
| Mouse | Striatum and substantia nigra | MPTP | Nicotine injection and minipump | (Janson et al., 1991) |
| Nicotine injection | (Janson et al., 1992) | |||
| Paraquat | Nicotine in drinking water | (Khwaja et al., 2006) | ||
| Striatum | MPTP | Smoke | (Carr and Rowell, 1990; Shahi et al., 1991) | |
| Nicotine injection | (Gao et al., 1998) | |||
| Methamphetamine | Nicotine injection | (Ryan et al., 2001) | ||
| Substantia nigra | MPTP | Smoke or nicotine injection | (Parain et al., 2001; Parain et al., 2003) | |
| No protection in striatum and/or substantia nigra | MPTP | Smoke | (Perry et al., 1987) | |
| Nicotine in drinking water | (Sershen et al., 1988) | |||
| Nicotine minipumps | (Behmand and Harik, 1992; Janson et al., 1992) | |||
| Nicotine injection | (Hadjiconstantinou et al., 1994; Ferger et al., 1998) |
All reported studies indicate that nicotine treatment protects against nigrostriatal damage using either the rat or monkey model. However, it should be noted that level of protection was dependent on the lesioning condition (severity) and nicotine treatment (timing and dose). Protection was observed with both intermittent and continuous nicotine dosing. In mice, however, nicotine attenuated nigrostriatal damage in some but not all studies. Moreover, protection was inconsistent with no clear relationship between protective effects and the nicotine and/or MPTP-lesioning protocols. In most of the studies nicotine was given a few hours or days prior to lesioning, and in one study at the time of lesioning. The outcome measures used to evaluate protection in the striatum include tyrosine hydroxylase, dopamine and/or dopamine transporter levels, while tyrosine hydroxylase positive cells were counted as a measure of protection in the substantia nigra.
A variety of different experimental approaches suggests that nicotine exerts its protective effect by acting at nicotinic receptors. The use of nicotinic receptor knockout mice and/or selective nicotinic receptor antagonists indicate that the α6β2* and α4β2* subtypes are most likely involved 73, 76. Stimulation of these receptors may lead to a protective activation via an increase in trophic factors and/or immune modulators 94-96. There is also some evidence that nicotine may act through non-receptor mediated mechanisms by reducing oxidative stress and/or enhancing mitochondrial function 87, 97.
Collectively, these data in culture systems and parkinsonian animal models have yielded a better understanding of the role of nicotine in neuroprotection against nigrostriatal damage. These results provide a firm basis for the suggestion that nicotine contributes to the lower incidence of Parkinson’s disease with tobacco use. Work remains to be done to determine whether nicotine is also protective when administered during nigrostriatal damage.
Nicotine and symptomatic improvement of Parkinson’s disease
Since nicotine can stimulate dopamine release in the striatum, an important question that arises is whether it can also improve motor symptoms associated with Parkinson’s disease. There is precedence for this possibility from rodent and nonhuman primate studies, which demonstrate that nicotinic receptor activation modulates motor function under normal physiological conditions 23. In addition, nicotine or nicotinic agonists given to parkinsonian rats or monkeys improved parkinsonian behaviors 98-100.
Several small studies have also evaluated the effect of nicotine on motor symptoms in Parkinson’s disease patients (Table 3). One of the first reports investigated the effect of smoking combined with the nicotine gum in 6 individuals with Parkinson’s disease; a transient beneficial response was observed on motor function 101. A single more recent case report also indicated that smoking transiently reduced symptoms in a patient with juvenile Parkinson’s disease 102. Attenuation of symptoms was also observed in two Parkinson’s disease patients receiving the nicotine gum and patch for variable periods over several months 103. These reports were followed by several studies that investigated the effect of acute nicotine exposure (1 day) with the nicotine patch or gum. Results were conflicting, with a worsening of motor performance in two of the studies 104, 105, and improvement in one 106. Small-scale clinical trials have also yielded variable outcomes. Improvements in cognitive measures and motor performance were obtained in a group of 15 Parkinson’s disease patients after treatment with intravenous nicotine and the nicotine patch, with beneficial effects persisting up to 1 month after nicotine was stopped 107. However in another trial involving 32 patients, positive effects were not observed with 3 weeks of patch application 108. It should be noted however that symptoms were only evaluated 3 weeks after patch cessation, at which time any improvements may have dissipated. In line with these results, however, is the study by Lemay and coworkers in which there were no improvement in motor or cognitive deficits with immediate testing of Parkinson’s disease symptoms after 3 to 4 weeks of treatment 109. A possible explanation for the lack of effect of nicotine in these studies may relate to inadequate dosing as Villafane et al recently showed that high dose nicotine resulted in a dramatic improvement in parkinsonism in a small open label study 110. An α4β2* CNS selective nicotinic agonist (SIB-1508Y) was also tested in one study; however, there was no observable effect on parkinsonism after 4 weeks of treatment 111. These latter data may suggest that other nicotinic receptor subtypes (α6β2* and α7) mediate effects against parkinsonism, that stimulation of multiple subtypes are required for an enhancement of motor function and/or may relate to pharmacokinetic issues with this specific drug. Further work with additional subtype selective nicotinic receptor drugs are required to understand the subtypes through which nicotine mediates its effects on motor control.
TABLE 3.
#. Summary of the effectiveness of nicotine or a nicotinic agonist in Parkinson’s disease patients
| Study | Test agent | Type of study | # patients | Outcome measure relating to movement | Duration | Final dose/day | Anti-parkinsonian effect | |
|---|---|---|---|---|---|---|---|---|
| Dose titration | Dose maintenance | |||||||
| Ishikawa and Miyatake, 1993 | Smoking and nicotine gum | Open-label | 6 | Tremor, rigidity, bradykinesia, posture | Chronic smoker | NA | Yes | |
| Fagerstrom et al., 1994 | Nicotine gum and patch | Double-blinded | 2 | Tremor, rigidity | ≥ 7 mo | 15 mg patch + 4×4 mg gum | Yes | |
| Clemens et al., 1995 | Nicotine gum | Double-blinded placebo-controlled | 48 | UPDRS | ND | 1 day | 3×2 mg | No |
| Ebersbach et al., 1999 | Nicotine patch | Double-blinded crossover | 16 | UPDRS | ND | 12 hours | 7 mg | No |
| Kelton et al., 2000 | Nicotine iv and patch | Open-label | 15 | Pronation/supination, finger dexterity, stand/walk/sit | 2 wk | 1-2 wk | 14 mg | Yes |
| Vieregge et al., 2001 | Nicotine patch | Double-blinded placebo-controlled | 32 | Columbia Univ rating scale, Schwab-England Fine motor testing | 1 wk | 2 wk | 14 mg | No |
| Mitsuoka et al., 2002 | Nicotine gum | Open-label | 8 | UPDRS | ND | 1 day | NA | Yes |
| Lemay et al., 2004 | Nicotine patch | Open-label | 22 | UPDRS | 22 days | 3 days | 21 mg | No |
| Shoulson et al., 2006 | SIB-1508Y | Double-blinded placebo controlled | 77 | UPDRS | 2 wk | 2 wk | 10 mg | No |
| Hanagasi et al., 2007 | Smoking | Open-label | 1 | UPDRS, Hoehn and Yahr | Chronic smoker | NA | Yes | |
| Villafane et al., 2007 | Nicotine patch | Open-label | 6 | UPDRS | 14 wk | 4 wk | Up to 105 mg | Yes |
Taken in modified form with permission from Quik et al., 2007b. In some of the above studies nicotine treatment appeared well tolerated with no side effects noted 101, 103, 107, whereas in others side effects were noted although similar side effects were often also observed with placebo 104, 105, 108. Reported side effects in the nicotine-treated groups include, itchiness at the site of the patch, dizziness, lightheadedness, headache, vivid dreaming, deterioration of tremor and balance, akinesia, sleep disturbances, nausea, vomiting, hypersalivation, dry mouth, hypertension, palpitations, and intestinal cramps 104, 105, 108, 136. Side effects in the placebo groups were similar and include itchiness at the site of the patch, dizziness, lightheadedness, fatigue, headache, vivid dreaming, deterioration of balance, tremor and gait, sleep disturbances, nausea, dry mouth and orthostatis 104, 105, 108. Since tolerance develops with continued nicotine use as readily evident with smoking, a more gradual dose escalation may circumvent the development of side effects.
NA - not available; ND – not determined.
In summary, beneficial effects of nicotine treatment were reported in about half the clinical studies (Table 3). These results may suggest that nicotine has the potential to ameliorate symptoms but that the conditions required for improvement remain to be clarified. One notable observation is that almost all of the double-blinded trials yielded negative results, while the open-label studies were positive, suggesting a placebo effect. However, it is also possible that the mode of administration of nicotine (patch versus gum), nicotine dose, the dosing regime or duration of nicotine treatment require optimization. The role of nicotine to reduce Parkinson’s disease symptoms thus currently remains unclear.
Nicotine reduces L-dopa-induced dyskinesias
As mentioned earlier, L-dopa often has a dramatic impact on motor deficits that arise with Parkinson’s disease and is one of the most effective therapies. However, chronic treatment is associated with significant side effects including on-off phenomena, neuropsychiatric problems and the development of abnormal involuntary movements or dyskinesias that may be very disabling. The development of dyskinesias may counterbalance L-dopa’s beneficial effects and adversely affect the quality of life of Parkinson’s disease patients 15, 54, 112-115,112, 113, 116-121. Current preventive strategies involve initial use of dopamine agonists, which induce dyskinesias much less readily. However, almost all patients require eventual use of L-dopa, which is then followed by dyskinesias in the majority of cases. Once present, dyskinesias are not easily eliminated. For most, pharmacological and surgical interventions result in only a partial decrease in dyskinesias, often at the expense of increased parkinsonian symptoms or other side effects. Better treatment is therefore urgently needed.
Novel drugs targeted to neurotransmitter and/or neuromodulatory systems linked to basal ganglia function are currently being tested for their ability to attenuate L-dopa-induced dyskinesias 118-120, 122-126. This includes nicotine, which was recently shown to reduce dyskinesias in parkinsonian nonhuman primates (Fig. 1)127. This effect of nicotine was most pronounced (50% decrease) in monkeys that were given nicotine before L-dopa treatment was initiated (L-dopa naïve). Dyskinesias were decreased to a somewhat lesser extent (~35%) in monkeys which first received L-dopa (L-dopa-primed animals) and then received nicotine together with L-dopa 127. This difference in effectiveness is consistent with findings that dyskinesias are generally more readily suppressed in L-dopa naïve versus L-dopa-primed animals 118, 119. Importantly, nicotine treatment did not worsen Parkinsonism either on or off L-dopa. Continued studies should help determine whether nicotine treatment may be useful for alleviating this debilitating consequence of L-dopa therapy in Parkinson’s disease patients. Subsequent work with selective nicotinic receptor agonists could then be initiated to determine which receptor subtypes are involved.
FIG. 1.
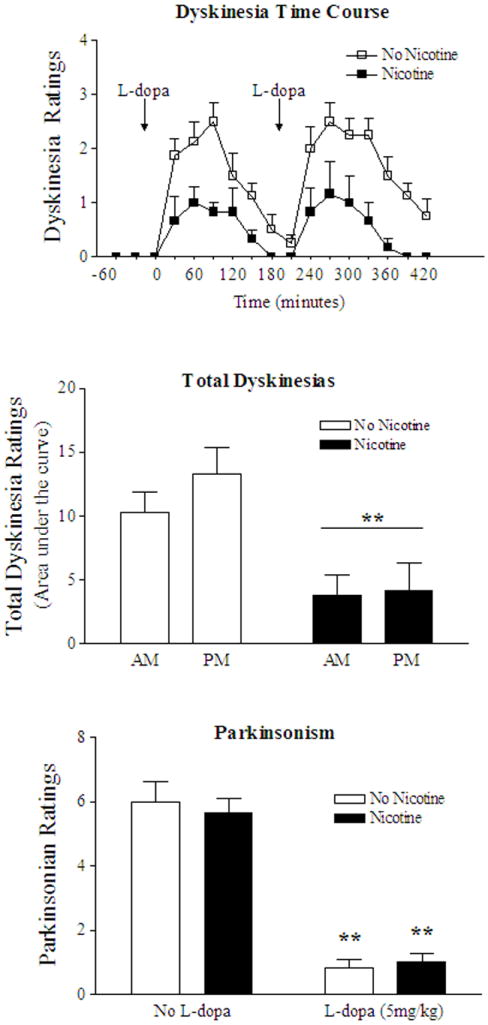
Nicotine treatment reduces L-dopa-induced dyskinesias in nonhuman primates 127. Time course of the nicotine-induced decline in levodopa-induced dyskinesias (top panel). MPTP-lesioned monkeys were given either nicotine or vehicle for several weeks and then gavaged with levodopa (L-dopa, 5 mg/kg) twice daily. Each symbol is the mean ± SE of 3 to 4 monkeys. Dyskinesias were significantly reduced in nicotine-treated animals compared to monkeys not receiving nicotine (p < 0.01) using a Mann-Whitney test. Total dyskinesias were significantly decreased by nicotine treatment after both the morning and afternoon dose of L-dopa (middle panel). **p < 0.01 depicts a main effect of nicotine by two-way ANOVA. Nicotine treatment did not affect parkinsonism either on or off L-dopa (lower panel). **p < 0.01 as compared to the same group with no levodopa treatment by a Mann-Whitney test.
NICOTINE TREATMENT REGIMENS
The preceding discussion suggests that nicotine or nicotine receptor ligands may be of therapeutic use in the management of Parkinson’s disease as neuroprotectants against continued nigrostriatal degeneration, for the treatment of Parkinson’s disease symptoms or to attenuate L-dopa-induced side effects. An important question that now arises is what is the optimal nicotinic treatment regimen with respect to dosing, mode of delivery and duration of administration. Numerous approaches are currently used in animal models including intermittent exposure via injection or self-administration, as well as continuous treatments via minipump or infusion 128. This distinction is important because continuous and intermittent administration may have significantly different impacts on nicotinic receptor function. Continuous administration yields steady nicotine plasma levels and most likely results in receptor desensitization. By contrast, intermittent or pulsatile treatment leads to a cycle of receptor activation, desensitization, and resensitization, with a potentially different repertoire of functional effects 50, 129-131. A multitude of nicotine formulations are currently available for use in humans as therapy for smoking cessation 128, which could potentially be used for Parkinson’s disease treatment. These include continuous dosing via the nicotine patch, or intermittent administration using nicotine gum, lozenge, sublingual pill, inhalant and spray. The specific manner in which nicotine is administered (pulsatile versus constant), the duration of treatment and the dose of nicotine are very important questions for appropriate management of Parkinson’s disease. Both pre-clinical studies and double-blinded placebo-controlled or crossover clinical trials will be required to address these issues. Parameters may differ for its use for neuroprotection or symptomatic treatment. Administration of nicotine via smoking would, of course, not be recommended because of the multitude of detrimental health-related effects associated with smoking.
Another very important point for consideration is that nicotine stimulates all nicotinic receptors in both the peripheral and central nervous systems. Its use may therefore be associated with side effects, in addition to its desired action. These may include alterations in cardiovascular measures (blood pressure, heart rate), the gastrointestinal system (cramping, nausea, vomiting), exocrine glands (dry mouth), as well as CNS effects including dizziness, headache and light-headedness. On the other hand, tolerance is well known to develop to systemic effects of nicotine, as evidenced by chronic smoking. However, the use of agonists that selectively target CNS nicotinic receptors, or better still the nigrostriatal system, such as the α6β2* and α4β2* receptor populations, would most likely be more beneficial from a therapeutic standpoint, with fewer peripherally-mediated side effects and greater specificity of central action. The use of selective nicotinic agonists that target the nicotinic receptor subtypes involved in motor control may also obviate potential problems with addiction 132-135. On the other hand, the detrimental effects of smoking do not appear to relate to the presence of nicotine but rather to the effects of a multitude of other toxic components in tobacco products.
Another important therapeutic consideration relates to the fact that most Parkinson’s disease patients are already treated with numerous medications anyone of which may interact with nicotine or nicotinic agonists and affect metabolism, metabolic pathways, the cardiovascular and other systems. However, as mentioned earlier, there is a large literature showing that Parkinson’s disease incidence is reduced with smoking, a behavior associated with a substantial nicotine intake. In addition, clinical trials have been done in Parkinson’s disease patients receiving nicotine (see Table 3). These observations suggest that nicotine treatment is most likely compatible with current Parkinson’s disease therapies.
CONCLUSION
These combined data indicate that nicotine or nicotinic agonists may be useful for Parkinson’s disease therapy from several perspectives, including neuroprotection against nigrostriatal damage, alleviation of symptoms and reduction of drug-induced side effects (L-dopa-induced dyskinesias). The use of such agents, in combination with existing treatments, may represent a new disease modifying approach for Parkinson’s disease.
Acknowledgments
This work was supported by National Institute of Health Grant NIH NS34886, NS42091, NS40467 and ES012077
References
- 1.Samii A, Nutt JG, Ransom BR. Parkinson’s disease. Lancet. 2004;363:1783–1793. doi: 10.1016/S0140-6736(04)16305-8. [DOI] [PubMed] [Google Scholar]
- 2.Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006;116:1744–1754. doi: 10.1172/JCI29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lang AE, Obeso JA. Challenges in Parkinson’s disease: restoration of the nigrostriatal dopamine system is not enough. Lancet Neurol. 2004;3:309–316. doi: 10.1016/S1474-4422(04)00740-9. [DOI] [PubMed] [Google Scholar]
- 4.Singh N, Pillay V, Choonara YE. Advances in the treatment of Parkinson’s disease. Prog Neurobiol. 2007;81:29–44. doi: 10.1016/j.pneurobio.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Hodaie M, Neimat JS, Lozano AM. The dopaminergic nigrostriatal system and Parkinson’s disease: molecular events in development, disease, and cell death, and new therapeutic strategies. Neurosurgery. 2007;60:17–28. doi: 10.1227/01.NEU.0000249209.11967.CB. discussion 28-30. [DOI] [PubMed] [Google Scholar]
- 6.Zhou FM, Wilson CJ, Dani JA. Cholinergic interneuron characteristics and nicotinic properties in the striatum. J Neurobiol. 2002;53:590–605. doi: 10.1002/neu.10150. [DOI] [PubMed] [Google Scholar]
- 7.Fuxe K, Hoekfelt T, Nilsson O. Observations on the Cellular Localization of Dopamine in the Caudate Nucleus of the Rat. Z Zellforsch Mikrosk Anat. 1964;63:701–706. doi: 10.1007/BF00339917. [DOI] [PubMed] [Google Scholar]
- 8.Dahlstroem A, Fuxe K, Olson L, Ungerstedt U. Ascending Systems of Catecholamine Neurons from the Lower Brain Stem. Acta Physiol Scand. 1964;62:485–486. doi: 10.1111/j.1748-1716.1964.tb10446.x. [DOI] [PubMed] [Google Scholar]
- 9.Nagatsu T, Levitt M, Udenfriend S. Tyrosine Hydroxylase. The Initial Step in Norepinephrine Biosynthesis. J Biol Chem. 1964;239:2910–2917. [PubMed] [Google Scholar]
- 10.Wonnacott S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- 11.MacDermott AB, Role LW, Siegelbaum SA. Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci. 1999;22:443–485. doi: 10.1146/annurev.neuro.22.1.443. [DOI] [PubMed] [Google Scholar]
- 12.Fonnum F. Recent developments in biochemical investigations of cholinergic transmission. Brain Res. 1973;62:497–507. doi: 10.1016/0006-8993(73)90714-2. [DOI] [PubMed] [Google Scholar]
- 13.Potter LT, Glover VA, Saelens JK. Choline acetyltransferase from rat brain. J Biol Chem. 1968;243:3864–3870. [PubMed] [Google Scholar]
- 14.Schwartz RD, McGee R, Jr, Kellar KJ. Nicotinic cholinergic receptors labeled by [3H]acetylcholine in rat brain. Mol Pharmacol. 1982;22:56–62. [PubMed] [Google Scholar]
- 15.Schapira AH, Bezard E, Brotchie J, et al. Novel pharmacological targets for the treatment of Parkinson’s disease. Nat Rev Drug Discov. 2006;5:845–854. doi: 10.1038/nrd2087. [DOI] [PubMed] [Google Scholar]
- 16.Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci. 2006;27:482–491. doi: 10.1016/j.tips.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 17.Court JA, Martin-Ruiz C, Graham A, Perry E. Nicotinic receptors in human brain: topography and pathology. J Chem Neuroanat. 2000;20:281–298. doi: 10.1016/s0891-0618(00)00110-1. [DOI] [PubMed] [Google Scholar]
- 18.Quik M. Smoking, nicotine and Parkinson’s disease. Trends Neurosci. 2004;27:561–568. doi: 10.1016/j.tins.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Dani JA, Bertrand D. Nicotinic Acetylcholine Receptors and Nicotinic Cholinergic Mechanisms of the Central Nervous System. Annu Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- 20.Salminen O, Murphy KL, McIntosh JM, et al. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol. 2004;65:1526–1535. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- 21.Kaiser S, Wonnacott S. alpha-bungarotoxin-sensitive nicotinic receptors indirectly modulate [(3)H]dopamine release in rat striatal slices via glutamate release. Mol Pharmacol. 2000;58:312–318. doi: 10.1124/mol.58.2.312. [DOI] [PubMed] [Google Scholar]
- 22.Quik M, McIntosh JM. Striatal alpha6* nicotinic acetylcholine receptors: potential targets for Parkinson’s disease therapy. J Pharmacol Exp Ther. 2006;316:481–489. doi: 10.1124/jpet.105.094375. [DOI] [PubMed] [Google Scholar]
- 23.Balfour DJ, Fagerstrom KO. Pharmacology of nicotine and its therapeutic use in smoking cessation and neurodegenerative disorders. Pharmacol Ther. 1996;72:51–81. doi: 10.1016/s0163-7258(96)00099-x. [DOI] [PubMed] [Google Scholar]
- 24.Wada E, Wada K, Boulter J, et al. Distribution of alpha 2, alpha 3, alpha 4, and beta 2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Comp Neurol. 1989;284:314–335. doi: 10.1002/cne.902840212. [DOI] [PubMed] [Google Scholar]
- 25.Zoli M, Moretti M, Zanardi A, et al. Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci. 2002;22:8785–8789. doi: 10.1523/JNEUROSCI.22-20-08785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Champtiaux N, Gotti C, Cordero-Erausquin M, et al. Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci. 2003;23:7820–7829. doi: 10.1523/JNEUROSCI.23-21-07820.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Champtiaux N, Han ZY, Bessis A, et al. Distribution and pharmacology of alpha 6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J Neurosci. 2002;22:1208–1217. doi: 10.1523/JNEUROSCI.22-04-01208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cui C, Booker TK, Allen RS, et al. The beta3 nicotinic receptor subunit: a component of alpha-conotoxin MII-binding nicotinic acetylcholine receptors that modulate dopamine release and related behaviors. J Neurosci. 2003;23:11045–11053. doi: 10.1523/JNEUROSCI.23-35-11045.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gotti C, Moretti M, Clementi F, et al. Expression of nigrostriatal alpha 6-containing nicotinic acetylcholine receptors is selectively reduced, but not eliminated, by beta 3 subunit gene deletion. Mol Pharmacol. 2005;67:2007–2015. doi: 10.1124/mol.105.011940. [DOI] [PubMed] [Google Scholar]
- 30.Salminen O, Whiteaker P, Grady SR, et al. The subunit composition and pharmacology of alpha-Conotoxin MII-binding nicotinic acetylcholine receptors studied by a novel membrane-binding assay. Neuropharmacology. 2005;48:696–705. doi: 10.1016/j.neuropharm.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 31.Quik M, Polonskaya Y, Kulak JM, McIntosh JM. Vulnerability of 125I-alpha-conotoxin MII binding sites to nigrostriatal damage in monkey. J Neurosci. 2001;21:5494–5500. doi: 10.1523/JNEUROSCI.21-15-05494.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quik M, Vailati S, Bordia T, et al. Subunit composition of nicotinic receptors in monkey striatum: effect of treatments with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine or L-DOPA. Mol Pharmacol. 2005;67:32–41. doi: 10.1124/mol.104.006015. [DOI] [PubMed] [Google Scholar]
- 33.Han ZY, Zoli M, Cardona A, et al. Localization of [3H]nicotine, [3H]cytisine, [3H]epibatidine, and [125I]alpha-bungarotoxin binding sites in the brain of Macaca mulatta. J Comp Neurol. 2003;461:49–60. doi: 10.1002/cne.10659. [DOI] [PubMed] [Google Scholar]
- 34.Le Novere N, Zoli M, Changeux JP. Neuronal nicotinic receptor alpha 6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci. 1996;8:2428–2439. doi: 10.1111/j.1460-9568.1996.tb01206.x. [DOI] [PubMed] [Google Scholar]
- 35.Whiteaker P, McIntosh JM, Luo S, et al. 125I-alpha-conotoxin MII identifies a novel nicotinic acetylcholine receptor population in mouse brain. Mol Pharmacol. 2000;57:913–925. [PubMed] [Google Scholar]
- 36.Quik M, Polonskaya Y, Gillespie A, et al. Localization of nicotinic receptor subunit mRNAs in monkey brain by in situ hybridization. J Comp Neurol. 2000;425:58–69. doi: 10.1002/1096-9861(20000911)425:1<58::aid-cne6>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 37.Bordia T, Grady SR, McIntosh JM, Quik M. Nigrostriatal damage preferentially decreases a subpopulation of {alpha}6{beta}2* nAChRs in mouse, monkey and Parkinson’s disease striatum. Mol Pharmacol. 2007;72:52–61. doi: 10.1124/mol.107.035998. [DOI] [PubMed] [Google Scholar]
- 38.Gotti C, Moretti M, Bohr I, et al. Selective nicotinic acetylcholine receptor subunit deficits identified in Alzheimer’s disease, Parkinson’s disease and dementia with Lewy bodies by immunoprecipitation. Neurobiol Dis. 2006;23:481–489. doi: 10.1016/j.nbd.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 39.Marchi M, Risso F, Viola C, et al. Direct evidence that release-stimulating alpha7* nicotinic cholinergic receptors are localized on human and rat brain glutamatergic axon terminals. J Neurochem. 2002;80:1071–1078. doi: 10.1046/j.0022-3042.2002.00805.x. [DOI] [PubMed] [Google Scholar]
- 40.McCallum SE, Parameswaran N, Bordia T, et al. Decrease in alpha3*/alpha6* nicotinic receptors but not nicotine-evoked dopamine release in monkey brain after nigrostriatal damage. Mol Pharmacol. 2005;68:737–746. doi: 10.1124/mol.105.012773. [DOI] [PubMed] [Google Scholar]
- 41.Quik M, Sum JD, Whiteaker P, et al. Differential declines in striatal nicotinic receptor subtype function after nigrostriatal damage in mice. Mol Pharmacol. 2003;63:1169–1179. doi: 10.1124/mol.63.5.1169. [DOI] [PubMed] [Google Scholar]
- 42.Kulak JM, McIntosh JM, Quik M. Loss of nicotinic receptors in monkey striatum after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine treatment is due to a decline in alpha-conotoxin MII sites. Mol Pharmacol. 2002;61:230–238. doi: 10.1124/mol.61.1.230. [DOI] [PubMed] [Google Scholar]
- 43.Schwartz RD, Lehmann J, Kellar KJ. Presynaptic nicotinic cholinergic receptors labeled by [3H]acetylcholine on catecholamine and serotonin axons in brain. J Neurochem. 1984;42:1495–1498. doi: 10.1111/j.1471-4159.1984.tb02818.x. [DOI] [PubMed] [Google Scholar]
- 44.Clarke PB, Pert A. Autoradiographic evidence for nicotine receptors on nigrostriatal and mesolimbic dopaminergic neurons. Brain Res. 1985;348:355–358. doi: 10.1016/0006-8993(85)90456-1. [DOI] [PubMed] [Google Scholar]
- 45.Quik M, Bordia T, Forno L, McIntosh JM. Loss of alpha-conotoxinMII- and A85380-sensitive nicotinic receptors in Parkinson’s disease striatum. J Neurochem. 2004;88:668–679. doi: 10.1111/j.1471-4159.2004.02177.x. [DOI] [PubMed] [Google Scholar]
- 46.Rinne JO, Myllykyla T, Lonnberg P, Marjamaki P. A postmortem study of brain nicotinic receptors in Parkinson’s and Alzheimer’s disease. Brain Res. 1991;547:167–170. doi: 10.1016/0006-8993(91)90588-m. [DOI] [PubMed] [Google Scholar]
- 47.Aubert I, Araujo DM, Cecyre D, et al. Comparative alterations of nicotinic and muscarinic binding sites in Alzheimer’s and Parkinson’s diseases. J Neurochem. 1992;58:529–541. doi: 10.1111/j.1471-4159.1992.tb09752.x. [DOI] [PubMed] [Google Scholar]
- 48.Perry EK, Morris CM, Court JA, et al. Alteration in nicotine binding sites in Parkinson’s disease, Lewy body dementia and Alzheimer’s disease: possible index of early neuropathology. Neuroscience. 1995;64:385–395. doi: 10.1016/0306-4522(94)00410-7. [DOI] [PubMed] [Google Scholar]
- 49.Bohr IJ, Ray MA, McIntosh JM, et al. Cholinergic nicotinic receptor involvement in movement disorders associated with Lewy body diseases. An autoradiography study using [(125)I]alpha-conotoxinMII in the striatum and thalamus. Exp Neurol. 2005;191:292–300. doi: 10.1016/j.expneurol.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 50.Gentry CL, Lukas RJ. Regulation of nicotinic acetylcholine receptor numbers and function by chronic nicotine exposure. Curr Drug Targets CNS Neurol Disord. 2002;1:359–385. doi: 10.2174/1568007023339184. [DOI] [PubMed] [Google Scholar]
- 51.Olanow CW. The scientific basis for the current treatment of Parkinson’s disease. Annu Rev Med. 2004;55:41–60. doi: 10.1146/annurev.med.55.091902.104422. [DOI] [PubMed] [Google Scholar]
- 52.LeWitt PA. Neuroprotection for Parkinson’s disease. J Neural Transm Suppl. 2006:113–122. [PubMed] [Google Scholar]
- 53.Ravina BM, Fagan SC, Hart RG, et al. Neuroprotective agents for clinical trials in Parkinson’s disease: A systematic assessment. Neurology. 2003;60:1234–1240. doi: 10.1212/01.wnl.0000058760.13152.1a. [DOI] [PubMed] [Google Scholar]
- 54.Olanow CW, Jankovic J. Neuroprotective therapy in Parkinson’s disease and motor complications: a search for a pathogenesis-targeted, disease-modifying strategy. Mov Disord. 2005;20(Suppl 11):S3–10. doi: 10.1002/mds.20457. [DOI] [PubMed] [Google Scholar]
- 55.Ritz B, Ascherio A, Checkoway H, et al. Pooled analysis of tobacco use and risk of Parkinson disease. Arch Neurol. 2007;64:990–997. doi: 10.1001/archneur.64.7.990. [DOI] [PubMed] [Google Scholar]
- 56.Morens DM, Grandinetti A, Reed D, et al. Cigarette smoking and protection from Parkinson’s disease: false association or etiologic clue? Neurology. 1995;45:1041–1051. doi: 10.1212/wnl.45.6.1041. [DOI] [PubMed] [Google Scholar]
- 57.Allam MF, Campbell MJ, Hofman A, et al. Smoking and Parkinson’s disease: systematic review of prospective studies. Mov Disord. 2004;19:614–621. doi: 10.1002/mds.20029. [DOI] [PubMed] [Google Scholar]
- 58.Gorell JM, Peterson EL, Rybicki BA, Johnson CC. Multiple risk factors for Parkinson’s disease. J Neurol Sci. 2004;217:169–174. doi: 10.1016/j.jns.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 59.Hernan MA, Takkouche B, Caamano-Isorna F, Gestal-Otero JJ. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann Neurol. 2002;52:276–284. doi: 10.1002/ana.10277. [DOI] [PubMed] [Google Scholar]
- 60.Ross GW, Petrovitch H. Current evidence for neuroprotective effects of nicotine and caffeine against Parkinson’s disease. Drugs Aging. 2001;18:797–806. doi: 10.2165/00002512-200118110-00001. [DOI] [PubMed] [Google Scholar]
- 61.Thacker EL, O’Reilly EJ, Weisskopf MG, et al. Temporal relationship between cigarette smoking and risk of Parkinson disease. Neurology. 2007;68:764–768. doi: 10.1212/01.wnl.0000256374.50227.4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benedetti MD, Bower JH, Maraganore DM, et al. Smoking, alcohol, and coffee consumption preceding Parkinson’s disease: a case-control study. Neurology. 2000;55:1350–1358. doi: 10.1212/wnl.55.9.1350. [DOI] [PubMed] [Google Scholar]
- 63.O’Reilly EJ, McCullough ML, Chao A, et al. Smokeless tobacco use and the risk of Parkinson’s disease mortality. Mov Disord. 2005;20:1383–1384. doi: 10.1002/mds.20587. [DOI] [PubMed] [Google Scholar]
- 64.Ellenberg JH. Differential postmorbidity mortality in observational studies of risk factors for neurologic disorders. Neuroepidemiology. 1994;13:187–194. doi: 10.1159/000110378. [DOI] [PubMed] [Google Scholar]
- 65.Menza M. The personality associated with Parkinson’s disease. Curr Psychiatry Rep. 2000;2:421–426. doi: 10.1007/s11920-000-0027-1. [DOI] [PubMed] [Google Scholar]
- 66.Tanner CM, Goldman SM, Aston DA, et al. Smoking and Parkinson’s disease in twins. Neurology. 2002;58:581–588. doi: 10.1212/wnl.58.4.581. [DOI] [PubMed] [Google Scholar]
- 67.Bharucha NE, Stokes L, Schoenberg BS, et al. A case-control study of twin pairs discordant for Parkinson’s disease: a search for environmental risk factors. Neurology. 1986;36:284–288. doi: 10.1212/wnl.36.2.284. [DOI] [PubMed] [Google Scholar]
- 68.Scott WK, Zhang F, Stajich JM, et al. Family-based case-control study of cigarette smoking and Parkinson disease. Neurology. 2005;64:442–447. doi: 10.1212/01.WNL.0000150905.93241.B2. [DOI] [PubMed] [Google Scholar]
- 69.Quik M, O’Neill M, Perez XA. Nicotine neuroprotection against nigrostriatal damage: importance of the animal model. Trends Pharmacol Sci. 2007 doi: 10.1016/j.tips.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 70.O’Neill MJ, Murray TK, Lakics V, et al. The role of neuronal nicotinic acetylcholine receptors in acute and chronic neurodegeneration. Curr Drug Target CNS Neurol Disord. 2002;1:399–411. doi: 10.2174/1568007023339166. [DOI] [PubMed] [Google Scholar]
- 71.Belluardo N, Mudo G, Blum M, Fuxe K. Central nicotinic receptors, neurotrophic factors and neuroprotection. Behav Brain Res. 2000;113:21–34. doi: 10.1016/s0166-4328(00)00197-2. [DOI] [PubMed] [Google Scholar]
- 72.Abin-Carriquiry JA, McGregor-Armas R, Costa G, et al. Presynaptic involvement in the nicotine prevention of the dopamine loss provoked by 6-OHDA administration in the substantia nigra. Neurotox Res. 2002;4:133–139. doi: 10.1080/10298420290015863. [DOI] [PubMed] [Google Scholar]
- 73.Costa G, Abin-Carriquiry JA, Dajas F. Nicotine prevents striatal dopamine loss produced by 6-hydroxydopamine lesion in the substantia nigra. Brain Res. 2001;888:336–342. doi: 10.1016/s0006-8993(00)03087-0. [DOI] [PubMed] [Google Scholar]
- 74.Soto-Otero R, Mendez-Alvarez E, Hermida-Ameijeiras A, et al. Effects of (-)-nicotine and (-)-cotinine on 6-hydroxydopamine-induced oxidative stress and neurotoxicity: relevance for Parkinson’s disease. Biochem Pharmacol. 2002;64:125–135. doi: 10.1016/s0006-2952(02)01070-5. [DOI] [PubMed] [Google Scholar]
- 75.Visanji NP, O’Neill MJ, Duty S. Nicotine, but neither the alpha4beta2 ligand RJR2403 nor an alpha7 nAChR subtype selective agonist, protects against a partial 6-hydroxydopamine lesion of the rat median forebrain bundle. Neuropharmacology. 2006;51:506–516. doi: 10.1016/j.neuropharm.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 76.Ryan RE, Ross SA, Drago J, Loiacono RE. Dose-related neuroprotective effects of chronic nicotine in 6-hydroxydopamine treated rats, and loss of neuroprotection in alpha4 nicotinic receptor subunit knockout mice. Br J Pharmacol. 2001;132:1650–1656. doi: 10.1038/sj.bjp.0703989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Janson AM, Fuxe K, Agnati LF, et al. Chronic nicotine treatment counteracts the disappearance of tyrosine-hydroxylase-immunoreactive nerve cell bodies, dendrites and terminals in the mesostriatal dopamine system of the male rat after partial hemitransection. Brain Res. 1988;455:332–345. doi: 10.1016/0006-8993(88)90092-3. [DOI] [PubMed] [Google Scholar]
- 78.Carr LA, Rowell PP. Attenuation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity by tobacco smoke. Neuropharmacology. 1990;29:311–314. doi: 10.1016/0028-3908(90)90019-n. [DOI] [PubMed] [Google Scholar]
- 79.Shahi GS, Das NP, Moochhala SM. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity: partial protection against striato-nigral dopamine depletion in C57BL/6J mice by cigarette smoke exposure and by beta-naphthoflavone-pretreatment. Neurosci Lett. 1991;127:247–250. doi: 10.1016/0304-3940(91)90804-3. [DOI] [PubMed] [Google Scholar]
- 80.Janson AM, Fuxe K, Agnati L, et al. The effect of chronic nicotine treatment on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced degneration of nigrostriatal dopamine neurons in the black mouse. Advances in Pharmacological Sciences. 1991;1:323–329. [Google Scholar]
- 81.Janson AM, Fuxe K, Goldstein M. Differential effects of acute and chronic nicotine treatment on MPTP-(1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced degeneration of nigrostriatal dopamine neurons in the black mouse. Clin Investig. 1992;70:232–238. doi: 10.1007/BF00184656. [DOI] [PubMed] [Google Scholar]
- 82.Khwaja M, McCormack A, McIntosh JM, et al. Nicotine partially protects against paraquat-induced nigrostriatal damage in mice; link to alpha6beta2* nAChRs. J Neurochem. 2007;100:180–190. doi: 10.1111/j.1471-4159.2006.04177.x. [DOI] [PubMed] [Google Scholar]
- 83.Gao ZG, Cui WY, Zhang HT, Liu CG. Effects of nicotine on 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine-induced depression of striatal dopamine content and spontaneous locomotor activity in C57 black mice. Pharmacol Res. 1998;38:101–106. doi: 10.1006/phrs.1998.0337. [DOI] [PubMed] [Google Scholar]
- 84.Parain K, Marchand V, Dumery B, Hirsch E. Nicotine, but not cotinine, partially protects dopaminergic neurons against MPTP-induced degeneration in mice. Brain Res. 2001;890:347–350. doi: 10.1016/s0006-8993(00)03198-x. [DOI] [PubMed] [Google Scholar]
- 85.Parain K, Hapdey C, Rousselet E, et al. Cigarette smoke and nicotine protect dopaminergic neurons against the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine Parkinsonian toxin. Brain Res. 2003;984:224–232. doi: 10.1016/s0006-8993(03)03195-0. [DOI] [PubMed] [Google Scholar]
- 86.Hadjiconstantinou M, Hubble JP, Wemlinger TA, Neff NH. Enhanced MPTP neurotoxicity after treatment with isoflurophate or cholinergic agonists. J Pharmacol Exp Ther. 1994;270:639–644. [PubMed] [Google Scholar]
- 87.Ferger B, Spratt C, Earl CD, et al. Effects of nicotine on hydroxyl free radical formation in vitro and on MPTP-induced neurotoxicity in vivo. Naunyn Schmiedebergs Arch Pharmacol. 1998;358:351–359. doi: 10.1007/pl00005264. [DOI] [PubMed] [Google Scholar]
- 88.Perry TL, Hansen S, Jones K. Exposure to cigarette smoke does not decrease the neurotoxicity of N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. Neurosci Lett. 1987;74:217–220. doi: 10.1016/0304-3940(87)90152-2. [DOI] [PubMed] [Google Scholar]
- 89.Sershen H, Hashim A, Wiener HL, Lajtha A. Effect of chronic oral nicotine on dopaminergic function in the MPTP-treated mouse. Neurosci Lett. 1988;93:270–274. doi: 10.1016/0304-3940(88)90094-8. [DOI] [PubMed] [Google Scholar]
- 90.Behmand RA, Harik SI. Nicotine enhances 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. J Neurochem. 1992;58:776–779. doi: 10.1111/j.1471-4159.1992.tb09786.x. [DOI] [PubMed] [Google Scholar]
- 91.Bordia T, Parameswaran N, Fan H, et al. Partial recovery of striatal nicotinic receptors in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-lesioned monkeys with chronic oral nicotine. J Pharmacol Exp Ther. 2006;319:285–292. doi: 10.1124/jpet.106.106997. [DOI] [PubMed] [Google Scholar]
- 92.Quik M, Parameswaran N, McCallum SE, et al. Chronic oral nicotine treatment protects against striatal degeneration in MPTP-treated primates. J Neurochem. 2006;98:1866–1875. doi: 10.1111/j.1471-4159.2006.04078.x. [DOI] [PubMed] [Google Scholar]
- 93.Quik M, Chen L, Parameswaran N, et al. Chronic oral nicotine normalizes dopaminergic function and synaptic plasticity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned primates. J Neurosci. 2006;26:4681–4689. doi: 10.1523/JNEUROSCI.0215-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jeyarasasingam G, Tompkins L, Quik M. Stimulation of non-alpha7 nicotinic receptors partially protects dopaminergic neurons from 1-methyl-4-phenylpyridinium-induced toxicity in culture. Neuroscience. 2002;109:275–285. doi: 10.1016/s0306-4522(01)00488-2. [DOI] [PubMed] [Google Scholar]
- 95.Belluardo N, Mudo G, Caniglia G, et al. The nicotinic acetylcholine receptor agonist ABT-594 increases FGF-2 expression in various rat brain regions. Neuroreport. 1999;10:3909–3913. doi: 10.1097/00001756-199912160-00034. [DOI] [PubMed] [Google Scholar]
- 96.Donnelly-Roberts DL, Xue IC, Arneric SP, Sullivan JP. In vitro neuroprotective properties of the novel cholinergic channel activator (ChCA), ABT-418. Brain Res. 1996;719:36–44. doi: 10.1016/0006-8993(96)00063-7. [DOI] [PubMed] [Google Scholar]
- 97.Cormier A, Morin C, Zini R, et al. Nicotine protects rat brain mitochondria against experimental injuries. Neuropharmacology. 2003;44:642–652. doi: 10.1016/s0028-3908(03)00041-8. [DOI] [PubMed] [Google Scholar]
- 98.Schneider JS, Van Velson M, Menzaghi F, Lloyd GK. Effects of the nicotinic acetylcholine receptor agonist SIB-1508Y on object retrieval performance in MPTP-treated monkeys: comparison with levodopa treatment. Ann Neurol. 1998;43:311–317. doi: 10.1002/ana.410430308. [DOI] [PubMed] [Google Scholar]
- 99.Meshul CK, Kamel D, Moore C, et al. Nicotine alters striatal glutamate function and decreases the apomorphine-induced contralateral rotations in 6-OHDA-lesioned rats. Exp Neurol. 2002;175:257–274. doi: 10.1006/exnr.2002.7900. [DOI] [PubMed] [Google Scholar]
- 100.Domino EF, Ni L, Zhang H. Nicotine Alone and in Combination with l-DOPA Methyl Ester or the D(2) Agonist N-0923 in MPTP-Induced Chronic Hemiparkinsonian Monkeys. Exp Neurol. 1999;158:414–421. doi: 10.1006/exnr.1999.7106. [DOI] [PubMed] [Google Scholar]
- 101.Ishikawa A, Miyatake T. Effects of smoking in patients with early-onset Parkinson’s disease. J Neurol Sci. 1993;117:28–32. doi: 10.1016/0022-510x(93)90150-w. [DOI] [PubMed] [Google Scholar]
- 102.Hanagasi HA, Lees A, Johnson JO, et al. Smoking-responsive juvenile-onset Parkinsonism. Mov Disord. 2007;22:115–119. doi: 10.1002/mds.21177. [DOI] [PubMed] [Google Scholar]
- 103.Fagerstrom KO, Pomerleau O, Giordani B, Stelson F. Nicotine may relieve symptoms of Parkinson’s disease. Psychopharmacology (Berl) 1994;116:117–119. doi: 10.1007/BF02244882. [DOI] [PubMed] [Google Scholar]
- 104.Ebersbach G, Stock M, Muller J, et al. Worsening of motor performance in patients with Parkinson’s disease following transdermal nicotine administration. Mov Disord. 1999;14:1011–1013. doi: 10.1002/1531-8257(199911)14:6<1011::aid-mds1016>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 105.Clemens P, Baron JA, Coffey D, Reeves A. The short-term effect of nicotine chewing gum in patients with Parkinson’s disease. Psychopharmacology (Berl) 1995;117:253–256. doi: 10.1007/BF02245195. [DOI] [PubMed] [Google Scholar]
- 106.Mitsuoka T, Kaseda Y, Yamashita H, et al. Effects of nicotine chewing gum on UPDRS score and P300 in early-onset parkinsonism. Hiroshima J Med Sci. 2002;51:33–39. [PubMed] [Google Scholar]
- 107.Kelton MC, Kahn HJ, Conrath CL, Newhouse PA. The effects of nicotine on Parkinson’s disease. Brain Cogn. 2000;43:274–282. [PubMed] [Google Scholar]
- 108.Vieregge A, Sieberer M, Jacobs H, et al. Transdermal nicotine in PD: a randomized, double-blind, placebo-controlled study. Neurology. 2001;57:1032–1035. doi: 10.1212/wnl.57.6.1032. [DOI] [PubMed] [Google Scholar]
- 109.Lemay S, Chouinard S, Blanchet P, et al. Lack of efficacy of a nicotine transdermal treatment on motor and cognitive deficits in Parkinson’s disease. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:31–39. doi: 10.1016/S0278-5846(03)00172-6. [DOI] [PubMed] [Google Scholar]
- 110.Villafane G, Cesaro P, Rialland A, et al. Chronic high dose transdermal nicotine in Parkinson’s disease: an open trial. Eur J Neurol. 2007 doi: 10.1111/j.1468-1331.2007.01949.x. [DOI] [PubMed] [Google Scholar]
- 111.Shoulson I. Randomized placebo-controlled study of the nicotinic agonist SIB-1508Y in Parkinson disease. Neurology. 2006;66:408–410. doi: 10.1212/01.wnl.0000196466.99381.5c. [DOI] [PubMed] [Google Scholar]
- 112.Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord. 2001;16:448–458. doi: 10.1002/mds.1090. [DOI] [PubMed] [Google Scholar]
- 113.Fabbrini G, Brotchie JM, Grandas F, et al. Levodopa-induced dyskinesias. Mov Disord. 2007 doi: 10.1002/mds.21475. [DOI] [PubMed] [Google Scholar]
- 114.Brotchie JM, Lee J, Venderova K. Levodopa-induced dyskinesia in Parkinson’s disease. J Neural Transm. 2005;112:359–391. doi: 10.1007/s00702-004-0251-7. [DOI] [PubMed] [Google Scholar]
- 115.Cenci MA. Dopamine dysregulation of movement control in l-DOPA-induced dyskinesia. Trends Neurosci. 2007;30:236–243. doi: 10.1016/j.tins.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 116.Muller T, Woitalla D, Russ H, et al. Prevalence and treatment strategies of dyskinesia in patients with Parkinson’s disease. J Neural Transm. 2007 doi: 10.1007/s00702-007-0718-4. [DOI] [PubMed] [Google Scholar]
- 117.Deogaonkar M, Subramanian T. Pathophysiological basis of drug-induced dyskinesias in Parkinson’s disease. Brain Res Brain Res Rev. 2005;50:156–168. doi: 10.1016/j.brainresrev.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 118.Olanow CW, Obeso JA, Stocchi F. Continuous dopamine-receptor treatment of Parkinson’s disease: scientific rationale and clinical implications. Lancet Neurol. 2006;5:677–687. doi: 10.1016/S1474-4422(06)70521-X. [DOI] [PubMed] [Google Scholar]
- 119.Brotchie JM. Nondopaminergic mechanisms in levodopa-induced dyskinesia. Mov Disord. 2005;20:919–931. doi: 10.1002/mds.20612. [DOI] [PubMed] [Google Scholar]
- 120.Fox SH, Lang AE, Brotchie JM. Translation of nondopaminergic treatments for levodopa-induced dyskinesia from MPTP-lesioned nonhuman primates to phase IIa clinical studies: keys to success and roads to failure. Mov Disord. 2006;21:1578–1594. doi: 10.1002/mds.20936. [DOI] [PubMed] [Google Scholar]
- 121.Guigoni C, Doudnikoff E, Li Q, et al. Altered D(1) dopamine receptor trafficking in parkinsonian and dyskinetic non-human primates. Neurobiol Dis. 2007 doi: 10.1016/j.nbd.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 122.Blanchet PJ, Calon F, Morissette M, et al. Relevance of the MPTP primate model in the study of dyskinesia priming mechanisms. Parkinsonism Relat Disord. 2004;10:297–304. doi: 10.1016/j.parkreldis.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 123.Jenner P. A2A antagonists as novel non-dopaminergic therapy for motor dysfunction in PD. Neurology. 2003;61:S32–38. doi: 10.1212/01.wnl.0000095209.59347.79. [DOI] [PubMed] [Google Scholar]
- 124.Guigoni C, Li Q, Aubert I, et al. Involvement of sensorimotor, limbic, and associative basal ganglia domains in L-3,4-dihydroxyphenylalanine-induced dyskinesia. J Neurosci. 2005;25:2102–2107. doi: 10.1523/JNEUROSCI.5059-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cenci MA, Lundblad M. Post- versus presynaptic plasticity in L-DOPA-induced dyskinesia. J Neurochem. 2006 doi: 10.1111/j.1471-4159.2006.04124.x. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 126.Samadi P, Bedard PJ, Rouillard C. Opioids and motor complications in Parkinson’s disease. Trends Pharmacol Sci. 2006;27:512–517. doi: 10.1016/j.tips.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 127.Quik M, Cox H, Parameswaran N, et al. Nicotine reduces levodopa-induced dyskinesias in lesioned monkeys. Annals of Neurology. 2007 doi: 10.1002/ana.21203. [DOI] [PubMed] [Google Scholar]
- 128.Matta SG, Balfour DJ, Benowitz NL, et al. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berl) 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- 129.Giniatullin R, Nistri A, Yakel JL. Desensitization of nicotinic ACh receptors: shaping cholinergic signaling. Trends Neurosci. 2005;28:371–378. doi: 10.1016/j.tins.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 130.Wang H, Sun X. Desensitized nicotinic receptors in brain. Brain Res Brain Res Rev. 2005;48:420–437. doi: 10.1016/j.brainresrev.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 131.Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol. 2002;53:457–478. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- 132.Rose JE. Multiple brain pathways and receptors underlying tobacco addiction. Biochem Pharmacol. 2007;74:1263–1270. doi: 10.1016/j.bcp.2007.07.039. [DOI] [PubMed] [Google Scholar]
- 133.Samaha AN, Robinson TE. Why does the rapid delivery of drugs to the brain promote addiction? Trends Pharmacol Sci. 2005;26:82–87. doi: 10.1016/j.tips.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 134.Wonnacott S, Sidhpura N, Balfour DJ. Nicotine: from molecular mechanisms to behaviour. Curr Opin Pharmacol. 2005;5:53–59. doi: 10.1016/j.coph.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 135.Buisson B, Bertrand D. Nicotine addiction: the possible role of functional upregulation. Trends Pharmacol Sci. 2002;23:130–136. doi: 10.1016/S0165-6147(00)01979-9. [DOI] [PubMed] [Google Scholar]
- 136.Lemay S, Blanchet P, Chouinard S, et al. Poor tolerability of a transdermal nicotine treatment in Parkinson’s disease. Clin Neuropharmacol. 2003;26:227–229. doi: 10.1097/00002826-200309000-00004. [DOI] [PubMed] [Google Scholar]