

Abstract
Hedgehog proteins, signaling molecules implicated in human embryo development and cancer, can be inhibited at the stage of autoprocessing by the trivalent arsenical phenyl arsine oxide (PhAsIII). The interaction (apparent Ki, 4×10−7M) is characterized by an optical binding assay and by NMR spectroscopy. PhAsIII appears to be the first validated inhibitor of hedgehog autoprocessing, which is unique to hedgehog proteins and essential for biological activity.
Keywords: active-site probe, arsenic, autoprocessing, cholesterol, hedgehog
Hedgehog (Hh) proteins represent a family of cell signaling ligands that influence human embryo development as well as multiple types of cancer.[1] In this work, we focus on the covalent modification of Hh proteins with cholesterol, a relatively unstudied event in Hh protein biosynthesis.[2] As shown in Scheme 1, cholesterol modification takes place autocatalytically in two chemical steps. First, a Hh precursor protein rearranges an internal peptide bond into a thioester; second, this intermediate is displaced by cholesterol. Activity for this “cholesterolysis” reaction[3] resides entirely in a C-terminal segment of the Hh precursor, referred to as HhC. Cholesterolysis is imperative for bioactivity, and appears to be specific to Hh proteins;[4] therefore, small molecules that can modulate this reaction are of fundamental and practical interest.[5]
Scheme 1.

Hedgehog precursor protein cholesterolysis. Autoprocessing begins with the central “HINT” domain’s first residue, cysteine, rearranging the adjacent amide bond to form a thioester. In step two, the C-terminal subdomain (dark gray) activates bound cholesterol for attack at the activated acyl group. Finally, the N-terminal domain, which possesses cell signaling activity, dissociates with its newly formed carboxy terminus esterified to cholesterol.
Hedgehog (Hh) precursor proteins across the animal kingdom contain three functional regions: an amino terminal domain with signaling activity (HhN) to which cholesterol is attached; a central autocatalytic “HINT” domain (Hedgehog/Intein) resembling self-splicing inteins; and a carboxy terminal subdomain with affinity for cholesterol (Scheme 1, white, gray, black, respectively).[4] The latter two segments comprise HhC and are essential for cholesterolysis. Structural studies thus far have focused on HhN signaling domains; only one 3D structure of the HINT domain has been obtained at atomic resolution,[6] and no structural data is available for the cholesterol-binding subdomain. Accordingly, compounds that target precursor Hh are scarce.[7]
Here, we report that the trivalent arsenical compound phenylarsine oxide (PhAsIII) is a potent active-site probe of precursor Hh (apparent Ki, ~4×10−7M). Binding of PhAsIII by Hh irreversibly blocks cholesterolysis. We probed this interaction using a novel optical assay that detects bimolecular association of PhAsIII and Hh by FRET. We also applied complementary NMR methods to map the PhAsIII binding site on Hh. Together, these techniques provide a general platform for identifying and characterizing inhibitors of Hh cholesterolysis. The inhibitory activity of PhAsIII reported here might also offer insight into arsenic’s teratogenic activity, as well as its anticancer properties.[8]
We were drawn to trivalent arsenic (AsIII) as a potential cholesterolysis inhibitor on the basis of this metalloid’s thiophilicity. PhAsIII and its organoarsenical analogues condense rapidly and covalently with dimercaptans and with closely spaced cysteines in peptides and proteins.[9] The products are presumed to be a cyclic S-AsIII(R)-S adduct and water. In the HINT domain of precursor Hh, there are two conserved cysteine residues, termed CysA and B. Although separated by >100 residues in primary sequence, CysA and B are brought together upon folding.[6a] The two residues are catalytically essential; single alanine point mutation at either position abolishes cholesterolsis.[6a, 10] CysA serves as the nucleophile in the first step of cholesterolysis (Scheme 1); recent studies suggest that the second cysteine can engage CysA in a disulfide bond, thereby imparting redox control over cholesterolysis.[10] If two cysteine residues are sufficiently close to engage in a disulfide bond, it seems conceivable that in the absence of steric or electrostatic constraints they would also prove reactive to AsIII.
Our initial experiments to test AsIII inhibition used a conventional gel-based cholesterolysis assay.[2a] Along with three AsIII compounds (meta-arsenite, arsenic trioxide, and PhAsIII), we also investigated representative AsV compounds (phenylarsonic acid and dimethylarsenic acid). Inhibitory activity was assessed against an engineered Hh precursor. The construct, ShhN-DHhC, contains the HhN of human sonic hedgehog protein fused to the HhC of Drosophila melanogaster Hh. This chimeric precursor shows robust expression in Escherichia coli and is catalytically competent. Expression and purification of native Hh precursors has proved exceedingly difficult. Cholesterolysis assays on ShhN-DHhC were performed under reducing conditions in Bis·Tris buffer (pH 7.1) with added TritonX-100 (0.4 %, v/v, final). Reactions were initiated by adding cholesterol to 500 μM. At selected intervals, aliquots of the reaction were quenched by boiling with added SDS-PAGE loading buffer. The extent of precursor cholesterolysis was then assessed by densitometry after resolving precursor and products by SDS-PAGE.
As shown in Figure 1A, PhAsIII produced marked inhibition of Hh precursor cholesterolysis, blocking production of the two processed fragments: cholesteroylated ShhN and DhhC to a level comparable to a control reaction lacking cholesterol (compare first and last lanes). Neither of the AsV compounds was capable of measurably inactivating cholesterolysis at a concentration of 80 μM. It is interesting to note that, under the present conditions, arsenic trioxide and (meta)arsenite also failed to inactivate cholesterolysis. These two AsIII compounds have been used interchangeably with PhAsIII as dithiol-modifying agents.[11] As arsenite, arsenic trioxide, and PhAsIII contain the same reactive AsIII atom, this apparent preference for the uncharged organoarsenical indicates a degree of specificity in the inactivation mechanism.
Figure 1.
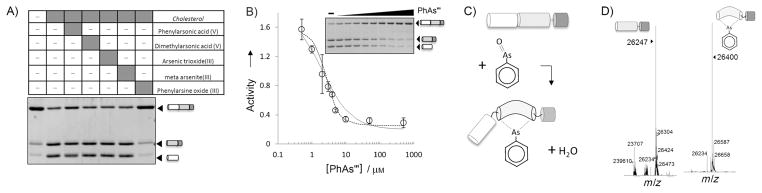
Identification of PhAsIII as a tight-binding inhibitor of Hh cholesterolysis. A) Screen of arsenical compounds as potential inhibitors of Hh cholesterolysis activity. Hh precursor (4 μM) was preincubated with indicated arsenical compounds (60 μm, final) before initiating reaction with cholesterol (500 μM). Samples were electrophoresed on 15% SDS-polyacrylamide gels, stained with Coomassie Brilliant Blue, and digitized with a BioRad imager. B) Titration of Hh activity by PhAsIII. Activity of Hh precursor (2 μM) was determined by the extent of processing as in (A). Data were fit to a standard dose–response curve (—) by using an IC50 value of 2 μM, as well as to a quadratic binding equation (····) by using a Ki value of 0.4 μM. Error bars indicate the data range (n=2). C) Proposed mechanism for covalent interation of PhAsIII and Hh precursor. D) Detection of Hh–PhAsIII adduct by mass spectrometry. Deconvoluted mass of autocatalytic HhC (10 μM) without (left) and with (right) 1 equiv of Hh–PhAsIII. Additional mass is consistent with a condensation mechanism (expected mass change: 154; observed mass change: 153).
To quantify inhibition, the Hh precursor (4×10−6M) was titrated with PhAsIII from 0.5 to 500 μM. The data fit reasonably well to a general dose–response equation (Figure 1B, solid line, and Methods section in the Supporting Information), with a calculated IC50 value of 2.2 μM. The correspondence of the IC50 value with 50% of the initial precursor concentration indicated that PhAsIII was bound stoichiometrically by the precursor. In view of the apparent tight binding, the data were analyzed with a quadratic equation for high-affinity inhibitors (Figure 1B, dotted line), yielding a better fit and an apparent Ki value of 0.4 μM. In two follow-up experiments, we found that 1) inhibition of cholesterolysis by PhAsIII remained unchanged after overnight incubation with a saturating amount of cholesterol, and 2) inhibition could not be reversed by extended dialysis (Figure S1 in the Supporting Information). Collectively, these observations, together with direct detection of a PhAsIII-HhC adduct by mass spectrometry, are consistent with covalent inhibition (Figure 1C, D).
To monitor the kinetics of the association of HhC with PhAsIII, we prepared a FRET-active Hh precursor. The construct, C-H-Y, has fluorescent proteins CFP and YFP fused to the amino and carboxy termini of the Drosophila melanogaster HhC, respectively (Figure 2A, inset). A similar construct was used to study the kinetics of self-cleaving inteins.[12] We tested C-H-Y on the speculation that HhC would undergo a structural change upon binding PhAsIII which could be detected by FRET. The spectra in Figure 2A are consistent with this hypothesis, as the addition of one equivalent of PhAsIII to a buffered solution of C-H-Y resulted in a substantial FRET change. Analysis of the PhAsIII-treated precursor by SDS-PAGE ruled out decomposition as a possible explanation for the observed FRET loss (Figure S2 A, inset). To assess specificity, two control constructs were used: C-Y, in which HhC was replaced with a peptide linker,[12] and, C-HCysA/A-Y, a mutant in which CysA was replaced with alanine. Neither of the control constructs exhibited an appreciable change in FRET in the presence of PhAsIII (Figure S2B and C). Thus, the altered FRET induced by added PhAsIII was consistent with a physical interaction with HhC that required its catalytically essential CysA residue.
Figure 2.
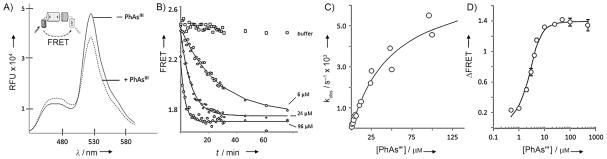
Real-time detection of PhAsIII binding by Hh using an optical reporter. A) Effect of PhAsIII on the fluorescence of FRET-active Hh precursor protein. Spectra of precursor, C-H-Y, in aqueous buffered solution (pH 7.1) with and without 1 equiv of added PhAsIII (λex=400 nm, λem=420–600 nm). Inset: Schematic of FRET-active construct with cyan fluorescent protein fused to the N terminus of the HhC domain and yellow fluorescent protein fused to the C terminus. B) Kinetics of PhAsIII binding by C-H-Y measured by FRET change. Time traces showing loss of FRET signal from C-H-Y after addition of the indicated concentration of PhAsIII. Solid lines show first-order exponential decay (kobs), calculated by using best-fit values. C) Bimolecular kinetics of PhAsIII binding by C-H-Y is saturable. Rate constants (kobs [s−1]) for FRET loss plotted as a function of increasing PhAsIII concentration. Solid line shows the expected behavior, assuming reversible formation of a noncovalent encounter complex, followed by irreversible formation of the PhAsIII/C-H-Y complex. D) Fluorometric titration of binding of PhAsIII to C-H-Y. Plot shows the overall FRET change (FRET○–FRETf) as a function of PhAsIII concentration. Solid line shows the expected behavior for an inhibitor with a EC50 value of 0.4 μM. Error bars indicate data range (n=3)
Next, we initiated kinetic studies with C-H-Y to determine the second-order rate constant for PhAsIII binding, as measured by FRET loss (Figure 2 B). Pseudo-first-order rate constants from experiments over a range of PhAsIII concentrations are plotted in Figure 2C. The curvature apparent in the plot is consistent with a two-step kinetic model in which a noncovalent encounter complex between PhAsIII and C-H-Y forms first, and then transitions irreversibly to a covalent adduct (Supporting Information). Accordingly, the data conform to a hyperbolic function yielding best fit values of 52 μM and 7×10−3 s−1 for the apparent dissociation constant of the encounter complex (Kd) and the rate of covalent modification (kinact), respectively. Expressed as a bimolecular rate constant (kinact/Kd), ~102M−1 s−1, the value is within the range of other AsIII–protein interactions.[9b] The apparent affinity of the stable PhAsIII–C-H-Y complex, obtained by plotting the final FRET change as a function of increasing PhAsIII concentration (Figure 2D), is nearly identical to the value obtained with the ShhN-HhC precursor described above. Thus, appending the fluorescent proteins to HhC did not seem to disrupt its structure or the interaction with PhAsIII.
As a final step aimed at mapping the site of interaction, we examined the HINT domain of Hh in its complex with PhAsIII by using two complementary NMR techniques. First, we titrated [U-15N]-labeled HINT domain with PhAsIII at molar ratios of 0.5:1, 1:1, and 4:1, respectively. The results of the titration were monitored by 1H{15N} HSQC. A cutoff of 80% signal reduction was used to identify residues close to PhAsIII in 3D structure (Figure S3). By using that filter with the 4:1 data set, ten residues were identified as comprising the PhAsIII binding site, with the majority forming a contiguous surface (Figure 3A) flanked by CysA and B. Many of the residues implicated in PhAsIII binding are conserved in the HINT domains of human Hh proteins (Figure S4), raising the possibility that the biogenesis of human Hh proteins would also be blocked by PhAsIII.
Figure 3.
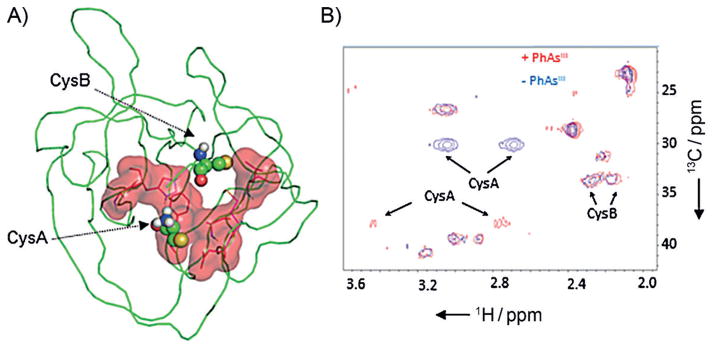
PhAsIII binding site on Hh located by NMR chemical shift mapping. A) Proposed binding site. Residues in 1H-/15N-labeled HINT domain of Hh whose resonances were suppressed by >80% following the addition of 4 equiv of PhAsIII are displayed in red (sticks), along with the calculated surface area. Catalytic cysteine residues (CysA and B) are shown as ball-and-stick. This figure was generated from PDB ID: 1ATO by using PyMOL (http://www.pymol.org). B) Chemical shift perturbation of CysA in the PhAsIII -bound HINT domain. 13C/1H spectra of the Hh HINT domain enriched with Cys-13C3 at CysA and B, acquired without (blue) and with (red) added PhAsIII.
The effect of PhAsIII on CysA and B, not resolved in the 1H{15N} HSQC spectra, were evaluated next by using an 1H{13C} HSQC experiment. CysA and CysB of the HINT domain were enriched with 13C by recombinant expression by using the cysteine auxotroph strain of E. coli, CysE(DE3).[13] In the presence of PhAsIII, along with a large chemical shift perturbation (Table S1), the peak intensity of CysA was markedly suppressed, consistent with conformational dynamics in the bound state (Figure 3B, compare blue and red). The chemical shift of CysB, however, appeared less sensitive to added PhAsIII. These NMR findings might be reconciled by proposing that an S-AsIII (R)-S complex requires minimal change in the conformation of CysB. Alternatively, the putative S- AsIII (R)-S complex could represent an intermediate state only, with CysB ultimately displaced by another nucleophilic amino acid side chain in the HINT domain. Structural studies to evaluate these possibilities are ongoing.
In summary, we have identified and characterized, to our knowledge, the first active-site directed inhibitor targeting cholesterolysis, a specialized autoprocessing event required for biological signaling by Hh proteins. In addition, the results could be relevant to the teratogenicity and medicinal properties of AsIII in the context of Hh. Inhibition of cholesterolysis is expected to suppress Hh function with effects in early life that could include severe developmental anomalies.[14] In the adult, however, Hh inhibitors can be therapeutic, as chronic overproduction of mature Hh can promote tumor growth and metastasis.[15] Recently, AsIII, in the form of arsenic trioxide, was reported to interfere indirectly with Hh through binding of Gli proteins, the Hh-responsive transcription factors.[11, 16] This discovery by Beachy et al. accords with the teratogenic activity of AsIII and has prompted efforts to explore arsenic trioxide as a therapeutic agent to treat Hh-driven cancers.[17] Our results expand the potential therapeutic applications of AsIII by suggesting PhAsIII as a means for direct antagonism of Hh itself.
Supplementary Material
Acknowledgments
This work was supported by start-up funds from Binghamton University and by the Office of the Assistant Secretary of Defense for Health Affairs, through the Prostate Cancer Research Program under Award No. W81XWH-14-1-0155 (B.P.C.). Opinions, interpretations, conclusions and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense. B.P.C. gratefully acknowledges Dr. Marlene Belfort for supporting the early stages of this work (grant R01 GM04484 to M.B.). The Regional NMR Facility (600 MHz instrument) at Binghamton University is supported by the National Science Foundation (NSF) (grant CHE-0922815).
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.201402421: Complete experimental details for the preparation of protein constructs, FRET assay, calculation of Ki values, isotopic labeling, and NMR experiments.
References
- 1.Theunissen JW, de Sauvage FJ. Cancer Res. 2009;69:6007–6010. doi: 10.1158/0008-5472.CAN-09-0756. [DOI] [PubMed] [Google Scholar]
- 2.a) Porter JA, Young KE, Beachy PA. Science. 1996;274:255–259. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]; b) Hulce JJ, Cognetta AB, Niphakis MJ, Tully SE, Cravatt BF. Nat Methods. 2013;10:259–264. doi: 10.1038/nmeth.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh C. Posttranslational Modification of Proteins: Expanding Nature’s Inventory. Roberts; Englewood: 2006. [Google Scholar]
- 4.Mann RK, Beachy PA. Annu Rev Biochem. 2004;73:891–923. doi: 10.1146/annurev.biochem.73.011303.073933. [DOI] [PubMed] [Google Scholar]
- 5.Heal WP, Jovanovic B, Bessin S, Wright MH, Magee AI, Tate EW. Chem Commun (Camb) 2011;47:4081–4083. doi: 10.1039/c0cc04710d. [DOI] [PubMed] [Google Scholar]
- 6.a) Hall TM, Porter JA, Young KE, Koonin EV, Beachy PA, Leahy DJ. Cell. 1997;91:85–97. doi: 10.1016/s0092-8674(01)80011-8. [DOI] [PubMed] [Google Scholar]; b) Xie J, Du Z, Callahan B, Belfort M, Wang C. Biomol NMR Assign. 2014;8:279–281. doi: 10.1007/s12104-013-9500-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang SQ, Paulus H. J Biomol Screening. 2010;15:1082–1087. doi: 10.1177/1087057110377498. [DOI] [PubMed] [Google Scholar]
- 8.Miller WH, Jr, Schipper HM, Lee JS, Singer J, Waxman S. Cancer Res. 2002;62:3893–3903. [PubMed] [Google Scholar]
- 9.a) Zahler WL, Cleland WW. J Biol Chem. 1968;243:716–719. [PubMed] [Google Scholar]; b) Shen S, Li XF, Cullen WR, Weinfeld M, Le XC. Chem Rev. 2013;113:7769–7792. doi: 10.1021/cr300015c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen X, Tukachinsky H, Huang CH, Jao C, Chu YR, Tang HY, Mueller B, Schulman S, Rapoport TA, Salic A. J Cell Biol. 2011;192:825–838. doi: 10.1083/jcb.201008090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, Lee JJ, Gardner D, Beachy PA. Proc Natl Acad Sci USA. 2010;107:13432–13437. doi: 10.1073/pnas.1006822107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amitai G, Callahan BP, Stanger MJ, Belfort G, Belfort M. Proc Natl Acad Sci USA. 2009;106:11005–11010. doi: 10.1073/pnas.0904366106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Strub MP, Hoh F, Sanchez JF, Strub JM, Bock A, Aumelas A, Dumas C. Structure. 2003;11:1359–1367. doi: 10.1016/j.str.2003.09.014. [DOI] [PubMed] [Google Scholar]; b) Arnesano F, Banci L, Bertini I, Felli IC, Losacco M, Natile G. J Am Chem Soc. 2011;133:18361–18369. doi: 10.1021/ja207346p. [DOI] [PubMed] [Google Scholar]
- 14.Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- 15.a) Rubin LL, de Sauvage FJ. Nat Rev Drug Discovery. 2006;5:1026–1033. doi: 10.1038/nrd2086. [DOI] [PubMed] [Google Scholar]; b) Chen M, Carkner R, Buttyan R. Expert Rev Endocrinol Metab. 2011;6:453–467. doi: 10.1586/EEM.11.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng JM, Curran T. Nat Rev Cancer. 2011;11:493–501. doi: 10.1038/nrc3079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J, Aftab BT, Tang JY, Kim D, Lee AH, Rezaee M, Chen B, King EM, Borodovsky A, Riggins GJ, Epstein EH, Jr, Beachy PA, Rudin CM. Cancer Cell. 2013;23:23–34. doi: 10.1016/j.ccr.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.