

Abstract
Rhizopus oryzae lipase (ROL) has a propeptide at its N-terminus that functions as an intramolecular chaperone and facilitates the folding of mature ROL (mROL). In this study, we successfully generated a functionally distinct imprinted mROL (mROLimp) through protein folding memory using a mutated propeptide. The mutated propeptide left its structural memory on mROL and produced mROLimp that exhibited different substrate specificities compared with mROLWT (prepared from the wild type propeptide), although the amino acid sequences of both mROLs were the same. mROLimp showed a preference for substrates with medium chain-length acyl groups and, noticeably, recognized a peptidase-specific substrate. In addition, ROLimp was more stable than mROLWT. These results strongly suggest that proteins with identical amino acid sequences can fold into different conformations and that mutations in intramolecular chaperones can dynamically induce changes in enzymatic activity.
Introduction
Most lipases and proteases are produced as precursor forms with propeptides and are subsequently processed into mature enzymes [1–4]. Some propeptides function as inhibitors that temporarily inhibit the enzymatic activities of their mature enzymes during cellular transport [5]. In addition, some propeptides play an essential role in protein maturation. These propeptides function as chaperones and facilitate the correct folding of their mature enzymes [6, 7]. Because propeptides are covalently bound to the mature enzyme, they are called “intramolecular chaperones,” distinct from “intermolecular chaperones.” Once the protein has been folded into its mature form, propeptides are removed by autolysis or exogenous proteases [5, 8].
In our previous study, we demonstrated that mutations in the propeptide of carboxypeptidase Y (CPY) generated a functionally distinct, mature CPY [9]. The mature form of CPY generated with the mutated propeptide had the same amino acid sequence as mature CPY prepared from the wild type propeptide, as both the wild type and mutated propeptides were completely cleaved off. This phenomenon, where the modified enzyme retains the “folding memory” of the mutated propeptide even after the propeptide digestion, has been called “protein folding memory [9]”. “Protein folding memory” contradicts Anfinsen’s dogma (Nobel Prize in 1972), which postulates that the structure of a protein is determined only by its amino acid sequence and that the native structure is a unique and most stable state. [10]. On the other hand, according to the “protein folding memory” theory, the structure of the mature enzyme is not only determined by the amino acid sequence but rather by the chaperoning function of the propeptide. Although “protein folding memory” is an attractive phenomenon, little is known about the underlying mechanism. “Protein folding memory” would have applications in the field of protein engineering and may be adopted to dynamically improve enzymatic functions through structural imprinting by propeptides.
Rhizopus oryzae lipase (ROL) is widely used in industrial applications [11]. ROL is initially produced as a precursor form, which comprises an N-terminal 69-amino acid propeptide and a 297-amino acid mature enzyme [12]. The propeptide of ROL (proROL) functions as an inhibitor and intramolecular chaperone for mature ROL (mROL) [13]. When ROL is produced in Saccharomyces cerevisiae and Pichia pastoris, proROL is cleaved by Kex2-like proteases, resulting in mROL [5, 14]. A previous study revealed that residues Ser20–Gly37 and Ser38–Glu57 in proROL were essential for the secretion and folding of mROL, respectively [6]. The active site of ROL is composed of the Ser242, Asp301, and His354 residues; the same three residues are also observed in the catalytic triad of serine proteases and peptidases [15]. Although there are only subtle differences between the active sites of fungal lipases and serine proteases, their substrate recognition mechanisms are different. The active centers of proteases and peptidases are exposed to the solvent, while those of lipases are not [16, 17]. The catalytic triads of lipases are buried under a short α-helix, called lid domain. Lid opening is accompanied by a concomitant structural change in the substrate-binding site that enables the binding of the substrate to the active site [18]. The differences in the activities and substrate recognition mechanisms between lipases and proteases may be partially attributed to the conformational characteristics of lipases. The conformation and activity of mROL would be altered by introducing mutations in an essential part of proROL for chaperoning function. This would allow conformational changes in the substrate-recognition domain of the mature protein and lead to the altered activity.
In this study, we demonstrated that mutations in proROL led to “protein folding memory” and generated a functionally distinct imprinted mROL (mROLimp) that had the same amino acid sequence as mROL prepared from wild-type proROL (mROLWT). Although mROLimp and mROLWT had the same amino acid sequences, mROLimp showed higher lipase activity and stability compared with mROLWT, and recognized a peptidase-specific substrate.
Materials and Methods
Strain and materials
Escherichia coli strain DH5α [F-, endA1, hsdR17(rk -/mK +), supE44, thi-1, λ-, deoR, recA1, gyrA96, phoA, φ80dlacZΔM15, Δ(lacZYA-argF)U169] (Toyobo, Osaka, Japan) was used as the host for recombinant DNA manipulation. P. pastoris strain GS115 [his4] (Life Technologies, CA, USA) was used as the host for protein production. S. cerevisiae strain BY4741/sed1Δ [MAT a, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0] (EUROSCARF, Frankfurt, Germany) was used for the cell surface display of mROLWT and mROLimp. E. coli transformants were grown in Luria-Bertani media (1% [w/v] tryptone, 0.5% [w/v] yeast extract, and 1% [w/v] sodium chloride) containing 50 μg/mL ampicillin. For protein production, P. pastoris transformants were pre-cultivated in buffered complex glycerol media (BMGY; 1% [w/v] yeast extract, 2% [w/v] peptone, 1.34% [w/v] yeast nitrogen base without amino acids, 4 × 10−5% [w/v] biotin, 1% [v/v] glycerol, and 100 mM potassium phosphate [pH 6.0]). To induce transcription, pre-cultivated transformants were grown in buffered complex methanol media (BMMY) (1% [w/v] yeast extract, 2% [w/v] peptone, 1.34% [w/v] yeast nitrogen base w/o amino acids, 4 × 10−5% [w/v] biotin, 0.5% [v/v] methanol, and 100 mM potassium phosphate [pH 6.0]). Yeast transformants were cultured in synthetic dextrose (SD) medium (0.67% [w/v] yeast nitrogen base w/o amino acids and 2% [w/v] glucose) supplemented with the appropriate amino acids.
Construction of plasmids
The ROL gene was amplified from the pWRL2 [3] plasmid using F proROL c-Myc primer (5'- TCTGTCTTCGCTCGAGAACAAAAGTTGATTTCTGAAGAAGATTTGGTTCCTGTTTCTGGTAAATCTGG -3') and R mROL His-tag primer (5'- AAGGATCCCGGGGAATTAATGATGATGATGATGATGCAAACAGCTTCCTTCGTTGATATC -3'), and inserted into the pHIL-S1 plasmid (Life Technologies) for protein production by P. pastoris. The resulting plasmid, labeled pHIL-proROL-mROL, was composed of the PHO1 secretion signal sequence, c-Myc-tag-encoding sequence, ROL gene, and His tag-encoding sequence. To construct the gene encoding the mutated propeptide, DNA fragments encoding the Val1–Met49 and Tyr56–Leu366 amino acid residues of ROL were amplified using the F proROL c-Myc primer and 3’ primer (5'- CATGTAGTAAGGTTCAGCTTGAAG -3'), and 5’ primer (5'- TCCCATGGTGGCAACCTGAC -3') and R mROL His-tag primer, respectively. These DNA fragments were fused with a DNA fragment (5'- TTCAAGCTGAACCTTACTACATGGTTGATGATGATGATAAATATGAGTCCCATGGTGGCAACCTGACATC -3') encoding the mutated sequence (VDDDDK) by overlap polymerase chain reaction (PCR) [19]. The resulting DNA fragment was inserted into the pHIL-S1 vector using the In-Fusion HD Cloning Kit (Clontech, CA, USA). The resultant plasmid was named pHIL-proROL-mut1-mROL. For the cell surface display of mROLWT and mROLimp, the genes encoding proROL-mROL and proROL-mut1-mROL were amplified using the primers F BglII-proROL (5'- GCCAGATCTGTTCCTGTTTCTGGTAAATCTGG -3') and R XhoI-mROL (5'- ACCCTCGAGCAAACAGCTTCCTTCGTTGATAAAG -3'), and inserted into the pUGD1 plasmid. A gene encoding proROL-mut2-mROL was constructed using the protocol described for the proROL-mut1-mROL and then inserted into the pUGD1 plasmid. proROL-mut2-mROL encoded another mutated proROL, in which residues Gln50–Glu57 were replaced with eight Asn residues, and mROL. The resulting pUGD1-based plasmid was composed of the GAPDH promoter, secretion signal sequence of the glucoamylase gene, ROL gene, FLAG tag-encoding gene, and 3’-half of α-agglutinin gene.
Production and purification of His-tagged mROL and mROLimp
pHIL-proROL-mROL, pHIL-proROL-mut1-mROL, and pHIL-S1 were digested with the restriction enzyme, SacI. P. pastoris GS115 cells were transformed with the linear plasmids using the Frozen-EZ Yeast Transformation II kit (Zymo Research, CA, USA). The P. pastoris transformants were grown in 10 mL of BMGY medium for 24 h at 30°C. The culture was subsequently centrifuged at 3000 ×g for 5 min. The cells were resuspended in BMMY medium and then grown for 24 h at 30°C for protein production. All purification steps were performed at 4°C. The supernatant of the culture was collected at 3000 ×g for 5 min and concentrated using a YM-10 filter device (Millipore, MA, USA). ROL proteins were purified from the concentrated supernatant using the Ni Sepharose High Performance medium (GE Healthcare, Little Chalfont, UK), according to manufacturer’s protocol. To remove propeptide-uncleaved precursors, the purified fraction was mixed with an Anti-c-Myc-Agarose Affinity Gel (Sigma-Aldrich, MO, USA) for 3 h. Because c-Myc tags were fused to the N-terminus of the wild type and mutated propeptides, propeptide-uncleaved precursors were removed by this process and only mature forms (mROLWT and mROLimp) were isolated. mROLWT and mROLimp were eluted by washing the resin four times with 500 μL of 50 mM potassium phosphate buffer (pH 6.5). Protein concentration was quantified using a Protein Assay Bicinchoninate Kit (Nacalai Tesque, Kyoto, Japan), which is based on Lowry assay [20].
SDS-PAGE and N-terminal sequencing
The purity and molecular weights of the isolated proteins were confirmed by SDS-PAGE with or without EndoH (New England Biolabs, MA, USA) treatment on a 5–20% gradient polyacrylamide gel. Protein bands were detected using the CBB Stain One kit (Nacalai Tesque). Amino acid sequencing of the purified mROLWT and mROLimp was performed by the Edman degradation method on a protein sequencing system PPSQ-33A (Shimadzu, Kyoto, Japan), using a Hybond-P membrane (GE Healthcare).
Activity assay
p-Nitrophenyl esters (2.5 mM) were used to determine the activities and substrate specificities of mROLWT and mROLimp. p-Nitrophenyl esters (p-nitrophenyl acetate [C2], p-nitrophenyl butyrate [C4], p-nitrophenyl hexanoate [C6], p-nitrophenyl caprylate [C8], p-nitrophenyl decanoate [C10], p-nitrophenyl laurate [C12], p-nitrophenyl myristate [C14], and p-nitrophenyl palmitate [C16]) were dissolved in 50 mM potassium phosphate buffer (pH 6.5) containing 0.5% (v/v) Triton X-100 and emulsified using an ultrasonic homogenizer (output 100 W) for 1 min at room temperature. Purified mROLWT and mROLimp were diluted to a concentration of 10 μg/mL, and 2 μL of the ROL solution was added to 98 μL of substrate solutions. After incubation for 30 min at 30°C, lipase activity was estimated by measuring the absorbance at 405 nm. For the competitive lipase activity assays with peptidase substrates, 4 μL of 10 mM Suc-Ile-Ile-Trp-MCA (Peptide Institute, Osaka, Japan), dissolved in dimethyl sulfoxide, was added to 94 μL of 2.5 mM p-nitrophenyl butyrate. After the addition of 2 μL of lipase solution (10 μg/mL), the mixtures were incubated for 40 min at 30°C and the absorbance was measured at 405 nm. p-Nitrophenyl butyrate was used, because its high solubility ensured the better reproducibility. Kinetic parameters were calculated from a Hanes-Woolf plot, using 4-methylumbelliferyl butyrate, which is a sensitive substrate to lipase activities and soluble at broad concentrations (10–300 μM). The activation energy was determined using the Arrhenius equation, E a = − RT ln(k/A), where A is the frequency factor, R is the universal gas constant (8.314 J·K−1·mol−1), T is the temperature, and k is the k cat at each temperature.
Determination of thermal stability and CD spectroscopy
The CD spectrum in the far-UV region (200–260 nm) was measured in a cell with an optical path length of 0.1 cm at 25°C using a JASCO J-720W spectropolarimeter (JASCO, Tokyo, Japan). The concentration of the protein was 50 μg/mL in 50 mM potassium phosphate buffer (pH 6.5). The fractions folded of mROLWT and mROLimp were measured by monitoring the negative ellipticities at 222 nm.
Cell surface display of mROLWT, mROLimp, and mROLimp2
S. cerevisiae strain BY4741/sed1Δ was transformed with the above-mentioned plasmids using the Frozen-EZ Yeast Transformation II kit. Transformants displaying mROLWT, mROLimp, or mROLimp2 were grown in 5 mL of SD medium containing the appropriate amino acids for 24 h at 30°C. The cells were harvested and resuspended in the same medium. After cultivation for 24 h at 30°C, the cells were used for immunofluorescence labeling and activity assays. For the immunofluorescence labeling, the cells were incubated with an anti-FLAG M2 mouse monoclonal antibody (Sigma-Aldrich) at room temperature for 1 h. The cells were then washed with phosphate buffer saline (pH 7.4) and mixed with the secondary antibody (Alexa Fluor-488 goat anti-mouse IgG [Life Technologies]) for 1 h at room temperature. After washing, fluorescence was measured at the excitation (λex) and emission (λem) wavelengths of 485 nm and 527 nm, respectively, using a Fluoroskan Ascent FL system (Labsystems, Helsinki, Finland). For the lipase activity assay, the cells were washed with 50 mM potassium phosphate buffer (pH 6.5) and the suspension was adjusted to OD600 of 3.0. After the addition of the substrate (100 μL) to the cell suspension (100 μL), the mixture was incubated for 20 min at 30°C with gentle shaking. For the peptidase activity assay, the cell suspension was adjusted to an OD600 of 20, and the cells (980 μL) were incubated with 10 mM Suc-Ile-Ile-Trp-MCA (20 μL) for 20 min at 30°C with gentle shaking. The amount of 7-amino-4-methylcoumarin was estimated at the excitation (λex) and emission (λem) wavelengths of 355 nm and 460 nm, respectively.
Results
Production and purification of mROLWT and mROLimp
Our previous study revealed that residues Ser38–Glu57 of proROL were essential for the folding of mROL [6]. This region is highly conserved among 4 homologous lipases (Fig 1), and contains three charged amino acids (Asp41, Glu45, and Lys51) and many hydrophilic residues, indicating that proROL interacts with mROL via electrostatic and hydrophilic interactions. In particular, residues Gln50–Trp55 are highly hydrophilic. We replaced these residues with a more hydrophilic and charged amino acid sequence (VDDDDK) to alter the chaperoning function of this region. The mutated propeptide was named proROL-mut1 and the mature ROL folded by proROL-mut1 was named mROLimp (imprinted ROL) to distinguish it from mROLWT (mROL folded by wild type proROL). The two distinct mROLWT and mROLimp with the same primary sequence were produced and purified from P. pastoris (S1 Fig) using a two-step purification protocol (see Materials and Method). No apparent decrease in molecular weight was observed after endoglycosidase H (EndoH) treatment, as analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), indicating that mROLWT and mROLimp were not N-glycosylated. To confirm that proROL and proROL-mut1 were cleaved at the same site and that the primary sequences of mROLWT and mROLimp were identical, the protein bands were excised and analyzed by N-terminal sequencing. The analysis confirmed that both sequences of mROLWT and mROLimp started with DDNLVGGMTLD, indicating that the propeptides were cleaved at the same site and that the primary sequences of mROLWT and mROLimp were the same.
Fig 1. Sequence alignment of Rhizopus oryzae lipase (ROL) and ROL-related lipases.

The full-length primary sequences of ROL, R. niveus lipase (RNL), R. stolonifer lipase (RSL), and R. chinensis lipase (RCL) are presented. Multiple-sequence alignments were generated using the ClustalW program (http://www.ebi.ac.uk/Tools/msa/clustalw2/). The underlined sequences in the propeptide of ROL (Ser20–Gly37 and Ser38–Glu57) indicate the regions that are essential for secretion and folding of mROL, respectively. The underlined sequences in the mature domain of ROL (Phe183–Asp189) indicate the lid domain. The shadowed region indicates residues that were replaced with hydrophilic amino acids (VDDDDK). In the original host, R. oryzae, the propeptide is also cleaved between the Ala97 and Ser98 residues [21]; however in P. pastoris and S. cerevisiae, the secondary cleavage has not been observed [14, 22]. Therefore, in this study, we defined the propeptide domain as the region between residues 1 and 69 and the mature domain as the region between residues 70 and 366 of ROL.
Lipase activity assay
The activities of mROLWT and mROLimp were measured using p-nitrophenyl esters as substrates to investigate their substrate specificities toward acyl groups of different chain lengths (C2–C16). Replacement of the hydrophilic residues of proROL with alternative residues had a considerable effect on the chaperoning function of the propeptide. The activity of mROLimp was higher than that of mROLWT, despite the fact that mROLimp and mROLWT had the same primary sequence (Fig 2A). The mutated propeptide not only generated mature protein with a higher activity but also altered the chain-length specificity. Compared with mROLWT, mROLimp showed a preference for substrates with medium-chain length acyl groups (Fig 2B). In particular, mROLimp exhibited 3.8-fold higher activity toward p-nitrophenyl decanoate (C10), and only 1.4-fold higher activity toward p-nitrophenyl palmitate (C16). To further characterize mROLimp, the enzyme kinetics of mROLWT and mROLimp were evaluated using 4-methylumbelliferyl butyrate (Table 1). The K M value of mROLimp was higher than that of mROLWT, indicating that the substrate specificity had been altered by proROL-mut1, resulting in a lower affinity of mROLimp for the substrate. However, mROLimp exhibited a significantly higher k cat value than mROLWT. The activation energy of mROLimp was also lower than that of mROLWT. These results suggest that the mutated propeptide affected the conformation of mROLimp not only around the substrate-binding site but also around the active site. A comparison of the activation energies indicated that the higher turnover rate of mROLimp than mROLWT may be attributed to the lower activation energy.
Fig 2. Lipase activity assay with purified ROL.

(A) Measurement of the lipase activities of mROLWT and mROLimp using p-nitrophenyl esters with various acyl chain lengths (C2–C16) as substrates. The resultant p-nitrophenol was quantified to estimate the lipase activities. (B) Relative lipase activities normalized with the values obtained for mROLWT. The values are presented as mean ± standard error of the mean (SEM) based on at least three independent measurements. (C) Competitive lipase activity assay with purified mROLWT and mROLimp. Lipase activities were determined in the presence of the peptidase substrate, Suc-Ile-Ile-Trp-MCA, dissolved in dimethyl sulfoxide (DMSO). The P-values were determined using the Student’s t-test. * P < 0.05.
Table 1. Kinetic parameters for mROL and mROLimp.
| K M (μM) | k cat (s-1) | k cat/K M (mM-1 s-1) | Ea (kJ mol-1) | |
|---|---|---|---|---|
| mROL | 24 ± 11 | 0.054 ± 0.003 | 2.9 ± 0.9 | 79 ± 10 |
| mROLimp | 79 ± 5 | 0.15 | 1.9 ± 0.2 | 46 ± 6 |
| P-value | 6.1 × 10–3 | 2.6×10–5 | 0.82 | 4.9×10–2 |
Results are show as mean ±sem.
Altered Substrate recognition
In order to further characterize mROLimp, we investigated whether the altered substrate binding site allowed mROLimp to recognize an alternative substrate that mROLWT could not recognize. The active sites of most lipases, including ROL, are mainly composed of three residues, Asp, His, and Ser [15, 23, 24]. These residues are also found in the active sites of serine proteases and peptidases [25]. Most serine protease and peptidases can be classified into three groups: type I, type II, and type III [26]. The disposition of the residues in the active site of ROL resembles that of type III serine protease (carboxypeptidase Y) closely (S2 Fig). Therefore, we investigated whether mROLWT and mROLimp recognized a peptidase-specific substrate by performing a lipase activity assay in the presence of a peptidase-specific substrate as a competitor. We used Suc-Ile-Ile-Trp-MCA as the peptidase-specific substrate; Suc-Ile-Ile-Trp-MCA is a hydrophobic substrate that is effectively cleaved by the type III serine peptidase, CPY [9]. Intriguingly, the peptidase substrate competed with the lipase substrate (p-nitrophenyl butyrate) for mROLimp, although it did not compete with the lipase substrate in the case of mROLWT (Fig 2C). This result further suggests that the mutated propeptide altered the substrate-binding site and allowed mROLimp to recognize the peptidase substrate.
Structural imprinting
We hypothesized that the mutated propeptide differently folded mROL and generated mROLimp which had distinct activities and substrate recognitions. The folding information of mROLimp were investigated using a far-ultraviolet (UV) circular dichroism (CD) spectrum. CD spectrum revealed that mROLimp obtained different secondary structures from mROLWT (Fig 3A). In addition, mROLimp was significantly more stable than mROLWT at higher temperatures (Fig 3B), according to the changes in negative ellipticity at 222 nm at each temperature. In contrast to mROLWT, mROLimp exhibited an intermediate state in the process of denaturation (60–80°C), indicating that the different denaturation pattern was attributed to the distinct secondary structure of mROLimp. The altered folding function of proROL-mut1 resulted in the generation of mROLimp which improved the lipase activities, altered the substrate recognitions, and had higher stability, in spite of having the same amino acid sequences as mROLWT.
Fig 3. Analysis of the structures and stabilities of mROLWT and mROLimp.

(A) Circular dichroism spectra of mROLWT and mROLimp. (B) The thermal stabilities of mROLWT and mROLimp. The negative ellipticities at 222 nm were measured to determine the fraction folded. The values are presented as mean ± SEM based on three independent measurements. The P-value was determined using a two-factor ANOVA with mROLs and temperatures as independent factors.
Discussion
In this study, we generated a functionally distinct mROL by mutating the propeptide of ROL. The mutated propeptide produced a distinct mROLimp with higher activity, and altered substrate recognition compared with mROLWT, even though the amino acid sequences of both lipases were the same. In addition, the structural stability of mROLimp was also improved compared with mROLWT. Because of the altered chaperoning function of proROL-mut1, mROLimp would be folded via an alternative folding pathway and reach a distinct minimum in the folding free energy landscape. The phenomenon of “protein folding memory” implies that proteins are not always folded into their most stable states, but can be folded into various states depending on their folding circumstances. “Protein folding memory” has also observed in CPY [9] and subtilisin (type II serine protease) [27, 28]. In these studies, mature domains were also folded into distinct states depending on their mutated propeptide. In this point, “protein folding memory” contradicts Anfinsen’s dogma.
The results of the lipase activity assay in the presence of peptidase-specific substrates revealed that purified mROLimp recognized the peptidase-specific substrate, while mROLWT did not. However, neither mROLWT nor mROLimp exhibited any peptidase activity. As an alternative method to evaluate peptidase activity, we adopted a cell-surface engineering technique, using the yeast Saccharomyces cerevisiae [29]. This method allows the display of a number of proteins on the cell surface of yeast and enables a simple and rapid activity assay. The productions and displays of mROLWT and mROLimp were confirmed by immunofluorescence labeling (S3 Fig). Remarkably, mROLimp displayed on the yeast cell surface exhibited significantly higher peptidase activity than mROLWT (Fig 4A). The displayed mROLimp also showed higher lipase activities, similar to that of purified mROLimp (Fig 4B). The mutated propeptide potentiated peptidase activity for mROL and immobilization on the cell wall enabled mROLimp to cleave the peptidase substrate.
Fig 4. Activity assay using yeast cell surface engineering.
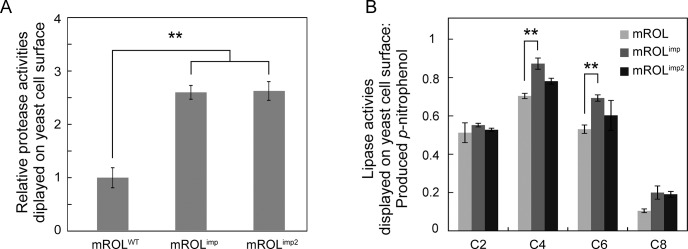
(A) Peptidase activity assay of mROLWT, mROLimp, and mROLimp2 displayed on the yeast cell surface. (B) Lipase activity assay of mROLWT, mROLimp, and mROLimp2 displayed on the yeast cell surface. The resultant p-nitrophenol was quantified to estimate the lipase activities. The peptidase and lipase activities were corrected by the number of displayed enzymes (S3 Fig). The values are presented as mean ± SEM based on three independent measurements. The P-values were determined using one-way analysis of variance followed by Tukey’s test for multiple comparisons. ** P < 0.01.
Most lipases have a lid domain that conceals the active site [16, 17]. In the case of Rhizomucor miehei lipase, which has 50% sequence homology to ROL, an open lid that forms a hydrophobic groove is important for substrate recognition [16]. The tertiary structure of mROLWT, modeled from the open-lid structure of R. miehei lipase, revealed that the open lid of mROLWT did not form a hydrophobic groove around the active site (Fig 5). The mutated propeptide probably affected the conformation of the lid domain and allowed the open lid to form a hydrophobic groove to interact with substrates composed of medium-chain length acyl groups and a hydrophobic peptidase-specific substrate. A previous study showed that mutations in the lid domain changed the substrate specificity of mROL [17], indicating that the open lid plays a critical role in substrate recognition. Because the lid domain and surrounding residues in ROL contain hydrophilic residues such as Arg184, Ser185, Thr188, and Asp189 (Fig 1), the additional hydrophilic residues introduced in the mutated propeptide might interact with the residues of the lid domain via hydrogen bonding. Interestingly, another propeptide mutant in which residues Gln50–Glu57 (which are essential for folding) were replaced with eight hydrophilic Asn residues also exhibited “protein folding memory” and induced peptidase activity in the mature domain of ROL (named mROLimp2; Fig 4A and 4B). These results strongly suggest that the hydrophilic residues in the propeptide interacted with the lid domain and surrounding residues in a different manner compared with wild type propeptide. CD spectrum analyses showed change in the secondary structure and thermal stability of mROLimp; however, it did not reveal detailed folding mechanisms of mROLWT and mROLimp. Crystallization analyses of mROLWT and mROLimp will provide further insight into the phenomenon of “protein folding memory”. Although we have attempted to crystallize ROL according to the crystallization methods for homologous lipases such as R. niveus lipase and R. miehei lipase, but it has been difficult. The elaborated structural characterizations of mROLs would be our subject of the future work.
Fig 5. Structure of mROLWT, modeled using the SWISS-MODEL program.
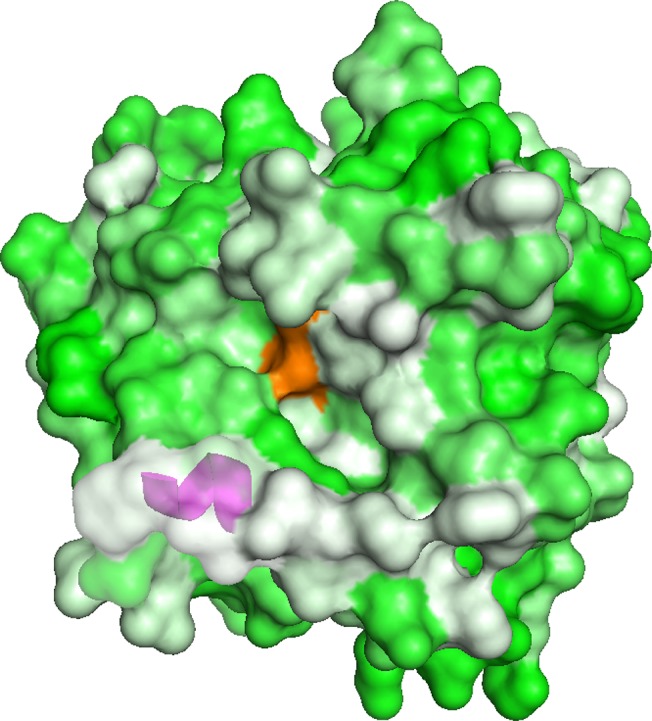
The structure of mROLWT was modeled based on the open-lid structure of Rhizomucor miehei lipase (Protein Data Bank [PDB] ID: 4TGL), and visualized using PyMOL. The active site residues, S242, D301, and H354, are colored orange. The magenta-colored α-helix represents the lid domain. Green residues indicate hydrophilic amino acids and white residues indicate hydrophobic amino acids.
Some peptide fragments have been reported to take both alpha- and beta- conformations depending on their context [30, 31]. Dalal et al. previously converted a predominantly β-sheet protein into four-helix bundle protein by redesigning the 50% of sequence in the original β-sheet protein [32]. Similarly, Alexander et al. designed two proteins that had 88% homology and completely distinct structures [33] and suggested that only 12% of the amino acids in the protein determined the whole structure. These studies clearly demonstrate that folding information for a protein structure does not reside equally in all residues in a protein, but rather resides in only a few residues. These studies, however, were limited to the folding information in mature domains. Our study further extends the idea. We suggest that the folding information is contained not only in a mature domain, but also in a propeptide domain which is cleaved off during maturation steps. In “protein folding memory”, the mature domain folds depending on the folding information in the propeptide and retains its folding memory even after the cleavage of the propeptide.
In conclusion, we demonstrated that mutated proROL exhibited “protein folding memory” and generated a functionally distinct and structurally more stable mROLimp that had the same amino acid sequence as mROLWT.
Supporting Information
N-glycosylation was not observed. *, mature form of ROL; **, EndoH; M, marker.
(TIF)
The active site of ROL is composed of three residues, S242, D301, and H354. These residues also form the catalytic triad in the active sites of serine proteases, bovine pancreas trypsin (PDB: 1S0Q), subtilisin (PDB: 2SIC), and carboxypeptidase Y (CPY; PDB: 1YSC). The steric conformation of the active site in ROL is particularly similar to that of the type III serine protease, CPY.
(TIF)
(A) The displays of mROLWT, mROLimp, and mROLimp2 were confirmed by immunofluorescence labeling. Positive control indicates yeast cells displaying only the FLAG tag and negative control indicates yeast cells displaying the strep-tag instead of the FLAG tag. Scale bar, 5 μm. (B) The number of displayed proteins was quantified by measuring fluorescence at the excitation (λex) and emission (λem) wavelengths of 355 and 460 nm, respectively.
(TIF)
Acknowledgments
We thank to Mr. Yoshihiro Yamamoto, Mr. Kiyo Hirooka, Mr. Naohiro Tomari, and Dr. Jun Wada from Kyoto Municipal Institute of Industrial Technology and Culture (Simogyo-ku, Kyoto, Japan) for N-terminal sequencing.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by a research fellowship from the Japan Society for the Promotion of Science for Young Scientists (grant 26-1413, http://www.jsps.go.jp/j-pd/index.html); Core Research for Evolutional Science and Technology, Japan Science and Technology Agency (http://www.jst.go.jp/kisoken/crest/); and a Grant-in-Aid for Scientific Research (B) (grant 25289293, http://www.jsps.go.jp/english/e-grants/grants01.html). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ikemura H, Takagi H, Inouye M. Requirement of pro-sequence for the production of active subtilisin E in Escherichia coli . J Biol Chem. 1987; 262: 7859–7864. [PubMed] [Google Scholar]
- 2. Inouye M. Intramolecular chaperone: the role of the pro-peptide in protein folding. Enzyme. 1990; 45: 314–321. [DOI] [PubMed] [Google Scholar]
- 3. Takahashi S, Ueda M, Atomi H, Beer HD, Bornscheuer UT, Schmid RD, et al. Extracellular production of active Rhizopus oryzae lipase by Saccharomyces cerevisiae . J Ferment Bioeng. 1998; 86: 164–168. [Google Scholar]
- 4. Boel E, Huge-Jensen B, Christensen M, Thim L, Fiil NP. Rhizomucor miehei triglyceride lipase is synthesized as a precursor. Lipids. 1988; 23: 701–706. [DOI] [PubMed] [Google Scholar]
- 5. Takahashi S, Ueda M, Tanaka A. Independent production of two molecular forms of a recombinant Rhizopus oryzae lipase by KEX2-engineered strains of Saccharomyces cerevisiae . Appl Microbiol Biotechnol. 1999; 52: 534–540. [DOI] [PubMed] [Google Scholar]
- 6. Takahashi S, Ueda M, Tanaka A. Function of the prosequence for in vivo folding and secretion of active Rhizopus oryzae lipase in Saccharomyces cerevisiae . Appl Microbiol Biotechnol. 2001; 55: 454–462. [DOI] [PubMed] [Google Scholar]
- 7. Ramos C, Winther JR, Kielland-Brandt M. Requirement of the propeptide for in vivo formation of active yeast carboxypeptidase Y. J Biol Chem. 1994; 269: 7006–7012. [PubMed] [Google Scholar]
- 8. Susanne OS, Van Den Hazel MC, Kielland-Brandt MC, Winther JR. pH-dependent processing of yeast procarboxypeptidase Y by proteinase A in vivo and in vitro . Eur J Biochem. 1994; 220: 19–27. [DOI] [PubMed] [Google Scholar]
- 9. Nagayama M, Maeda H, Kuroda K, Ueda M. Mutated intramolecular chaperones generate high-activity isomers of mature enzymes. Biochemistry. 2012; 51: 3547–3553. doi: 10.1021/bi3001159 [DOI] [PubMed] [Google Scholar]
- 10. Anfinsen CB. The formation and stabilization of protein structure. Biochem J. 1972; 128: 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ghosh B, Ray RR. Current commercial perspective of Rhizopus oryzae: a review. J Appl Sci. 2011; 11: 2470–2486. [Google Scholar]
- 12. Haas MJ, Bailey DG, Baker W, Berka TR, Cichowicz DJ, Derewenda ZS, et al. Biochemical and molecular biological characterization of a lipase produced by the fungus Rhizopus delemar . Eur J Lipid Sci Tech. 1999; 101: 364–369. [Google Scholar]
- 13. Beer HD, Wohlfahrt G, Schmid RD, McCarthy JE. The folding and activity of the extracellular lipase of Rhizopus oryzae are modulated by a prosequence. Biochem J. 1996; 319: 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Minning S, Schmidt-Dannert C, Schmid RD. Functional expression of Rhizopus oryzae lipase in Pichia pastoris: high-level production and some properties. J Biotechnol. 1998; 66: 147–156. [DOI] [PubMed] [Google Scholar]
- 15. Shibamoto H, Matsumoto T, Fukuda H, Kondo A. Molecular engineering of Rhizopus oryzae lipase using a combinatorial protein library constructed on the yeast cell surface. J Mol Catal B: Enzym. 2004; 28: 235–239. [Google Scholar]
- 16. Brzozowski AM, Derewenda U, Derewenda ZS, Dodson GG, Lawson DM, Turkenburg JP, et al. A model for interfacial activation in lipases from the structure of a fungal lipase-inhibitor complex. Nature. 1991; 351: 491–494. [DOI] [PubMed] [Google Scholar]
- 17. Shiraga S, Ishiguro M, Fukami H, Nakao M, Ueda M. Creation of Rhizopus oryzae lipase having a unique oxyanion hole by combinatorial mutagenesis in the lid domain. Appl Microbiol Biotechnol. 2005; 68: 779–785. [DOI] [PubMed] [Google Scholar]
- 18. Derewenda U, Brzozowski AM, Lawson DM, Derewenda ZS. Catalysis at the interface-the anatomy of a conformational change in a triglyceride lipase. Biochemistry. 1992; 31: 1532–1541. [DOI] [PubMed] [Google Scholar]
- 19. Horton RM, Cai Z, Ho S, Pease L. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. BioTechniques. 1990; 8: 528–535. [PubMed] [Google Scholar]
- 20. Smith P, Krohn RI, Hermanson G, Mallia A, Gartner F, Provenzano M, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985; 150: 76–85. [DOI] [PubMed] [Google Scholar]
- 21. Haas MJ, Allen J, Berka TR. Cloning, expression and characterization of a cDNA encoding a lipase from Rhizopus delemar . Gene. 1991; 109: 107–113. [DOI] [PubMed] [Google Scholar]
- 22. Ueda M, Takahashi S, Washida M, Shiraga S, Tanaka A. Expression of Rhizopus oryzae lipase gene in Saccharomyces cerevisiae . J Mol Catal B: Enzym. 2002; 17: 113–124. [Google Scholar]
- 23. Derewenda ZS, Derewenda U, Dodson GG. The crystal and molecular-structure of the Rhizomucor miehei triacylglyceride lipase at 1.9-angstrom resolution. J Mol Biol. 1992; 227: 818–839. [DOI] [PubMed] [Google Scholar]
- 24. Emmerich J, Beg O, Peterson J, Previato L, Brunzell J, Brewer H, et al. Human lipoprotein lipase. Analysis of the catalytic triad by site-directed mutagenesis of Ser-132, Asp-156, and His-241. J Biol Chem. 1992; 267: 4161–4165. [PubMed] [Google Scholar]
- 25.Brady L, Brzozowski AM, Derewenda ZS, Dodson E, Dodson G, Tolley S, et al. A serine protease triad forms the catalytic centre of a triacylglycerol lipase. 1990. [DOI] [PubMed]
- 26. Robertus JD, Kraut J, Alden RA, Birktoft JJ. Subtilisin. Stereochemical mechanism involving transition-state stabilization. Biochemistry. 1972; 11: 4293–4303. [DOI] [PubMed] [Google Scholar]
- 27. Shinde UP, Liu JJ, Inouye M. Protein memory through altered folding mediated by intramolecular chaperones. Nature. 1997; 389: 520–522. [DOI] [PubMed] [Google Scholar]
- 28. Shinde U, Fu X, Inouye M. A pathway for conformational diversity in proteins mediated by intramolecular chaperones. J Biol Chem. 1999; 274: 15615–15621. [DOI] [PubMed] [Google Scholar]
- 29. Kuroda K, Ueda M. Arming technology in yeast—novel strategy for whole-cell biocatalyst and protein engineering. Biomolecules. 2013; 3: 632–650. doi: 10.3390/biom3030632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mezei M. Chameleon sequences in the PDB. Protein engineering. 1998; 11: 411–414. [DOI] [PubMed] [Google Scholar]
- 31. Ghozlane A, Joseph AP, Bornot A, de Brevern AG. Analysis of protein chameleon sequence characteristics. Bioinformation. 2009; 3: 367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dalal S, Balasubramanian S, Regan L. Protein alchemy: Changing beta-sheet into alpha-helix. Nat Struct Biol. 1997; 4: 548–552. [DOI] [PubMed] [Google Scholar]
- 33. Alexander PA, He Y, Chen Y, Orban J, Bryan PN. The design and characterization of two proteins with 88% sequence identity but different structure and function. Proc Natl Acad Sci USA. 2007; 104: 11963–11968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
N-glycosylation was not observed. *, mature form of ROL; **, EndoH; M, marker.
(TIF)
The active site of ROL is composed of three residues, S242, D301, and H354. These residues also form the catalytic triad in the active sites of serine proteases, bovine pancreas trypsin (PDB: 1S0Q), subtilisin (PDB: 2SIC), and carboxypeptidase Y (CPY; PDB: 1YSC). The steric conformation of the active site in ROL is particularly similar to that of the type III serine protease, CPY.
(TIF)
(A) The displays of mROLWT, mROLimp, and mROLimp2 were confirmed by immunofluorescence labeling. Positive control indicates yeast cells displaying only the FLAG tag and negative control indicates yeast cells displaying the strep-tag instead of the FLAG tag. Scale bar, 5 μm. (B) The number of displayed proteins was quantified by measuring fluorescence at the excitation (λex) and emission (λem) wavelengths of 355 and 460 nm, respectively.
(TIF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.