

Abstract
Trypanosoma brucei , the causative agent for African trypanosomiasis, possesses a single mitochondrion that imports hundreds of proteins from the cytosol. However, the parasite only possesses a few homologs of the canonical protein translocases found in fungi and animals. We recently characterized a homolog of the translocase of the mitochondrial inner membrane, Tim50, in T. brucei. TbTim50 knockdown (KD) moderately reduced cell growth, decreased the mitochondrial membrane potential, and inhibited import of proteins into mitochondria. In contrast to Tim50 KD, we show here that TbTim50 overexpression (OE) increased the mitochondrial membrane potential as well as increased the production of cellular reactive oxygen species (ROS). Therefore, TbTim50 OE also inhibits cell growth. In addition, TbTim50 OE and KD cells showed different responses upon treatment with H2O2. Surprisingly, TbTim50 KD cells showed a greater tolerance to oxidative stress. Further analysis revealed that TbTim50 KD inhibits transition of cells from an early to late apoptotic stage upon exposure to increasing concentrations of H2O2. On the other hand TbTim50 OE caused cells to be in a pro-apoptotic stage and thus they underwent increased cell death upon H2O2 treatment. However, externally added H2O2 similarly increased the levels of cellular ROS and decreased the mitochondrial membrane potential in both cell types, indicating that tolerance to ROS is mediated through induction of the stress-response pathway due to TbTim50 KD. Together, these results suggest that TbTim50 acts as a stress sensor and that down regulation of Tim50 could be a survival mechanism for T. brucei exposed to oxidative stress.
Keywords: TbTim50, Mitochondria, Membrane potential, T. brucei, Oxidative stress, Apoptosis
1. Introduction
Trypanosoma brucei, a group of hemoflagellated parasitic protozoa, is the causative agent of a deadly disease known as African trypanosomiasis [1, 2]. The disease is spread among humans and domestic animals by an insect vector, the tsetse fly. During its digenetic life cycle within the fly and the mammalian host the parasite faces dramatic changes in its environment such as available nutrients, temperature, pH and oxygen concentration. To adapt to these changes T. brucei undergoes a series of developmental modulations involving its morphology, ultrastructure, metabolic patterns, and in particular its mitochondrial activities [3–6]. In eukaryotes, mitochondria play a central role in cellular homeostasis and cell death when exposed to various stresses by regulating different cellular functions such as, energy metabolism, redox balance, autophagy and apoptosis [7–10]. However, the role of mitochondria in a cellular stress response is less understood in parasitic protozoa like T. brucei.
Similar to higher eukaryotes, the majority of mitochondrial proteins in T. brucei are nuclear-encoded. Therefore these proteins are imported into mitochondria in order to perform their functions. In fungi and higher eukaryotes, there are three major protein translocases in mitochondria, the translocase of the mitochondrial outer membrane (TOM), and two translocases of the mitochondrial inner membrane, TIM23 and TIM22 [11, 12]. TOM and TIMs are multi-subunit protein complexes that have been extensively characterized in fungi and later in animals and plants. Interestingly, trypanosomatids do not have any homologs of the TOM subunits. Instead, an archaic β-barrel protein, ATOM, serves the function of Tom40, the major component of the fungal TOM complex [13]. Similarly, homologs of the TIM22 components could not be detected in this parasite, but it possesses a few homologs of the fungal TIM23 subunits, such as Tim17 and Tim50 [14, 15].
We have shown previously that T. brucei Tim17 (TbTim17) is essential for cell survival and is critical for mitochondrial protein import [14]. TbTim17 is present in a larger molecular mass protein complex and it is associated with several novel trypanosome-specific proteins [16]. We also have characterized Tim50 in T. brucei (TbTim50), which interacts with TbTim17 in vivo [15]. TbTim50 possesses a mitochondrial-targeting signal (MTS) at its N-terminus and a characteristic phosphatase motif at its C-terminus, which shows similarity to the transcription factor II (TFII)-stimulated RNA polymerase II C-terminal domain (CTD)-phosphatase. Knockdown (KD) of TbTim50 inhibits import of proteins into mitochondria that contain an N-terminal MTS, while the recombinant TbTim50 possesses a dual-specificity phosphatase activity [15].
Increasing evidence indicates that besides mitochondrial protein import, these Tom and Tim proteins are also involved in other functions, such as maintaining mitochondrial morphology, regulation of fission and fusion of the organelle and recruitment of anti-apoptotic and autophagy proteins [17–19]. Tom proteins are phosphorylated by cytosolic kinases to control mitochondrial protein biogenesis as a means of regulating mitochondrial activities [20–22]. In addition Tim50 is also known to be involved in developmental regulation and apoptosis in zebra fish, drosophila, and human [23, 24], although the mechanisms responsible for these actions are not well understood.
Here we discovered that TbTim50 plays a role in the stress response pathway in T. brucei. We found that maintenance of a particular level of TbTim50 is required in order to maintain cell growth and mitochondrial membrane potential at normal levels. Down regulation of TbTim50 inhibited the transition of cells from an early to a late apoptotic stage and thus increased the tolerance of T. brucei to peroxide treatment. Whereas, TbTim50 overexpression hyperpolarized the mitochondrial inner membrane, increased ROS production and promoted cell apoptosis. These results further indicate the important role of the T. brucei mitochondrion and its protein translocator in the maintenance of cellular homeostasis.
2. Materials and methods
2.1. Strains, media, cell growth and isolation of mitochondria
The procyclic form of T. brucei 427 cells was grown in SDM-79 medium containing 10% heat inactivated fetal bovine serum [15, 16]. T. brucei cells expressing TbTim50 RNAi (TbTim50 KD) and TbTim50 with a C-terminally 3x hemagglutinine (HA) tag (TbTim50 OE) were developed as previously reported [15]. These cell lines were maintained in the same medium supplemented with hygromycin (50 μg/ml), G418 (15 μg/ml) and phleomycin (2.5 μg/ml). Cell growth was assessed by inoculating the procyclic form at a cell density of 2 × 106/ml in fresh medium containing antibiotics in the presence or absence of doxycycline (1 μg/ml). Cells were counted at different growth time points with a Neubauer hemocytometer. The log of the cumulative cell number was plotted against time of incubation in culture.
Mitochondria were isolated from the parasite after lysis by nitrogen cavitation in isotonic buffer [14, 15]. The isolated mitochondria were stored at a protein concentration of 10 mg/ml in SME buffer (250 mM sucrose, 20 mM MOPS/KOH, 2 mM EDTA, pH 7.4) containing 50% glycerol at −70°C. Before use, mitochondria were washed twice with 9 volumes of SME buffer to remove glycerol.
2.2. Isolation of RNA and northern analyses
Total RNA was isolated from the procyclic form of T. brucei using Trizol (Invitrogen) according to the manufacturer’s protocol. Northern blot analysis was performed as described [14, 15]. Specific probes were made using a random primer labeling protocol (Invitrogen) from cDNA fragments, which were generated by PCR using sequence-specific primers designed from the coding regions of TbTim50 and actin.
2.3. SDS-PAGE and immunoblot
Proteins from isolated mitochondria were resolved on a 10% SDS/PAGE gel and then transferred to nitrocellulose membranes (Bio-Rad) [14, 15]. The antigens were visualized by using an enhanced chemiluminescence kit (ECL; Amersham). Antibodies against TbTim50 [15], HA-epitope (Clone 12CA5, Roche Applied Science), and T. brucei β-tubulin [25] were used as probes. The secondary antibodies used were either anti-rabbit or anti-mouse immunoglobulins linked to horseradish peroxidase (Amersham Life Science).
2.3. Hydrogen peroxide treatment
TbTim50 RNAi and TbTim50-3HA T. brucei were induced for four days with doxycycline (1 μg/ml). Parental T. brucei 427 cells were also grown in parallel for 4 days. Cells were then exposed to different concentration of hydrogen peroxide (0–800 μM) for 2 h. Treated cells were immediately used for further analysis.
2.4. Live/dead assay
T. brucei parental 427, TbTim50 RNAi and TbTim50-3HA cells (2 x 107) were harvested, washed once with 1x PBS and resuspended in 200 μl of 1 x PBS. C12-resazurin and SYTOX Green dyes (Molecular Probes®) were then added to each 100 μl cell suspension at a final concentration of 500 nM. Cells were incubated at 27 °C for 15 min. Next, 400 μl of the 1x PBS was added to the cells and the mixtures kept on ice. The samples were analyzed immediately by flow cytometry, with excitation at 488 nm and measurement of the fluorescence emission at 530 nm and 575 nm.
2.5. Mitotracker staining
Cells were harvested and suspended in fresh culture medium at a density of 4–5 × 106 /ml. Cells were stained with MitoTracker® Red, fixed and permeabilized as previously described [14, 15]. Briefly, MitoTracker® Red CMXROS (Molecular Probe®) was dissolved in DMSO at a concentration of 1 mM and added to the cell suspension at a final concentration of 0.5 μM. The mixtures were incubated at 27°C for 10 min. Cells were then washed and incubated in fresh culture medium for an additional 30 min. Cells were washed twice with 1x PBS and fixed in 0.37% paraformaldehyde at 4°C for 5 min. Next, cells were centrifuged, washed and resuspended in cold PBS and stored at 4 °C until FACS analysis. Fluorescence intensity (Geom. Mean) was measured with a FACSCalibur (Becton Dickinson) analytical flow cytometer using absorption at 578 nm and emission at 599 nm.
2.6. Measurement of ROS
T. brucei cells (1 x 107/ml) were incubated with 10 μM 2′,–7′ dichlorodihydrofluorescin diacetate (DCFH-DA) for 30 min at 27 °C. After incubation, cells were washed once with 1x PBS and analyzed by flow cytometry, by measuring the fluorescence emission at 520 nm using the excitation wavelength at 492 nm.
2.7. Annexin staining
T. brucei cells were stained with Alexa FluorR 488 conjugated annexin V and propidium iodide (PI) using an apoptosis assay kit (Molecular Probe®) according to the manufacturer’s protocol. Briefly, cells (2 x 107) were harvested and washed in cold PBS. The washed cells were resuspended in 1x annexin-binding buffer (200 μl). Alexa FluorR 488 annexin V and propidium iodide were added to the cell suspensions as directed in the protocol and cells were incubated at room temperature for 15 min. After the incubation period, 400 μl 1x annexin-binding buffer was added, mixed gently and kept on ice. The stained and unstained cells were analyzed by flow cytometry, by measuring the fluorescence emission at 530 nm and 575 nm using the excitation wavelength at 488 nm.
2.8. TUNEL assay
T. brucei cells (1 x 107) were harvested, washed with 1x PBS and resuspended in 500 μl of the same buffer. Cells were fixed in para-formaldehyde (0.37%) and permeabilized uxing Triton X-100 (0.1%). Cells were stained with fluorescin-12-dUTP using the DeadEndTM fluorometric TUNEL system (Promega) according to manufacturer’s protocol. Cells were then analyzed by flow cytometry by measuring the green fluorescence of fluorescein-12-dUTP at 520 nm using the excitation wavelength at 488 nm.
2.8. Flow cytometry
Samples were analyzed on a Beckman Coulter Epics XL-MCL and results were analyzed using FlowJo software (Treestar software). Compensation of spectral overlap was performed using single-stained control cells. Compensation was manually corrected as needed to ensure that single-stained cells did not fall below the axis. All flow cytometry experiments were performed at least three times for each condition.
3. Results
3.1. TbTim50 OE reduces T. brucei cell growth
A stable cell line of T. brucei containing an inducible construct for TbTim50 with 3xHA epitope tag at the C-terminal was developed [15]. After induction with doxycycline, TbTim50 OE cells produced an ectopic copy of the TbTim50 transcript, which was smaller in size than the endogenous copy because of having different 3′ and 5′ untranslated regions. The endogenous TbTim50 transcript was also expressed at similar levels in the presence and absence of doxycycline (Fig. 1A). Immunoblot analysis using ant-HA antibody detected TbTim50-3HA (~54 kDa) in the mitochondria samples isolated from TbTim50 OE cells but not from those in the parental wild type control (Fig. 1B). A pair of smaller molecular size proteins (~52 kDa and ~50 kDa) was also detected by anti-HA antibody from TbTim50 OE mitochondrial proteins, which could represent the degradation products of TbTim50-3xHA. Reprobing the same blot with anti-TbTim50 antibody recognized both the endogenous and ectopically expressed TbTim50 in TbTim50 OE samples and only the endogenous protein in the mitochondrial samples isolated from the parental T. brucei. There was about a 2-fold increase of the TbTim50 protein levels in TbTim50 OE compared to parental controls.
Fig. 1.

TbTim50 overexpression and its effect on cell growth. (A) Northern blot analysis of total RNA isolated from T. brucei with a genome-integrated copy of TbTim50-3x hemaglutinin antigen (HA). Cells were grown in the presence (+) or absence (−) of doxycycline for 4 days. Northern blot was probed using radiolabelled TbTim50 and tubulin cDNAs. The ectopic (ecto) and the endogenous (endo) TbTim50 transcripts in TbTim50 OE are indicated. (B) Western blot analysis of mitochondrial proteins from the parental control and TbTim50 OE T. brucei after induction for 4 days with doxycycline were performed using antibodies for HA-epitope, TbTim50, and T. brucei β-tubulin. One hundred and 50 μg of mitochondrial proteins was loaded per sample. (C) Growth kinetics of T. brucei parental, and TbTim50 OE cells. The TbTim50 OE cells were grown either in the absence (Uninduced) or presence (Induced) of doxycycline. Cell numbers were counted every alternate day post-induction. Parasite cultures were diluted when the cell number reached 1 x 107/ml. The log of the cumulative cell number was plotted versus time. Results are representatives of three independent experiments. Standard errors were less than 0.1%.
Interestingly, we found that overexpression of TbTim50 did not favor cell growth of T. brucei (Fig. 1C). Induced cells grew at a moderately lower rate than uninduced controls. However, when compared to the parental controls both uninduced and induced cells grew at a slower rate. The cellular doubling times calculated at the exponential phases of growth (2–6 days) were 10±0.5, 13.6±1.6, and 19.4±0.15 for parental, uninduced, and induced T. brucei, respectively. Previously, we reported that TbTim50 KD caused a moderate reduction in cell growth [15]. Together, these results suggest that an optimal level of TbTim50 is required for normal cell growth. We did not observe any growth inhibition due to overexpression of TbTim17, another mitochondrial protein translocator in T. brucei [16], indicating that TbTim50 may play an additional role in cell function other than translocation of proteins into mitochondria.
3.2. TbTim50 OE increases the mitochondrial membrane potential
In fungi, Tim50 maintains the permeability barrier across the MIM by closing the TIM23 channel in the absence of precursor proteins [26]. In order to investigate the effect of TbTim50 OE on the mitochondrial membrane potential, cells were stained with Mitotracker red which accumulates in mitochondria in a membrane potential-dependent manner. The parental and TbTim50 KD cells were also used in parallel to compare. We found that compared to the parental controls, TbTim50 OE increased the mitochondrial membrane potential ~20% and ~50% on day 2 and day 4, respectively (Fig. 2). This is in contrast to TbTim50 KD, which reduced the mitochondrial membrane potential by ~30% on day 2 and ~40% on day 4 post-induction compared to parental controls (Fig. 2) [15]. The change in the membrane potential due to alteration of TbTim50 levels progressed with the term of the induction period. As expected, pretreatment of cells with carbonyl cyanide m-chlorophenyl hydrazine (CCCP) at a final concentration of 50 μM reduced the intensity of staining more than 80%. Therefore, these results indicate that similar to fungi [26] and Drosophila [24], TbTim50 plays a role in maintaining the mitochondrial inner membrane permeability barrier in T. brucei.
Fig. 2.

Effects of TbTim50 up- and down-regulations on the mitochondrial membrane potential. The TbTim50 KD and OE cells were induced with doxycycline and cells (2 x 107) were harvested at day 2 (A) and day 4 (B). Live cells were stained with Mitotracker Red as described in the materials and methods. Cells were fixed with para-formaldehyde, washed and resuspended in cold PBS for FACS analysis. Fluorescence intensity was measured with a FACSCalibur (Becton Dickinson) analytical flow cytometer using absorption at 578 nm and emission at 599 nm. Flowjo software was used to analyze the results. Parental cells were also pre-treated with carbonyl cyanide m-chlorophenyl hydrazine (CCCP) at a final concentration of 50 μg/ml to disrupt mitochondrial membrane potential. (B) Quantitation of the fluorescence intensity from triplicate samples was performed using the same software. Fluorescence intensity of TbTim50 OE and KD cells were calculated as percent of that in the parental control in respective days. Values shown are mean ±standard errors (*p <0.05).
3.3. TbTim50 OE increases cellular ROS
Mitochondria are the primary site for the production of cellular ROS. To investigate the effect of an alteration of TbTim50 levels on cellular ROS production, we treated cells with the fluorogenic probe, DCFH-DA, which diffuses into cells and is deacetylated by cellular esterases to non-fluorescent DCFH. Inside cell, DCFH is rapidly oxidized to highly fluorescent DCF by ROS. After measurement of the fluorescent intensity of the DCFH-DA treated cells revealed that the levels of cellular ROS was increased about 4 fold on day 2 and 12 fold on day 4 post-induction in TbTim50 OE cells in comparison to parental controls (Fig. 3). Whereas, the ROS level in TbTim50 KD cells was about the same level at day 2, and was slightly (~2 fold) increased on day 4 in comparison to that in parental T. brucei controls. Therefore, TbTim50 OE caused more distress in cells than TbTim50 KD. We did not see a similar change in cellular ROS due to over-production of Tim17 (data not shown), suggesting that the effect was specific for TbTim50.
Fig. 3.

Effects of TbTim50 up- and down-regulations on the levels of cellular reactive oxygen species (ROS). The TbTim50 KD and OE cells were induced with doxycycline and cells (2 x 107) were harvested at day 2 (A) and day 4 (B). Cells were washed with 1x PBS and stained with DCFH-DA as described in the materials and methods. Fluorescence intensity was measured with a FACSCalibur (Becton Dickinson) analytical flow cytometer at 520 nm. Flowjo software was used to analyze the results. (C) Fluorescence intensity was calculated from triplicate samples using the same software and plotted as percent of the parental control at the respective time periods. Values shown are mean ± standard errors (*p <0.05).
3.4. TbTim50 KD increases T. brucei tolerance to oxidative stress
In order to investigate the effect of externally added oxidative stress on TbTim50 KD and OE T. brucei, we treated cells with various concentrations of H2O2 and assayed cell viability on the basis of incorporation of the two dyes sytox and C12-resazurin, followed by flow-cytometry analyses (Fig. 4). Sytox is cell impermeable, thus it only stains dead or necrotic cells, whereas C12-resazurin is cell permeable, thus it is capable of penetrating living cells [27, 28]. Within metabolically active cells, the nonfluorescence C12-resazurin is converted to fluorescent C12-resorufin, thus fluorescence intensity is directly proportional to the number of live cells in population. Cells that were unstained or stained with resorufin alone and those that were heat-killed and treated with sytox were used as controls. Flow-cytometry results revealed that with increasing concentrations of H2O2, the parental cells became more and more injured as indicated by the number of cells present in the upper right corner of each plot (Fig. 4A). A similar situation was observed for Tim50 OE cells. However, Tim50 KD cells were less injured as seen with the increasing concentrations of H2O2. Even after treatment with 800 μM H2O2 about 80–90 % of the cells were alive and healthy as shown by the number present in the lower right corner of the plot. Comparing the live versus injured ratio of each cell type at different concentrations of H2O2 (from three independent experiments) clearly confirmed that Tim50 KD cells were relatively more resistant to exposure to an oxidative stress than either parental or Tim50 OE cells (Fig. 4B).
Fig. 4.

Effects of TbTim50 up- and down-regulations on cell injury during H2O2 treatment. TbTim50 KD and TbTim50 OE cells were grown in the presence of doxycycline for 4 days. Parental control T. brucei were also grown in parallel. (A) Cells were treated with different concentration of H2O2 (0–800 μM) for 2 h, washed with PBS and stained with sytox green and resorufin as described. FACS analysis was performed immediately and data were analyzed by Flowjo software. FACS analysis of the unstained, heat killed (Dead) and live parasites were performed in parallel. (B) Ratios of the live and injured cell (%) were calculated from three independent experiments and plotted versus the different cell type at different concentrations of H2O2. Values shown are mean ± standard errors from triplicate samples.
3.5. Treatment with H2O2 decreases the mitochondrial membrane potential and increases the levels of ROS in the parental, TbTim50 OE and TbTim50 KD T. brucei
As we observed that TbTim50 KD cells were more tolerant to H2O2 treatment than TbTim50 OE and parental T. brucei, we wanted to investigate the effect of such treatment on the mitochondrial membrane potential and levels of ROS in these cells. Results showed that the mitochondrial membrane potential dropped over 75% in all three types of cells after H2O2 treatment (Fig. 5A–D). A reduction in mitochondrial membrane potential was observed even at the lowest concentration of H2O2 (200 μM). This indicated that oxidative stress similarly depolarized the mitochondrial membrane in parental, TbTim50 OE and KD cells. These results also explained our observation that longer incubation times (> 4h) killed most of these cells even at 200 μM H2O2. Therefore an increased tolerance in Tim50 KD cells is due to an increase in the immediate adaptive response of T. brucei upon H2O2 treatment.
Fig. 5.

Effects of hydrogen peroxide treatment on mitochondrial membrane potential and cellular ROS. T. brucei parental (A and E), TbTim50 OE (B and F), and TbTim50 KD (C and G) cells were treated with different concentrations of H2O2 (0, 200, 400, and 800 μM) for 2h. Before treatment, TbTim50 OE and KD cells were induced for 4 days in the presence of doxycycline. Cells were washed once with 1x PBS and immediately used for Mitotracker (A–C) and DCFH-DA (E–G) staining as described. FACS analysis results are shown as histograms. Fluorescence intensity from Mitotracker (D) and DCFH (H) staining were measured using Flowjo software and plotted for different types of cells. Values for intensity were presented in arbitrary unit (A.U.). Standard errors were calculated from three independent experiments.
In addition to mitochondrial membrane potential, we found that the levels of ROS increased gradually with increasing concentrations of H2O2 in parental and Tim50 KD cells (Fig. 5E, G, and H). Therefore, these results showed that although the level of ROS increased during peroxide treatment, TbTim50 KD cells were able to partially resist that stress. Peroxide treatment also increased the level of ROS in TbTim50 OE cells however the results were more adverse (Fig. 5F and H). With an increasing concentration of H2O2, the number of TbTim50 OE cells with higher DCF-staining was reduced and a sub-population of cells stained at a lower intensity increased (Fig. 5F). This is because those TbTim50 OE cells that were injured/dead due to H2O2 treatment were weakly stained with DCF. Some weakly stained cells also accumulated in parental and Tim50 KD T. brucei after treatment with higher concentrations (400–800 μM) of H2O2, however, the number of cells in these populations was relatively lower in comparison to that found for TbTim50 OE. Therefore, these results further indicate that oxidative stress caused more injury to TbTim50 OE cells in comparison to the parental and Tim50 KD T. brucei. The tolerance of TbTim50 KD cells to oxidative stress is not likely due to an increased capacity of these cells to neutralize ROS. Rather it could be due to an increase in a signaling ability of these cells, which mediates cell survival upon oxidative stress. On the other hand, TbTim50 OE cells were more vulnerable to oxidative stress because these cells already have more ROS due to OE of TbTim50.
3.6. TbTim50 KD reduces the transition of cells from early to late apoptotic stages upon treatment with an increasing concentration of H2O2
Next we determined if Tim50 KD or OE had any effect on the development of an apoptotic phenotype in cells treated with H2O2. For this purpose we simultaneously stained cells with propidium iodide (PI) and annexin V and then analyzed these cells by flow-cytometry (Fig. 6). Cells that were either unstained or treated with annexin V alone, and heat-killed cells treated only with PI were used as positive and negative controls. FACS analysis showed that parental cells not treated with H2O2 were sorted in the lower-left quadrant, indicating that these were live cells minimally stained with both dyes (Fig. 6A). With increasing concentration of H2O2, cells became more apoptotic, thus more cells sorted to the upper- and lower-right quadrants. Cells in the upper-right quadrant (Annexin+ /PI+) are at the late-apoptotic stage while those in the lower-right quadrant (Annexin+ /PI-) of the plot are in the early-apoptotic stage. We found that a large proportion (50–60%) of Tim50 OE cells were already in the early apoptotic stage without any H2O2 treatment. This could be due to increased ROS levels and hyperpolarization of the mitochondrial membrane in this cell type due to Tim50 OE. After treatment with a low dose (200 μM) of H2O2, these cells were sorted as being dead. With increasing concentrations of H2O2, the numbers of Tim50 OE cells sorted into the late apoptotic stage were relatively greater in comparison to the parental cell type (Fig. 6A). In contrast to OE, most of the Tim50 KD cells were alive to begin with. After treatment with H2O2 these cells were primarily in an early apoptotic stage and only 2–3% of cells were in the late apoptotic stage even after treatment with the highest concentration of H2O2 (Fig. 6A). Comparing the ratio of the early versus late apoptotic cell populations for different types of T. brucei tested at different concentrations of H2O2 revealed that the parental and Tim50 OE cells quickly reached the late apoptotic stage as H2O2 concentrations were raised (Fig. 6B). However, transition from the early to the late apoptotic stage upon exposure to oxidative stress was somehow blocked in Tim50 KD cells (Fig. 6B).
Fig. 6.
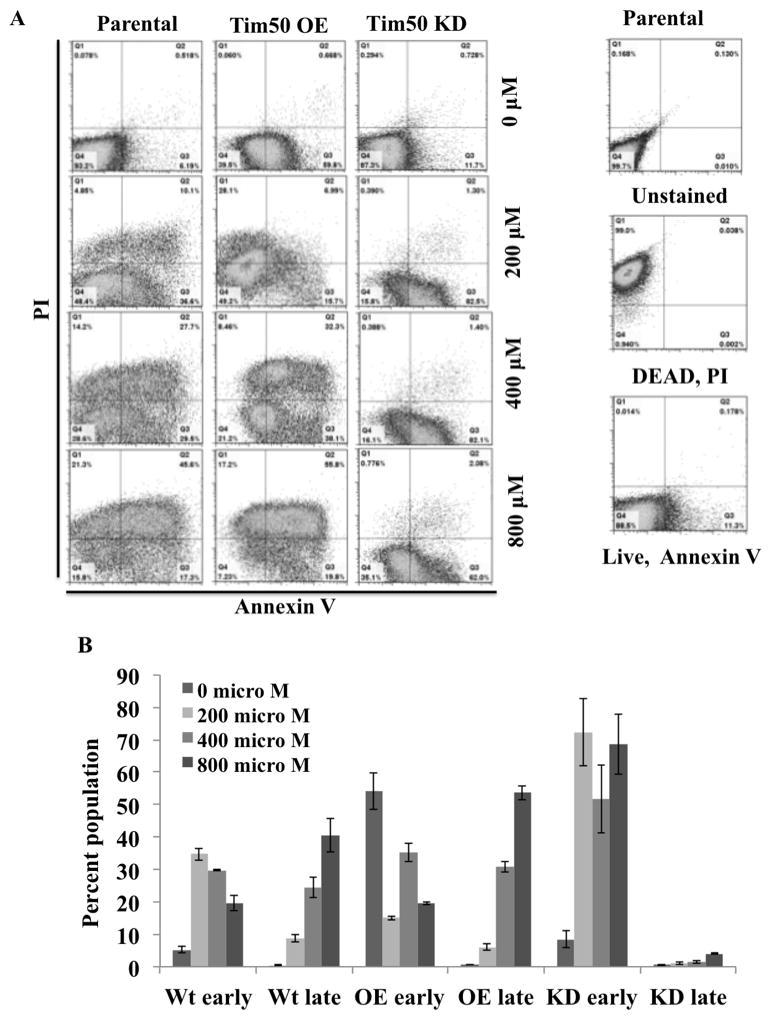
Effects of TbTim50 up- and down-regulations on apoptosis upon oxidative stress. The TbTim50 KD and TbTim50 OE cells were grown in the presence of doxycycline for 4 days. Parental control T. brucei were also grown in parallel. (A) Cells were treated with different concentration of H2O2 (0–800 μM) for 2 h, washed with PBS and stained with Annexin V and Propidium iodide (PI) as described. FACS analysis was performed immediately and data were analyzed by Flowjo software. FACS analysis of the unstained, heat killed (Dead) and live parasites of each type were performed in parallel. (B) Cell populations in the lower-right and upper-right quadrants were considered to be the late- and early-apoptotic stages, respectively. The populations of early- and late- apoptotic cells (%) were calculated from three independent experiments and plotted for different cell types [parental (Wt), TbTim50 OE (OE) and TbTim50 KD (KD)] and for different H2O2 concentrations.
3.7. TbTim50 OE increases nuclear DNA fragmentation upon treatment with an increasing concentration of H2O2
Nuclear DNA fragmentation is a hallmark for the late stage of apoptosis. To assess nuclear DNA fragmentation, TUNEL assays were performed after treating the parental, Tim50 OE, and Tim50 KD cells with different concentrations of H2O2. Unstained cells were used as the negative controls for FACS analysis. The TUNEL system measures the fragmented DNA of apoptotic cells by catalytically incorporating fluorescin-12-dUTP at 3′-OH DNA ends using the recombinant enzyme terminal deoxynucleotidyl transferase (rTDT) [29]. Results showed that TUNEL-positive cell populations increased with increasing concentration of peroxide due to Tim50 OE (Fig. 7A and B). The shift in fluorescence intensity was maximum when Tim50 OE cells were treated with 800 μM H2O2 (Fig. 7B). In contrast, the fluorescence intensities were comparable in Tim50 KD cells when treated with 200–800 μM concentration of H2O2. At 800 μM H2O2, parental cells were stained relatively more than Tim50 KD T. brucei. Together these results support the concept that Tim50 is required for transition of T. brucei cells to late apoptotic stages in the presence of oxidative stress.
Fig. 7.

Effect of TbTim50 up- and down-regulations on nuclear DNA degradation during H2O2 treatment. TbTim50 KD and TbTim50 OE cells were grown in the presence of doxycycline for 4 days. Parental control T. brucei were also grown in parallel. (A) Cells were treated with different concentrations of H2O2 (0–800 μM) for 2 h, washed with 1x PBS and prepared for TUNEL assay as described. Incorporation of BrdU into nicked DNA was assessed by FACS analysis. Fluorescence intensity was measured with a FACSCalibur (Becton Dickinson) analytical flow cytometer at 520 nm. Flowjo software was used to analyze the results. (B) Quantitation of the fluorescence intensity from triplicate samples was performed using the same software. Fluorescence intensity in arbitrary units (A.U.) of the parental, TbTim50 OE and KD cells was plotted with respect to H2O2 concentrations (0–800 μM). Values shown are mean ± standard errors (*p <0.05).
4. Discussion
Although the major function of mitochondria is cellular energy production, this organelle is intimately involved in various other cellular processes, such as cell death, aging, and stress [30–32]. Therefore, communication between mitochondria and the rest of the cell is critical to maintaining cell homeostasis during normal stages and during various challenges due to environmental stimuli [33–35]. This communication is critical in the case of T. brucei, which faces various environmental challenges during its digenetic life cycle and it is supported by the fact that the parasite extensively modulates its mitochondrial activities to cope with such assaults. Here we show that the mitochondrial protein translocator Tim50 in T. brucei is associated with the cellular stress-response pathway. TbTim50 works as a sensor for mediating stress signals during oxidative assault. Therefore, both up and down regulation of this protein hampered cellular homeostasis. TbTim50 OE increased the level of cellular ROS and hyperpolarized the mitochondrial membrane leading to cellular apoptosis upon externally added ROS. In contrast, TbTim50 KD induced an unknown signaling event that protected T. brucei during oxidative stress.
In fungi, it has been reported that Tim50 controls the mitochondrial membrane potential by closing the import channel formed by Tim23 in the absence of any presequence-containing protein to avoid proton leakage [26]. The N-terminal targeting signal or presequence of mitochondrial proteins interact with the Tim50 C-terminal domain, which is topologically positioned in the inter-membrane space of mitochondria [36, 37]. This interaction then activates the TIM23 channel for protein translocation [26]. Therefore it can be assumed that TbTim50 has a similar function by closing the protein import channel that is possibly formed by TbTim17 (T. brucei lacks an equivalent of Tim23). We have shown that TbTim50 interacts in vivo with TbTim17 [15]. Although the membrane topology of TbTim50 has not been determined, the predicted primary and the 3D-structure analyses revealed that TbTim50 does possess a unique region within amino acid residues 259–274, which could be responsible for its interaction with TbTim17 [15]. Therefore this unique region of TbTim50 may play a role in keeping the channel closed in the absence of incoming precursor proteins. However, further investigation is required to verify these assumptions. In any case it is most likely that TbTim50 OE hyperpolarized the mitochondrial inner membrane by tightly closing the import channel. It is known in other systems that hyperpolarization increases swelling of mitochondria, which in turn induce mitochondrial permeability transition pores formed by the mitochondrial ADP/ATP carrier and VDAC, thus inducing apoptosis [38, 39]. A similar mechanism may exist in T. brucei. In a unicellular parasitic protozoan like T. brucei, the path to apoptotic cell death has not been well defined [40, 41]. However, typical hallmarks of apoptosis such as mitochondrial membrane depolarization, cytochrome C release, phosphatidyl serine exposure and fragmentation of nuclear DNA are induced in this parasite upon induction of oxidative stress or treatment with staurosporin, concanavalin A, and prostaglandin A2 [40–43]. We found that a large proportion of the TbTim50 OE cells (~60%) were annexin-positive even before addition of H2O2, which shows that TbTim50 OE is apoptotic in T. brucei. In addition, we found that after H2O2 treatment, more cells became TUNEL positive due to TbTim50 OE than TbTim50 KD, indicating that Tim50 OE enhances apoptotic events under such conditions in T. brucei.
In contrast to Tim50 OE, Tim50 KD cells are somehow resistant to progression to the late stage of apoptosis during oxidative assault. Since TbTim50 is critical for mitochondrial protein import, Tim50 KD alters mitochondrial protein homeostasis (or proteostasis). We speculate that this change induces the mitochondrial unfolded protein response (UPRmt) pathway. It has been reported recently in human cells that chemical stress induces degradation of Tim17A, a membrane integral component of the TIM23 translocase [44]. Degradation of Tim17A by mitochondrial proteases is adequate to activate the UPRmt pathway in these cells. Furthermore, in Caenorhabditis elegans, shRNA mediated down regulation of Tim17 increased resistance to oxidative stress [44]. In addition, down regulation of Tim23 in the same organism caused an accumulation of ATFS in the nucleus. ATFS is a bZip transcription factor, which translocates to the mitochondria and degrades quickly via mitochondrial protease in the absence of oxidative stress [45]. Once accumulated in the nucleus under stress, ATFS induces expression of various stress response proteins, which includes mitochondrial chaperones and protein translocators, to restore mitochondrial functions [45]. It is not clear yet how UPRmt works in T. brucei, however, it can be assumed that inhibition of mitochondrial protein import by down regulation of TbTim50 induces a stress response pathway that increases the levels of the stress-response proteins post-transcriptionally (since transcriptional regulation is rarely found in trypanosomatids [46]), thus increases tolerance of T. brucei to oxidative stress.
We reported previously that TbTim50 up- and down-regulation inversely regulate the steady state levels of the mitochondrial voltage-dependent anion channel (VDAC) [15]. VDAC is a β-barrel protein and is the most abundant protein in the mitochondrial outer membrane (MOM) [47]. VDAC forms an aqueous channel that mediates the major metabolite flux between mitochondria and the cytosol [48, 49]. In T. brucei down regulation of VDAC reduces cell growth, mitochondrial ATP production, and alters the respiratory phenotype [47, 50]. In metazoans, VDAC plays a crucial role in programmed cell death (PCD) [51–53]. VDAC interacts with various pro- and anti-apoptotic proteins and regulates the fate for cell death/survival [54–57]. Here, we speculated that an increased level of VDAC in TbTim50 KD cells inhibits apoptosis by recruiting some uncharacterized anti-apoptotic proteins in T. brucei. Therefore, these cells appear more tolerant to oxidative stress. However, further investigations are required to prove this hypothesis.
Overall our results showed that there is a well-orchestrated regulatory process in T. brucei to help maintain cellular homeostasis upon extrinsic stress signals; mitochondrial proteins like Tim50 play a crucial role in this process. Future investigations will explore the communication between mitochondrial and other cellular functions in T. brucei.
Highlights.
Tim50 acts as a stress-sensor
Tim50 OE increases ROS and the mitochondrial membrane potential
Tim50 KD inhibits transition of cells from an early to late apoptotic stage
Tim50 KD increases T. brucei’s tolerance to oxidative stress
Tim50 KD could be a survival mechanism for T. brucei exposed to oxidative stress.
Acknowledgments
We thank George Cross for Procyclic 427 (29-13), Keith Gull for p2T7-177 RNAi vector and anti-tubulin antibody, and Xiaoming Tu for modified pLEW100-3HA vector. We thank Julia Liu for assistance with flow cytometry. We also thank Diana Marver for critically reviewing the manuscript. This work was supported, in whole or in part, by National Institute of Health grant 2SC1GM081146 and training grants 5T32HL007737, 5T32AI007281 and 2R25GM059994. The Flow Cytometry Core Facility was supported by National Institutes of Health grant G12RR003032.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature cited
- 1.Sternberg JM, Maclean L. A spectrum of disease in human African trypanosomiasis: the host and parasite genetics of virulence. Parasitology. 2010;137:2007–15. doi: 10.1017/S0031182010000946. [DOI] [PubMed] [Google Scholar]
- 2.Barrett MP, Burchmore RJ, Stich A, Lazzari JO, Frasch AC, Cazzulo JJ, Krishna S. The trypanosomiases. Lancet. 2003;362:1469–80. doi: 10.1016/S0140-6736(03)14694-6. [DOI] [PubMed] [Google Scholar]
- 3.Hendriks E, van Deursen FJ, Wilson J, Sarkar M, Timms M, Timms M, Matthews KR. Life-cycle differentiation in Trypanosoma brucei: molecules and mutants. Biochem Soc Trans. 2000;28:531–6. doi: 10.1042/bst0280531. [DOI] [PubMed] [Google Scholar]
- 4.Tomas AM, Castro H. Redox metabolism in mitochondria of trypanosomatids. Antioxid Redox Signal. 2013;19:696–707. doi: 10.1089/ars.2012.4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chaudhuri M, Ott RD, Hill GC. Trypanosome alternative oxidase: From molecule to function. Trends Parasitol. 2006;22:484–91. doi: 10.1016/j.pt.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Lukes J, Hashimi H, Zikova A. Unexplained complexity of the mitochondrial genome and transcriptome in kinetoplastid flagellates. Curr Genet. 2005;48:277–99. doi: 10.1007/s00294-005-0027-0. [DOI] [PubMed] [Google Scholar]
- 7.Schmeisser S, Priebe S, Groth M, Monajembashi S, Hemmerich P, Guthke R, Platzer M, Ristow M. Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Mol Metab. 2013;2:92–102. doi: 10.1016/j.molmet.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grenier K, McLelland GL, Fon EA. Parkin and PINK1-dependent mitophagyin neurons: will the real pathway please stand up? Front Neurol. 2013;4:100. doi: 10.3389/fneur.2013.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilles LA, Kuwana T. Apoptosis regulation at the mitochondrial outer membrane. J Cell Biochem. 2014;115:632–40. doi: 10.1002/jcb.24709. [DOI] [PubMed] [Google Scholar]
- 10.Boland ML, Chourasia AH, Macleod KF. Mitochondrial Dysfunction in cancer. Front Oncol. 2013;3:292. doi: 10.3389/fonc.2013.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010;11:655–67. doi: 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- 12.Neupert W. Protein import into mitochondria. Annu Rev Biochem. 1997;66:863–917. doi: 10.1146/annurev.biochem.66.1.863. [DOI] [PubMed] [Google Scholar]
- 13.Pusnik M, Schmidt O, Perry AJ, Oelijeklaus S, Niemann M, Warcheid B, Lithgow T, Meisinger C, Schneider A. Mitochondrial preprotein translocase of trypanosomatids has a bacterial origin. Curr Biol. 2011;21:1738–43. doi: 10.1016/j.cub.2011.08.060. [DOI] [PubMed] [Google Scholar]
- 14.Singha UK, Peprah E, Williams S, Walker R, Saha L, Chaudhuri M. Characterization of the mitochondrial inner membrane protein translocator Tim17 from Trypanosoma brucei. Mol Biochem Parasitol. 2008;159:30–43. doi: 10.1016/j.molbiopara.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duncan M, Fullerton M, Chaudhuri M. Tim50 in Trypanosoma brucei possesses a dual-specificity phosphatase activity and is critical for mitochondrial protein import. J Biol Chem. 2013;288:3184–97. doi: 10.1074/jbc.M112.436378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singha UK, Hamilton V, Duncan MR, Weems E, Tripathi MK, Chaudhuri M. The translocase of mitochondrial inner membrane in Trypanosoma brucei. J Biol Chem. 2012;287:14480–93. doi: 10.1074/jbc.M111.322925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colin J, Garibal J, Mignotte B, Guenal I. The mitochondrial TOM complex modulates bax-induced apoptosis in Drosophila. Biochem Biophys Res Commun. 2009;379:939–43. doi: 10.1016/j.bbrc.2008.12.176. [DOI] [PubMed] [Google Scholar]
- 18.Grad LI, Descheneau AT, Neupert W, Lill R, Nargang FE. Inactivation of the Neurospora crassa mitochondrial outer membrane protein TOM70 by repeat-induced point mutation (RIP) causes defects in mitochondrial protein import and morphology. Curr Genet. 1999;36:137–46. doi: 10.1007/s002940050483. [DOI] [PubMed] [Google Scholar]
- 19.Meisinger C, Rissler M, Chacinska A, Szklarz LK, Milenkovic D, Kozjak V, Schonfisch B, Lohaus C, Meyer HE, Yaffe HP, Guiard B, Eiedemann N, Pfanner N. the mitochondrial morphology protein Mdm10 functions in assemblyof the preprotein translocase of the outer membrane. Dev Cell. 2004;7:61–71. doi: 10.1016/j.devcel.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 20.Rao S, Schmidt O, Harbauer AB, Schönfisch B, Guiard B, Pfanner N, Meisinger C. Biogenesis of the preprotein translocase of the outer mitochondrial membrane: protein kinase A phosphorylates the precursor of Tom40 and impairs its import. Mol Biol Cell. 2012;23:1618–1627. doi: 10.1091/mbc.E11-11-0933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rao S, Gerbeth C, Harbauer A, Mikropoulou D, Meisinger C, Schmidt O. Signaling at the gate: phosphorylation of the mitochondrial protein import machinery. Cell Cycle. 2011;10:2083–90. doi: 10.4161/cc.10.13.16054. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt O, Harbauer AB, Rao S, Eyrich B, Zahedi RP, Stojanovski D, Schonfisch B, Guiard B, Sickmann A, Pfanner N, Mesinger C. Regulation of mitochondrial protein import by cytosolic kinases. Cell. 2011;144:227–39. doi: 10.1016/j.cell.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 23.Guo Y, Cheong N, Zhang Z, De Rose R, Deng Y, Farber SA, Fernandes-Alnemri T, Alnemri ES. Tim50, a component of the mitochondrial translocator, regulates mitochondrial integrity and cell death. J Biol Chem. 2004;279:24813–25. doi: 10.1074/jbc.M402049200. [DOI] [PubMed] [Google Scholar]
- 24.Sugiyama S, Moritoh S, Furukawa Y, Mizuno T, Lim YM, Tsuda L, Nishida Y. Involvement of the mitochondrial protein translocator component tim50 in growth, cell proliferation, and the modulation of respiration in Drosophila. Genetics. 2007;176:927–36. doi: 10.1534/genetics.107.072074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woods A, Sherwin T, Sasse R, MacRae TH, Baines AJ, Gull K. Definition of individual components within the cytoskeleton of Trypanosoma brucei by a library of monoclonal antibodies. J Cell Sci. 1989;93:491–500. doi: 10.1242/jcs.93.3.491. [DOI] [PubMed] [Google Scholar]
- 26.Meinecke M, Wagner R, Kovermann P, Guiard B, Mick DU, Hutu DP, Voos W, Truscott KN, Chacinska A, Pfanner N, Rehling P. Tim50 maintains the permeability barrier of the mitochondrial inner membrane. Science. 2006;312:1523–26. doi: 10.1126/science.1127628. [DOI] [PubMed] [Google Scholar]
- 27.Zhi-Jun Y, Sriranganathan N, Vaught T, Arasta SK, Ahmed SA. A dye-based lymphocyte proliferation assay that permits multiple immunological analyses: mRNA, cytogenetics, apoptosis and immunophenotyping studes. J Immun Methods. 1997;210:25–39. doi: 10.1016/s0022-1759(97)00171-3. [DOI] [PubMed] [Google Scholar]
- 28.Nociari MM, Shaler A, Benias P, Russo C. A novel one-step, highly sensitive assay to evaluate cell-mediated toxicity. J Immun Methods. 1998;213:157–67. doi: 10.1016/s0022-1759(98)00028-3. [DOI] [PubMed] [Google Scholar]
- 29.Oberhammer F, Wilson JW, Dive C, Morris ID, Hickman JA, Wakeling AE, Walker PR, Sikorska M. Apoptotic death in epithelial cells: cleavage of DNA to 300 and/or 50 kb fragments prior to or in the absence of internucleosomal fragmentation. EMBO J. 1993;12:3679–84. doi: 10.1002/j.1460-2075.1993.tb06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghosh T, Panday T, Maitra A, Brahmachari, Pillai B. Role for voltage-dependent anion channel VDAC1 in polyglutamine-mediated neuronal cell death. PLoS One. 2007;2:e1170. doi: 10.1371/journal.pone.0001170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dancy BM, Sedansky MM, Morgan PG. Effects of the mitochondrial respiratory chain on longevity in C. elegans. Exp Gerontol. 2014;56:245–55. doi: 10.1016/j.exger.2014.03.028. [DOI] [PubMed] [Google Scholar]
- 32.Esseiva AC, Chanez AL, Bochud-Allemann N, Martinou JC, Hemphill A, Schneider A. Temporal dissection of Bax-induced events leading to fission of the single mitochondrion in Trypanosoma brucei. EMBO Report. 2014;5:268–73. doi: 10.1038/sj.embor.7400095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Candas D, Li JJ. MnSOD in oxidative stress response-potential regulation via mitochondrial protein influx. Antioxid Redox Signal. 2014;20:1599–617. doi: 10.1089/ars.2013.5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sripathi SR, He W, Atkinson CL, Smith JJ, Liu Z, Elledge BM, Jahng WJ. Mitochondrial-nuclear communication by prohibitin shuttling under oxidative stress. Biochemistry. 2011;50:8342–51. doi: 10.1021/bi2008933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shoshan-Barmatz V, Israelson A, Brdiczka D, Sheu SS. The voltage-dependent anion channel (VDAC): function in intracellular signaling, cell life and cell death. Curr Pharm Des. 2006;12:2249–70. doi: 10.2174/138161206777585111. [DOI] [PubMed] [Google Scholar]
- 36.Mokranjac D, Paschen SA, Kozany C, Prokisch H, Hoppins SC, Nargang FE, Neupart W, Hell K. Tim50, a novel component of the TIM23 preprotein translocase of mitochondria. EMBO J. 2003;22:816–25. doi: 10.1093/emboj/cdg090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamamoto H, Esaki M, Kanamori T, Tamura Y, Nishikawa S, Endo T. Tim50 is a subunit of the TIM23 complex that links protein translocation across the outer and inner mitochondrial membranes. Cell. 2002;111:519–28. doi: 10.1016/s0092-8674(02)01053-x. [DOI] [PubMed] [Google Scholar]
- 38.Siemen D, Ziemer M. What is the nature of the mitochondrial permeability transition pore and what is it not? IUBMB Life. 2013;65:255–62. doi: 10.1002/iub.1130. [DOI] [PubMed] [Google Scholar]
- 39.Abu-Hamad S, Arbel N, Calo D, Arzoine L, Israelson A, Keinan N, Ben-Romano R, Friedman O, Shoshan-Barmatz V. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J Cell Sci. 2009;122:1906–16. doi: 10.1242/jcs.040188. [DOI] [PubMed] [Google Scholar]
- 40.Welburn SC, Macleod E, Figarella K, Duzensko M. Programmed cell death in African trypanosomes. Parasitology. 2006;132:S7–S18. doi: 10.1017/S0031182006000825. [DOI] [PubMed] [Google Scholar]
- 41.Proto WR, Coombs GH, Mottram JC. Cell death in parasitic protozoa: regulated or incidental? Nat Rev Microbiol. 2013;11:58–66. doi: 10.1038/nrmicro2929. [DOI] [PubMed] [Google Scholar]
- 42.Moss CX, Westrop GD, Juliano L, Coombs GH, Mottram JC. Metacaspase 2 of Trypanosoma brucei is a calcium-dependent cysteine peptidase active without processing. FEBS Lett. 2007;581:5635–39. doi: 10.1016/j.febslet.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 43.Figarella K, Uzcategui NL, Beck A, Schoenfeld C, Kubata BK, Lang F, Duszenko M. Prostaglandin-induced programmed cell death in Trypanosoma brucei involves osidative stress. Cell Death Differ. 2006;13:1802–14. doi: 10.1038/sj.cdd.4401862. [DOI] [PubMed] [Google Scholar]
- 44.Rainbolt TK, Atanassova N, Genereux JC, Wiseman L. Stress-regulated translational attenuation adapts mitochondrial protein import through Tim17A degradation. Cell Metab. 2013;18:908–19. doi: 10.1016/j.cmet.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337:587–90. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clayton C. The regulation of trypanosome gene expression by RNA binding proteins. PLoS Pathogen. 2013;9:e1003680. doi: 10.1371/journal.ppat.1003680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singha UK, Sharma S, Chaudhuri M. Down regulation of mitochondrial porin inhibits cell growth and alters respiratory phenotype in Trypanosoma brucei. Eukaryot Cell. 2009;8:1418–28. doi: 10.1128/EC.00132-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pinto VD, Reina S, Guarino F, Messina A. Structure of the voltage dependent anion channel: state of the art. J Bioenerg Biomembr. 2008;40:139–47. doi: 10.1007/s10863-008-9140-3. [DOI] [PubMed] [Google Scholar]
- 49.Shoshan-Barmatz V, Israelson A, Brdiczka D, Sheu SS. The voltage-dependent anion channel (VDAC): function in intracellular signaling, cell life and cell death. Curr Pharm Des. 2006;12:2249–70. doi: 10.2174/138161206777585111. [DOI] [PubMed] [Google Scholar]
- 50.Pusnik M, Charrière F, Mäser P, Waller RF, Dagley MJ, Lithgow T, Schneider A. The single mitochondrial porin of Trypanosoma brucei is the main metabolite transporter in the outer mitochondrial membrane. Mol Biol Evol. 2009;26:671–80. doi: 10.1093/molbev/msn288. [DOI] [PubMed] [Google Scholar]
- 51.Tan W. VDAC blockage by phosphorothioate oligonucleotides and its implication in apoptosis. Biochim Biophys Acta. 2012;1818:1555–56. doi: 10.1016/j.bbamem.2011.12.032. [DOI] [PubMed] [Google Scholar]
- 52.Abu-Hamad S, Sivan S, Shoshan-Barmatz V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc Natl Acad Sci, USA. 2006;103:5787–92. doi: 10.1073/pnas.0600103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siemen D, Ziemer M. What is the nature of the mitochondrial permeability transition pore and what is it not? IUBMB Life. 2013;65:255–62. doi: 10.1002/iub.1130. [DOI] [PubMed] [Google Scholar]
- 54.Abu-Hamad S, Arbel N, Calo D, Arzoine L, Israelson A, Keinan N, Ben-Romano R, Friedman O, Shoshan-Barmatz V. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J Cell Sci. 2009;122:1906–16. doi: 10.1242/jcs.040188. [DOI] [PubMed] [Google Scholar]
- 55.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion chnnels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–55. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shore GC. Apoptosis: it’s BAK to VDAC. EMBO Rep. 2009;10:1311–13. doi: 10.1038/embor.2009.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arbel N, Ben-Hail D, Shoshan-Barmatz V. Mediation of antiapoptotic activity of Bcl-xL protein upon interaction withVDAC1 protein. J Biol Chem. 2012;287:23152–56. doi: 10.1074/jbc.M112.345918. [DOI] [PMC free article] [PubMed] [Google Scholar]