

Abstract
Genetic investigation of inherited skin disorders has informed understanding of skin self-renewal, differentiation, and barrier function. Erythrokeratodermia variabilis et progressiva (EKVP) is a rare, inherited skin disease characterized by transient figurate patches of erythema, localized or generalized scaling, and frequent palmoplantar keratoderma. By employing exome sequencing, we show that de novo missense mutations in GJA1 (gap junction protein alpha 1) cause EKVP. The severe, progressive skin disease in EKVP subjects with GJA1 mutations is distinct from limited cutaneous findings rarely found in the systemic disorder oculodentodigital dysplasia, also caused by dominant GJA1 mutations. GJA1 encodes connexin 43 (Cx43), the most widely expressed gap junction protein. We show that the GJA1 mutations in EKVP subjects lead to disruption of Cx43 membrane localization, and aggregation within the Golgi. These findings reveal a critical role for Cx43 in epidermal homeostasis, and provide evidence of organ-specific pathobiology resulting from different mutations within GJA1.
INTRODUCTION
Erythrokeratodermia variabilis (EKV [MIM 133200]) is a rare, congenital skin disorder characterized by transient figurate patches of erythema on a background of localized or generalized scaling, with palmoplantar keratoderma present in nearly 50% of cases (Richard, 2000). EKV is primarily inherited in an autosomal dominant fashion, though rare autosomal recessive inheritance has been reported (Fuchs-Telem et al., 2011; Gottfried et al., 2002), and it shows marked phenotypic heterogeneity, even within kindreds bearing the same disease-causing mutation (van Steensel et al., 2009). The term erythrokeratodermia variabilis et progressiva (EKVP) has been proposed to encompass the diversity of phenotypes, ranging from limited hyperkeratotic plaques and erythematous patches to severe progressive symmetric erythrokeratodermia which can feature more generalized cutaneous involvement (van Steensel, 2004). Mutations in GJB3 and GJB4, encoding connexins 31 and 30.3 respectively, have been reported to cause EKV/EKVP (Macari et al., 2000; Richard et al., 1998), though there is evidence of further genetic heterogeneity (Common et al., 2005; Wei et al., 2011).
Connexin proteins are named for their molecular weight in kDa (Pfenniger et al., 2011), and are phylogenetically classified into alpha and beta subgroups (Martin et al., 2014), encoded by GJA and GJB genes. All connexin proteins have a conserved structure and topology, consisting of a cytoplasmic N-terminus and C-terminus, and four transmembrane domains connected by two extracellular loops and one cytoplasmic loop. After translation, connexins oligomerize to form hexameric connexons in the endoplasmic reticulum (ER) and Golgi, and are transported to the membrane where they can either function as non-junctional hemichannels between the cytoplasm and extracellular space, or dock with partners in neighboring cells to form intercellular gap junctions. These channels provide both critical communication pathways, allowing passage of ions and small molecules, and structural support to tissues, accumulating to create large plaques. A diversity of potential permeabilities are possible via homomeric or heteromeric connexon configuration, and homotypic or heterotypic interactions between cells (Meşe et al., 2007). Gap junctions play diverse and important roles in normal physiology, and mutations in connexin genes cause a variety of pathological phenotypes including cardiovascular disease, myelin-related disease, skin disease, craniofacial disorders, and hearing loss (Pfenniger et al., 2011; Scott et al., 2012).
In recruiting subjects with EKVP-spectrum phenotypes, we identified a cohort without mutation in GJB3 or GJB4, including kindreds 101–103. Each kindred consists of one affected subject born to unaffected parents.
RESULTS
Subject 101-1 is a 32-month-old boy who had no clinical phenotype until the age of 5 months. He then developed thick brown-gray scale on frictional surfaces (Figure 1a), as well as progressive darkening of his dorsal hands, arms, legs, and face. His palmoplantar surfaces and digits became pink and then markedly hyperkeratotic (Figure 2a), and migrating areas of transient figurate erythema overlay the generalized scaling (Figure 3a).
Figure 1. Clinical and histologic features of EKVP due to GJA1 mutation.
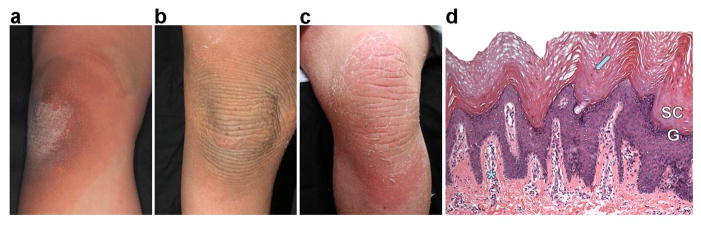
(a) The knee of 101-1 was normal at birth, but became thickened and scaly when he began to crawl; shown at age 30 months. (b) The knee of 6-year-old 102-1 shows progression to corrugated, thickened, scaly skin which has extended beyond sites of friction. (c) The knee of 30-year-old 103-1 shows marked hyperkeratosis with peeling scale; a patch of figurate erythema is present inferiorly. (d) Histology of affected leg skin of 102-1 shows a compact, thickened stratum corneum (SC) with retained nuclei (arrow), papillomatosis, a thickened granular layer (G), and a perivascular lymphocytic infiltrate (asterisk).
Figure 2. Prominent white nails and progressive keratoderma in EKVP due to GJA1 mutation.

All subjects show prominent porcelain white proximal nails without dystrophy. Honeycombed thick hyperkeratosis is present on the palms, and similar thick hyperkeratosis is present on the feet. Keratoderma is progressive with 30-month-old 101-1 (a), showing less hyperkeratosis than 6-year-old 102-1 (b) or 30-year-old 103-1 (c). Subject 103-1 is on a systemic retinoid, accounting for peeling seen at lateral aspects of hand and foot and less exuberant hyperkeratosis.
Figure 3. Figurate erythema and darkening of the skin in EKVP due to GJA1 mutation.
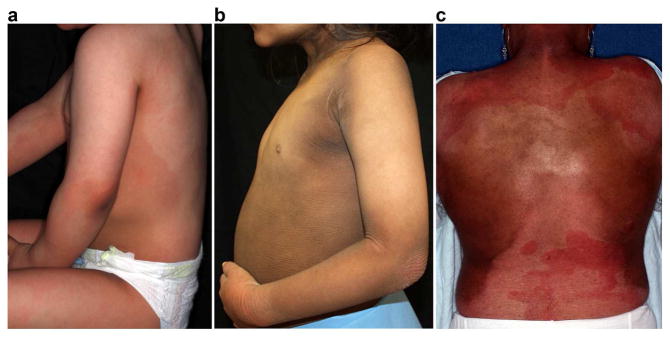
All subjects report figurate erythema and have experienced progressive skin darkening. (a) In subject 101-1, figurate erythema is present on the back, flank, arm, and upper thigh. (b) Subject 102-1 shows thick, corrugated keratoderma with darkening prominent on the neck, forearm, axilla, and abdomen. There is a patch of erythema on the wrist. (c) In subject 103-1, figurate erythema is present on the upper and lower back. Notably, subject 103-1 reports that figurate erythema became more frequent and prominent at age 10 years but was present throughout childhood. Treatment with acitretin has reduced scaling, but hyperpigmentation is prominent on the flanks, shoulders, and neck. In all cases, erythema is induced by stress and warm conditions and can be accompanied by a stinging or burning sensation.
Subject 102-1 is a 6-year-old girl who was adopted from Guatemala at 6 months of age without notable skin disease (Supplementary Figure 1a). At 8 months, she developed darkening and scaling of frictional surfaces (Figure 1b) and the dorsal hands, and progressive thickening of the palms and soles (Figure 2b). By age 13 months, hyperpigmented, hyperkeratotic plaques appeared, affecting the axillae, elbows, and inner thighs; these became confluent by age 2, with sparing of cheeks and upper chest (Figure 3b). She has intermittent annular red patches which fade within hours after onset.
Subject 103-1 is a 30-year-old woman who had normal skin through 6 months of age (Supplementary Figure 1b), after which darkening of the inner thighs was noted, followed by development of thick scale which began on the knees, elbows, hands, and feet and then progressively spread up the legs and arms (Figure 1c, Figure 2c). At the age of 10, pink to deep red transient erythematous patches which were associated with a burning sensation became prominent (Figure 3c). Oral acitretin therapy has reduced scale and improved keratoderma.
All subjects exhibit enlarged porcelain-white lunulae (Figure 2, Supplementary Figure 2) and darkening of periorificial areas (Supplementary Figure 3). Histology of the most severely affected skin in each case shows papillomatosis, acanthosis, hypergranulosis, and compact orthohyperkeratosis with retained nuclei (Figure 1d and Supplementary Figure 4). In less severely affected skin, acanthosis, papillomatosis, orthohyperkeratosis, and follicular plugging is found (Supplementary Figure 4).
Exome sequencing was performed from peripheral blood DNA of affected subjects (Supplementary Table 1). Sequences were aligned to the reference human genome, variants annotated, and data examined to identify novel coding mutations. Analysis revealed the same heterozygous missense mutation in GJA1, E227D, in two affected subjects, 101-1 and 102-1. Another heterozygous GJA1 mutation, A44V, was found in a third affected subject, 103-1 (Supplementary Figure 5). Sanger sequencing confirmed these mutations, and revealed that E227D is not present in either parent of 101-1, and A44V is not present in either parent of 103-1; thus these mutations arose de novo (Figure 4). Because subject 102-1 is adopted, we have no parental DNA, but her biological parents are documented to be unaffected by any skin disease. Neither E227D nor A44V were found in ~2500 control exomes or in public databases of human genetic variation. E227 is highly conserved in both orthologous and paralogous genes; A44 is highly conserved in orthologs (Figure 5, Supplementary Figure 6). GJA1 encodes connexin 43 (Cx43), a widely expressed gap junction protein.
Figure 4. GJA1 mutations in EKVP kindreds.

(a) Affected and unaffected subjects are denoted with black and white symbols, respectively; adoption is shown with a dashed line. GJA1 alleles determined by sequencing of genomic DNA are denoted as ‘+’ (wild-type) or by the amino acid substitution in red (mutant). To the right of each pedigree, Sanger sequence traces at GJA1 mutation sites are shown for each subject and parents from whom DNA was available (mutant bases indicated with *). Amino acid sequences are shown at the top of each trace (mutant residues in red). Mutations are p.E227D, c.A681T (subjects 101-1 and 102-1) and p.A44V, c.C131T (subject 103-1) (NCBI RefSeq NM_000165). (b) Schematic model of Cx43 shows locations of mutations in EKVP patients reported here (red), and those reported in ODDD (blue), ODDD with palmoplantar hyperkeratosis (purple), and ODDD with Clouston syndrome (green).
Figure 5. Cx43 and paralogs.

Transmembrane domains are outlined; conserved residues are shaded gray. Disease-causing Cx43 mutations are shown above the alignment: EKVP reported here (red shade), ODDD (blue), ODDD with palmoplantar hyperkeratosis (purple), ODDD with Clouston syndrome (green shade); frameshifts (‘fs’), deletions (‘d’), insertions (‘i’). Mutation sites in paralogs reported to cause skin disease are in orange.
The GJA1 E227D mutation in subjects 101-1 and 102-1 is at the intracellular boundary of the Cx43 fourth transmembrane domain, and is within the region reported to be necessary for Cx43 phosphorylation and interaction with partner protein CIP150 (amino acids 227–242); Cx43 mutant protein lacking this region fails to localize to the membrane (Akiyama et al., 2005). The observation of two identical, independent E227D mutations in unrelated EKVP subjects suggests a critical role for this site in the epidermal function of Cx43.
The A44V mutation in subject 103-1 is at the extracellular boundary of the first transmembrane domain. In contrast to most other paralogs, which have valine this position and alanine at a position three residues N-terminal, in Cx43 these residues are exchanged, with valine at aa41 and alanine at aa44. In Cx26, an A40V mutation causes keratitis-ichthyosis-deafness syndrome (Montgomery et al., 2004), despite valine being wild-type at this site in Cx43 (Figure 5). These data suggest that these valine-alanine configurations are required for normal function across paralogs, including the role of Cx43 in the skin.
To examine the consequence of these mutations, we immunostained tissue sections from wild-type skin and affected skin of individuals 102-1 and 103-1 with an antibody to Cx43. In contrast to normal skin, in which Cx43 primarily localizes to intercellular junctions with faint, uniform cytoplasmic staining, both E227D (102-1) and A44V (103-1) mutants do not localize to the membrane, demonstrating cytoplasmic localization (Figure 6a–c). To further explore this Cx43 localization defect, we generated expression constructs of wild-type, E227D, and A44V mutant Cx43 with a C-terminal hemagglutinin (HA) tag. Constructs were transfected into HeLa cells and immunostained with an anti-HA antibody and an antibody to the cis-Golgi marker GM130. While the wild-type protein primarily localized to intercellular junctions, both the E227D and A44V mutants aggregated in a subcellular compartment partly co-localizing with GM130, suggesting retention in the Golgi (Fig. 6d–f).
Figure 6. Cx43 is mis-localized in EKVP.
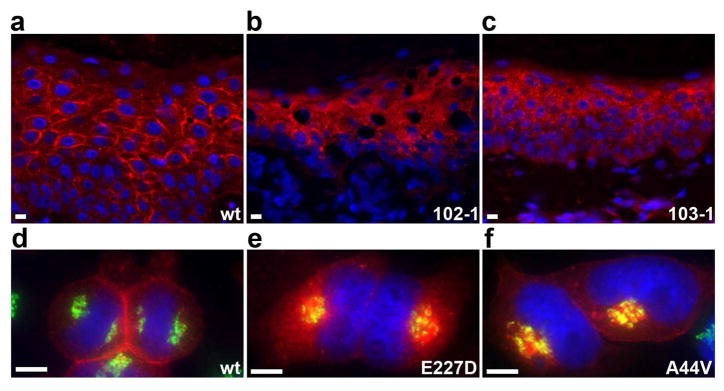
(a–c) Cx43 immunolocalization was performed on tissue sections from wild-type (wt) and affected patient skin (102-1: E227D, 103-1: A44V), with Cx43 antibody in red and DAPI nuclear counterstain in blue. (a) Wild-type tissue shows primarily intercellular membrane localization of Cx43. (b–c) Affected skin shows primarily cytoplasmic localization. (d–f) Wild-type (wt) and mutant Cx43 (E227D, A44V) were HA-tagged and expressed in HeLa cells. Immunolocalization of Cx43 is red, cis-Golgi marker GM130 is green, and DAPI nuclear counterstain is blue. (d) Wild-type Cx43 localizes to intercellular junctions and does not co-localize with GM130. (e and f) The E227D and A44V mutants do not localize to intercellular junctions and accumulate in a subcellular compartment, partly co-localizing with GM130. Scale bars are 20 μm in all panels.
DISCUSSION
There are 21 connexin-encoding genes identified in the human genome (Martin et al., 2014). Given the variety of possible heteromeric connexon configurations, and heterotypic gap junction interactions between cells, a mutation in a single connexin gene has the potential to exert trans-dominant effects on a number of other connexins expressed in the same cell type. This may contribute to the spectrum of phenotypes seen with different mutations within a single connexin-encoding gene. Mutations in GJB2 (Cx26) can cause sensorineural hearing loss, palmoplantar keratoderma (with and without deafness), and keratitis-ichthyosis-deafness syndrome. Phenotypes are correlated with mutation consequence, with pure sensorineural deafness resulting from loss of Cx26 channel function, and skin phenotypes from mechanisms including mislocalizaton, ER-stress leading to abnormal epidermal differentiation, pathologically altered hemichannel or gap junction activity, and trans-dominant effects on localization or function of other connexins (Meşe et al., 2007).
Previously described mutations in GJA1 cause oculodentodigital dysplasia (ODDD) (Laird, 2014; Paznekas et al., 2003), characterized by distinct facial anomalies including micocephaly, microphthalmia, and a thin nose with hypoplastic alae nasi, small anteverted nares, and a prominent bridge and columella. Features with wider phenotypic variability include other abnormalities of the eyes (microcornea, porous spongy irises, cataracts, glaucoma, optic atrophy), dentition (mandibular overgrowth, cleft palate, small or absent teeth, enamel hypoplasia, multiple caries, early tooth loss), and digits (syndactyly, short, bent, or flexed fingers and toes). Brittle nails and hypotrichosis may be present, and there are frequently neurological symptoms which can include hearing loss, lower body weakness, spasticity, and incontinence, often not presenting until adulthood. Mild mental retardation, cardiac abnormalities, and palmoplantar hyperkeratosis occur rarely. Our EKVP subjects with GJA1 mutations exhibit none of these ODDD phenotypes, save mild dental enamel defects in 102-1 which are not present in 101-1, who shares an identical mutation, or 103-1 (Figure 2, Supplementary Figures 1 and 3), and mild clinodactyly (Figure 2) which can be found in ODDD but is also present in the general population (Skvarilova and Smahel, 1984).
Over 70 different mutations in GJA1 cause ODDD, and almost all are heterozygous missense mutations or small in-frame deletions or insertions occurring prior to the C-terminal tail (Laird, 2014). Two kindreds have been reported with severe ODDD due to homozygous or compound heterozygous early-termination mutations (Jamsheer et al., 2010; Richardson et al., 2006), suggesting a dominant-negative mechanism for the majority of ODDD cases. There are two frameshift mutations within the Cx43 tail (Y230fsX236 and C260fsX307) and two missense mutations (L11P and K134E) which are inconsistently associated with the additional feature of mild palmoplantar hyperkeratosis (Alao et al., 2010; Kelly et al., 2006; Paznekas et al., 2003; van Steensel et al., 2005; Vreeburg et al., 2007), and a single case report describes a subject with a GJA1 missense mutation (V41L) and a GJB2 mutation who exhibits features of ODDD and Clouston syndrome with alopecia, nail dystrophy, and well-demarcated, limited hyperkeratosis of extensor surfaces (Kellermayer et al., 2005). Neither transient figurate erythema nor generalized erythrokeratodermia have been reported in ODDD subjects with GJA1 mutations.
We have shown that previously unreported, dominant, de novo missense mutations in GJA1 cause a consistent clinical phenotype of normal skin at birth which develops hyperpigmentation and scale at sites of friction in childhood, with progression to near-confluent corrugated hyperkeratosis, palmoplantar keratoderma, and transient figurate erythema. While mild palmoplantar hyperkeratosis and limited hyperkeratosis of extensor surfaces has been found in a small subset of ODDD patients, the widespread, severe phenotype described here is distinct. Subjects are without features of ODDD caused by over 70 other GJA1 mutations, and the E227D mutation was observed in two out of three unrelated EKVP subjects, suggesting that EKVP pathology may be restricted to a small number of GJA1 mutation sites.
Cx43 is the most widely expressed connexin (Laird, 2014), is expressed throughout the epidermis, and is the predominant connexin in basal proliferating cells (Martin et al., 2014). Yet Cx43 itself is not required for epidermal homeostasis, given that a human homozygous null allele causes ODDD without skin disease (Richardson et al., 2006), though Cx43 is up-regulated in chronic wounds, and knock-down of Cx43 speeds wound healing (Becker et al., 2012; Martin et al., 2014; Scott et al., 2012). In this context, the induction of initial skin findings in our affected subjects at sites of friction with subsequent generalization is intriguing.
Our immunostaining shows that GJA1 mutations A44V and E227D lead to Cx43 mislocalization. In normal skin, Cx43 localizes to intracellular junctions, whereas in the skin of our EKVP subjects Cx43 does not localize to the membrane, and cells transfected with either A44V or E227D mutant Cx43 demonstrate aggregation of Cx43 in the Golgi. This mislocalization of Cx43 with an A44V or E227D mutation stands in stark contrast to the localization observed with dominant ODDD mutations in Cx43. Of the twenty ODDD mutations which have been experimentally examined, the vast majority are expressed at the cell surface, but form functionally impaired gap junctions, with a dominant-negative effect on wild-type Cx43 (Shao et al., 2012). Whether the observed retention of A44V and E227D mutant Cx43 in the Golgi contributes to the previously unreported skin-specific EKVP phenotype we describe, either by trans-dominant effects on connexins or other proteins as observed for GJB2 mutations in KID syndrome (Meşe et al., 2007), or by other mechanisms, will require further experimental investigation.
MATERIALS AND METHODS
Human subjects
The Yale Human Investigation Committee approved the study protocol, consistent with the Declaration of Helsinki guidelines, and subjects or their parents provided verbal and written informed consent, and consented to the publication of images as displayed. Each subject provided a blood sample and punch biopsies of affected skin from the upper thigh. De-identified, site-matched normal skin tissue discarded at the time of skin cancer excisions was obtained for immunostaining controls. Genomic DNA was isolated from blood using a standard phenol-chloroform protocol.
Whole exome sequencing
Bar-coded DNA libraries were prepared and whole exome capture was performed (EZ Exome 2.0, Roche, Basel, Switzerland) by the Yale Center for Genome Analysis. Illumina HiSeq 2000 and 2500 instruments were used for sequencing samples pooled 6 per lane, with 75 bp paired-end reads. Resulting reads were aligned to the human reference sequence (hg18 or hg19) with Efficient Large-scale Alignment of Nucleotide Databases software (ELAND, Illumina, San Diego, CA) and processed via a SAMtools-based Perl script to trim sequence to targeted intervals and remove PCR duplicates. Single nucleotide variants (SNVs) and deletions and insertions (indels) were identified using SAMtools software. Variants were annotated for functional impact using a Perl script, and filtered in Excel to exclude frequent variants present in dbSNP (Build 140), 1000 Genomes, the NHLBI exome database (release ESP6500SI-V2) and in 2577 control exomes, and to examine coding mutations (missense, nonsense, and splice site SNVs and indels) with SAMtools quality scores ≥50 and coverage ≥8. Aligned reads were examined with the Broad Institute Integrative Genomics Viewer (IGV) to exclude SNVs resulting from alignment error.
Sanger sequencing
Verification of GJA1 mutations and sequencing of parental DNA was performed via PCR using Kapa 2G Fast polymerase (Kapa Biosystems, Woburn, MA) and Sanger sequencing. Primers were designed with ExonPrimer and SNPmasker to specifically amplify GJA1 and not its pseudogene.
Expression construct generation
A GJA1 cDNA clone was obtained (HsCD00332741, Harvard PlasmID, Brookline, MA) and subcloned into pcDNA3.1(-) Zeo (Invitrogen, Carlsbad, CA). Site-directed mutagenesis to introduce the A44V or E227D mutations into the wild-type cDNA was performed with QuikChange (Agilent, Santa Clara, CA). The entire cDNA sequence of resulting clones (wild-type, A44V, and E227D) was sequenced to ensure absence of secondary mutation.
Transfections
HeLa cells (CCL-2, ATCC, Manassas, VA) were seeded at a density of 2 × 104 cells per well on 8-well culture slides (Thermo Fisher Scientific, Waltham, MA) in DMEM + 10% fetal bovine serum. Cells were transfected using Lipofectamine 2000 (Invitrogen) per standard protocols and fixed for immunofluorescence studies at 24 hours post-transfection.
Immunohistochemistry
5μm sections from FFPE tissue were deparaffinized using a xylene-ethanol gradient, rehydrated, and rinsed in phosphate-buffered saline (PBS). Antigen retrieval was performed by immersion in a modified pH 6.0 citrate buffer for 20 minutes in a steamer. Slides were cooled and rinsed in PBS. Tissue sections or transfected cells were fixed with 1:1 acetone:methanol for 10 minutes at −20°C. Blocking was performed with 10% donkey serum/1% BSA for 1 hour at room temperature. Slides were incubated 1 hour with primary antibody, washed 3 times with 1X PBS, incubated 1 hour with secondary antibody, and again washed 3 times with 1X PBS before mounting with Mowiol/1% n-propyl gallate (Polysciences, Warrington, PA). Primary antibodies: 1:100 rabbit anti-Cx43, 1:100 goat anti-HA, 1:70 mouse anti-GM130 (ab11370, ab9134, ab169276; Abcam, Cambridge, England). Secondary antibodies: 1:10000 Cy3 donkey anti-rabbit IgG, 1:10000 Cy3 donkey anti-goat IgG, 1:200 Cy2 donkey anti-mouse IgG (711-165-152, 711-165-003, 711-225-150; Jackson ImmunoResearch, West Grove, PA).
Supplementary Material
Acknowledgments
We thank the EKVP subjects and their families for their invaluable contribution to this work; Jonathan Levinsohn, Young Lim, Leonard Milstone, and Haris Mirza for critical review of the manuscript; and Christopher Castaldi, Vincent Klump, Shrikant Mane, Peggy Myung, Carol Nelson-Williams, and Irina Tikhonova for technical assistance. This work was supported by a Doris Duke Charitable Foundation Clinical Scientist Development Award and a Research Grant from the Foundation for Ichthyosis & Related Skin Types, Inc. (FIRST) funded by the Lennox Foundation to KAC; a Dermatology Foundation Pediatric Dermatology Fellowship, FIRST funds, and National Institutes of Health funds (NIH T32 AR007016) to BGC; the Yale Center for Mendelian Genomics (NIH U54 HG006504); and the Yale Center for Clinical Investigation (NIH UL1 TR000142).
Abbreviations
- Cx43
connexin 43
- EKV
erythrokeratodermia variabilis
- EKVP
erythrokeratodermia variabilis et progressiva
- ER
endoplasmic reticulum
- GJA1
gap junction protein alpha 1
- GJIC
gap junction intercellular communication
- HA
hemagglutinin
- ODDD
oculodentodigital dysplasia
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
References
- Akiyama M, Ishida N, Ogawa T, et al. Molecular cloning and functional analysis of a novel Cx43 partner protein CIP150. Biochem Biophys Res Commun. 2005;335:1264–71. doi: 10.1016/j.bbrc.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Alao MJ, Bonneau D, Holder-Espinasse M, et al. Oculo-dento-digital dysplasia: lack of genotype-phenotype correlation for GJA1 mutations and usefulness of neuro-imaging. Eur J Med Genet. 2010;53:19–22. doi: 10.1016/j.ejmg.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Becker DL, Thrasivoulou C, Phillips AR. Connexins in wound healing; perspectives in diabetic patients. Biochim Biophys Acta. 2012;1818:2068–75. doi: 10.1016/j.bbamem.2011.11.017. [DOI] [PubMed] [Google Scholar]
- Common JE, O’Toole EA, Leigh IM, et al. Clinical and genetic heterogeneity of erythrokeratoderma variabilis. J Invest Dermatol. 2005;125:920–7. doi: 10.1111/j.0022-202X.2005.23919.x. [DOI] [PubMed] [Google Scholar]
- Fuchs-Telem D, Pessach Y, Mevorah B, et al. Erythrokeratoderma variabilis caused by a recessive mutation in GJB3. Clin Exp Dermatol. 2011;36:406–11. doi: 10.1111/j.1365-2230.2010.03986.x. [DOI] [PubMed] [Google Scholar]
- Gottfried I, Landau M, Glaser F, et al. A mutation in GJB3 is associated with recessive erythrokeratodermia variabilis (EKV) and leads to defective trafficking of the connexin 31 protein. Hum Mol Genet. 2002;11:1311–6. doi: 10.1093/hmg/11.11.1311. [DOI] [PubMed] [Google Scholar]
- Jamsheer A, Badura-Stronka M, Sowińska A, et al. A severe progressive oculodentodigital dysplasia due to compound heterozygous GJA1 mutation. Clin Genet. 2010;78:94–7. doi: 10.1111/j.1399-0004.2010.01412.x. [DOI] [PubMed] [Google Scholar]
- Kellermayer R, Keller M, Ratajczak P, et al. Bigenic connexin mutations in a patient with hidrotic ectodermal dysplasia. Eur J Dermatol. 2005;15:75–9. [PubMed] [Google Scholar]
- Kelly SC, Ratajczak P, Keller M, et al. A novel GJA 1 mutation in oculo-dento-digital dysplasia with curly hair and hyperkeratosis. Eur J Dermatol. 2006;16:241–5. [PubMed] [Google Scholar]
- Laird DW. Syndromic and non-syndromic disease-linked Cx43 mutations. FEBS Lett. 2014;588:1339–48. doi: 10.1016/j.febslet.2013.12.022. [DOI] [PubMed] [Google Scholar]
- Macari F, Landau M, Cousin P, et al. Mutation in the gene for connexin 30.3 in a family with erythrokeratodermia variabilis. American journal of human genetics. 2000;67:1296–301. doi: 10.1016/s0002-9297(07)62957-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PE, Easton JA, Hodgins MB, et al. Connexins: sensors of epidermal integrity that are therapeutic targets. FEBS Lett. 2014;588:1304–14. doi: 10.1016/j.febslet.2014.02.048. [DOI] [PubMed] [Google Scholar]
- Meşe G, Richard G, White TW. Gap junctions: basic structure and function. J Invest Dermatol. 2007;127:2516–24. doi: 10.1038/sj.jid.5700770. [DOI] [PubMed] [Google Scholar]
- Montgomery JR, White TW, Martin BL, et al. A novel connexin 26 gene mutation associated with features of the keratitis-ichthyosis-deafness syndrome and the follicular occlusion triad. J Am Acad Dermatol. 2004;51:377–82. doi: 10.1016/j.jaad.2003.12.042. [DOI] [PubMed] [Google Scholar]
- Paznekas WA, Boyadjiev SA, Shapiro RE, et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72:408–18. doi: 10.1086/346090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfenniger A, Wohlwend A, Kwak BR. Mutations in connexin genes and disease. Eur J Clin Invest. 2011;41:103–16. doi: 10.1111/j.1365-2362.2010.02378.x. [DOI] [PubMed] [Google Scholar]
- Richard G. Connexins: a connection with the skin. Exp Dermatol. 2000;9:77–96. doi: 10.1034/j.1600-0625.2000.009002077.x. [DOI] [PubMed] [Google Scholar]
- Richard G, Smith LE, Bailey RA, et al. Mutations in the human connexin gene GJB3 cause erythrokeratodermia variabilis. Nat Genet. 1998;20:366–9. doi: 10.1038/3840. [DOI] [PubMed] [Google Scholar]
- Richardson RJ, Joss S, Tomkin S, et al. A nonsense mutation in the first transmembrane domain of connexin 43 underlies autosomal recessive oculodentodigital syndrome. J Med Genet. 2006;43:e37. doi: 10.1136/jmg.2005.037655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CA, Tattersall D, O’Toole EA, et al. Connexins in epidermal homeostasis and skin disease. Biochim Biophys Acta. 2012;1818:1952–61. doi: 10.1016/j.bbamem.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Shao Q, Liu Q, Lorentz R, et al. Structure and functional studies of N-terminal Cx43 mutants linked to oculodentodigital dysplasia. Mol Biol Cell. 2012;23:3312–21. doi: 10.1091/mbc.E12-02-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skvarilová B, Smahel Z. Clinodactyly--frequency and morphological implications. Acta Chir Plast. 1984;26:72–8. [PubMed] [Google Scholar]
- van Steensel M. Does progressive symmetric erythrokeratoderma exist? Br J Dermatol. 2004;150:1043–5. doi: 10.1111/j.1365-2133.2004.05965.x. [DOI] [PubMed] [Google Scholar]
- van Steensel MA, Oranje AP, van der Schroeff JG, et al. The missense mutation G12D in connexin30.3 can cause both erythrokeratodermia variabilis of Mendes da Costa and progressive symmetric erythrokeratodermia of Gottron. Am J Med Genet A. 2009;149A:657–61. doi: 10.1002/ajmg.a.32744. [DOI] [PubMed] [Google Scholar]
- van Steensel MA, Spruijt L, van der Burgt I, et al. A 2-bp deletion in the GJA1 gene is associated with oculo-dento-digital dysplasia with palmoplantar keratoderma. Am J Med Genet A. 2005;132A:171–4. doi: 10.1002/ajmg.a.30412. [DOI] [PubMed] [Google Scholar]
- Vreeburg M, de Zwart-Storm EA, Schouten MI, et al. Skin changes in oculo-dento-digital dysplasia are correlated with C-terminal truncations of connexin 43. Am J Med Genet A. 2007;143:360–3. doi: 10.1002/ajmg.a.31558. [DOI] [PubMed] [Google Scholar]
- Wei S, Zhou Y, Zhang TD, et al. Evidence for the absence of mutations at GJB3, GJB4 and LOR in progressive symmetrical erythrokeratodermia. Clin Exp Dermatol. 2011;36:399–405. doi: 10.1111/j.1365-2230.2010.03974.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.