

Abstract
Background
Opening of the Mitochondrial Permeability Transition pore is the underlying cause of cellular dysfunction during diverse pathological situations. Although this bioenergetic entity has been studied extensively, its molecular componentry is constantly debated. Cyclophilin D is the only universally accepted modulator of this channel and its selective ligands have been proposed as therapeutic agents with the potential to regulate pore opening during disease.
Scope of review
This review aims to recapitulate known molecular determinants necessary for Cyclophilin D activity regulation and binding to proposed pore constituents therefore regulating the mitochondrial permeability transition pore.
Major conclusions
While the main target of Cyclophilin D is still a matter of further research, permeability transition is finely regulated by post-translational modifications of this isomerase and its catalytic activity facilitates pore opening.
General significance
Complete elucidation of the molecular determinants required for Cyclophilin D-mediated control of the mitochondrial permeability transition pore will allow the rational design of therapies aiming to control disease phenotypes associated with the occurrence of this unselective channel.
Keywords: Mitochondrial permeability transition, cyclophilin-D, peptidyl-prolyl cis-trans isomerase
1. Introduction
Ever since the first protocols for isolating intact mitochondria were published, the conditions required to maximize the yield and functionality of these isolated organelles have been subject of extensive empirical improvements. Among these conditions, perhaps the addition of Ca2+-sequestrating agents has been the most recurrent feature of many buffers used to isolate mitochondria from cell homogenates [1]. The re-addition of Ca2+ to Ca2+-depleted isolated beef heart mitochondria initially led Hunter and Haworth to propose the presence of ‘a nonspecific increase in the permeability of the inner membrane that resulted in ATP hydrolysis, uncoupling of oxidative phosphorylation and the consequent loss of respiratory control [2]. Subsequent work by the same scientists evidenced the presence of a Ca2+-induced proteinaceous pore today known as the Mitochondrial Permeability Transition (MPT) Pore [3-5]. The MPT pore wreaks havoc on mitochondria by depleting ion gradients across the inner mitochondrial membrane. If the ability of the mitochondrion to replenish these gradients is surpassed by the MPT pore uncoupling activity, the cell will develop a pro-necrotic phenotype (for a review see [1,6]).
The molecular structure of the MPT pore is still an enigma, however its modulation is a topic that has been extensively addressed [2,7]. Although the MPT pore can be either activated or blocked by many chemicals with disparate identities, perhaps its most notable modulator has been Cyclosporin A (CsA) [3-5,8]. Initially used to inhibit the immune response following organ transplantations [9], CsA has remained the choice “control drug” to determine the putative involvement of the MPT pore in a given treatment or condition (see [10] ).
It was Fournier et al. who first described the enhanced capacity of CsA-treated mitochondria to accumulate substantially higher loads of Ca2+ [11]. However, Martin Crompton determined that CsA was actually inhibiting the Ca2+-induced MPT pore [8]. Further work from Halestrap’s group determined that CsA efficiently inhibited mitochondrial peptidyl proline cistrans-isomerase activity from pure mitochondrial extracts with a ki closely matching the ki for MPT pore closure. Further studies using covalent labeling with photoactive CsA determined that the immunosuppressant potently interacted with human mitochondrial Cyclophilin D [12]. As a historical fact, this protein was first termed “Cyclophilin D” and later on, it had to be annotated as Cyclophilin F (gene name Ppif) given that the original Cyclophilin-D gene Ppid encodes for a cytoplasmic cyclophilin, cyclophilin-40. We believe this term ambiguity can represent a potential source of confusion between cytosolic (Ppid) and mitochondrial (Ppif) cyclophilins. For coherence with previous and current studies on mitochondrial cyclophilins and the MPT pore, we refer here to mitochondrial cyclophilin as Cyclophilin D (CypD).
Pharmacological evidence strongly suggested CsA was inhibiting the MPT pore by binding to CypD [13]. In addition, initial working models suggested CypD would bind the Adenine Nucleotide Translocator (ANT) and possibly the Voltage Dependent Anion Channel (VDAC) to form the MPT pore [14]. These hypotheses were challenged later on as mice deficient in either ANT or VDAC still displayed an MPT response [15,16]. It is noteworthy to mention however, that the pore detected in ANT-less animals was remarkably resistant to Ca2+ and oxidative stress [15]. A subsequent hypothesis proposed CypD was rather binding the mitochondrial phosphate carrier (PiC) instead of ANT [17,18]. This hypothesis was also challenged as cell lines and mice with altered levels of PiC displayed normal MPT pore readings with a minimally modified response to Ca2+ only upon PiC deletion [19-21].
In 2005, four different groups published studies on mice lacking CypD [22-25]. In all cases, MPT pore opening was desensitized to Ca2+ and reactive oxygen species (ROS) but not to other inducers such as diamide or phenylarsine oxide. These results suggested a regulatory role of CypD in a mechanism likely involving its enzymatic activity. Given the unknown identity of the MPT pore, it is not an easy task to determine how CypD exactly interacts with the pore. Recent models suggest that CypD binds to ATP synthase subunit OSCP to modulate the MPT pore [26]: potentially formed by dynamic relaxation of ATP synthase c subunit oligomers [27]. However, this hypothesis still awaits further confirmation and mechanistic validation since little is known about the molecular mechanisms by which CypD can activate MPT pore opening. Therefore, it is the purpose of this article to review the current state-of-the-art structural and mechanistic insight into CypD-induced opening of the MPT pore.
2. Structure-Function Relationships of Mitochondrial Cyclophilin D
Human CypD is a cytoplasm-translated globular protein of 206 residues and ~22 kDa. Upon its transport to the mitochondria, its mitochondrial targeting sequence is cleaved thus resulting in a mature protein with a theoretical MW of 18.9 kDa. According to the available PDB model 3QYU, CypD consists of 8 antiparallel β sheets and 2 well defined α-helices enclosing the sheets (Fig. 1A). CypD also presents a conserved short α-helical turn containing a tryptophan (W121) required for CsA sensitivity as well as for catalysis. This peculiarity is also true for CypA [28]. Previous studies suggest the active site of most cyclophilins consists of a highly conserved patch of aminoacids encompassing 150 Å2 in the so-called CsA-binding domain (CsABD). Structural insight into the PPIase family of proteins reveals the presence of a remarkably conserved pair of pockets that can contribute to substrate selectivity and turnover of the enzymes [28]. The proline interaction surface pocket (S1) constitutes the docking interface for the target proline to be isomerized (Fig. 1B) and the substrate interaction surface (S2) provides a relatively deep scaffold binding structure where ligands with disparate chemical moieties can be accommodated. Nonetheless, a cluster of “gatekeeper” aminoacids, whose sidechains provide sterical access to potential substrates, restricts substrate binding to S2 [28]. Less is known about the opposite side of the CsABD also known as the “backface” of CypD. Previous studies suggest that this portion of most cyclophilins mediates protein:protein interactions [29,30], and CypD can bind to target proteins through its backface [20]. This can explain why conservation between CypD and CypA is less evident at the backface of CypD (Fig. 1B).
Figure 1. Structural elements of Cyclophilin D.
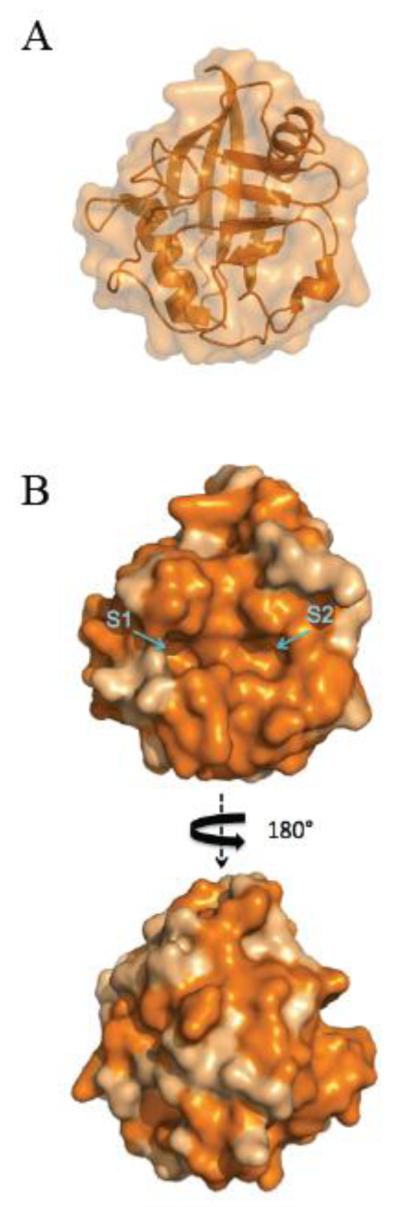
(A) Secondary structure-surface representation denotes a canonical cyclophilin family structure with 8 antiparallel β sheets and 2 well defined α-helices enclosing the sheets. (B) CypD homology rendering comparison between human CypA and CypD using ProtSkin shows high conservation (in orange). Homology is remarkably high in the CsABD, which also encompasses S1 and S2 pockets. Models were retrieved from the Protein Data Bank (PDB ID: 3QYU ) and rendered using Pymol [65].
The molecular mechanism of CypD-mediated peptidyl-proline isomerization has remained relatively unstudied. However, given its striking identity with cytosolic CypA (78%), which reaches almost 100% in the catalytic site (Fig. 1B) it is possible to infer a working model for CypD-mediated proline isomerization based on the published catalytic mechanism of CypA. This working model may also be extrapolated to other mitochondrial cyclophilins given its remarkable conservation (Fig. 1B). In such model, the proline residue to be isomerized remains immovable relative to the catalytic site of CypA located close to the highly conserved R55. Next, the oxygen of the adjacent N-terminal residue rotates 180° clockwise from cis to trans. Apparently, formation of a hydrogen bond between Arg55 and the proline to be isomerized enables the formation of a pyramidal sp3 state for the proline’s nitrogen atom and this deconjugates the resonance of the prolyl amide bond [31,32] which facilitates the isomerization step. Rat CypD catalytic arginine is located at the conserved residue 96 [33]. In mutant CypD R96G, substrates are still able to bind to CypD but are not isomerized (loss-of-function mutation). The H167Q mutation on the CsA-binding domain of rat CypD provides a steric hindrance for substrates and therefore proline isomerization does not take place. These results support a potential catalytic mechanism where substrates bind to CypD active site and the guanidinium group of R96 facilitates isomerization by anchoring the substrate proline oxygen. This would finally result in the hybridization of the proline nitrogen atom in sp3 during the cistrans transition state.
The high affinity of CypD for CsA (KD=13.4 nM) has allowed the determination of the crystal structure of CypD in the presence of this undecapeptide [34]. A close look at the binding geometry between both molecules shows that the interaction involves both hydrophobic contacts and hydrogen bonding. For hydrophobic contacts, F60, M61, A101, A103, L122, Q111, F113 and H126 play a major role in the tight interaction. Hydrogen bonding involves residues W121, R55, Q63, 102, and G72 (Fig. 2A). On the side of CsA, binding occurs at residues Bmt1, Aba2 and Mle9-Mva11. Importantly, hydrogen bonding occurs only at CsA main-chain N and O atoms (Fig. 2B). Many other CsA derivatives have been shown to specifically bind to CypD such as NMe-Ala-6- cyclosporine A and N-Me-Val-4-cyclosporin A 3 [35-37]. Sangliferin A, another immunosuppressant drug which is chemically unrelated to CsA, does inhibit CypD and MPT pore opening while preserving isomerase activity and binding to the pore regulator ANT [38].
Figure 2. Structural elements of the CsABD of human Cyclophilin D.

(A) Residues involved in the interaction with CsA have hydrophobic (yellow) and polar (green) interactions with CsA. (B) High affinity binding of CsA (orange) completely occludes S1 and S2 pockets and effectively hampers PPIase activity. Models were retrieved from the Protein Data Bank (PDB ID: 3QYU ) and rendered using Pymol [65].
2.1 Cyclophilin D Post Translational Modifications
Although the preceding section suggests a working model for CypD-mediated isomerization of proline residues, little is known about the physiological activation of CypD. On this, Linard et al. proposed that CypD enzymatic activity could be modulated under conditions of oxidative stress [39]. This group further proposed that PPIase activity of CypD is not dependent on the presence of C203 but is decreased in the C82S, C104S and C157S CypD mutants. Of all these residues, C82 and C104 are contiguous to the CsABD and consequently a mutation to serine could affect putative redox-dependent isomerization activity. According to this model, C203 and C157 could form an intramolecular disulfide bridge whereas in the absence of such adduct, CypD was slightly more active [39].
A possible physiological switch by which these cysteines are protected by signaling ROS during non-stress conditions is by posttranslational modifications (PTMs) such as S-nitrosylation. Indeed, Kohr and colleagues detected S-nitrosylation on Cysteines 103, 156 and 203 of CypD [40]. The relevance of these PTMs is beginning to be understood. For instance, Nguyen et al. demonstrated that cysteine 203 is required for redox-induced activation of the MPT pore [41]. Mutation of the S-nitrosylation site on C203 for S203 still resulted in opening of the MPT pore but with enhanced resistance to H2O2 and Ca2+. In addition, the C203S mutation rendered Mouse Embryo Fibroblasts resistant to reactive oxygen species-induced cell death. When analyzing human CypD, it is possible to locate the relative C203 on the “backface” of the protein (Fig. 3B). It is consequently reasonable to assume that the protection elicited in the C203S mutant CypD does not involve PPIase activity given the CsABD is sterically unaffected. This assumption is also supported by the fact that CypD S-nitrosylation with GSNO does not affect PPIase activity of recombinant CypD [41]. Overall these results strongly suggest that the C203 of CypD is rather required for adequate binding to its target or for the interaction with another modulatory factor(s). Indeed, previous studies have proposed that the backface of cyclophilins mediate protein:protein interactions [29,30]. Two proposed signaling factors include GSK3-β and p53.
Figure 3. Cartoon sequences showing sequential rotations of human CypD.

All serines (green) and threonines (yellow) are represented in sphere projection. Residues C203 (magenta) and K175 (white) are located in the backface of CypD, whereas conserved S123 (purple) is located in the CsABD. Previously proposed residues of CypD involving PTMs are pointed with an arrow. Models were retrieved from the Protein Data Bank (PDB ID: 3QYU ) and rendered using Pymol [65].
Based on a previous work by Juhaszova et al. showing that GSK3-β mediates regulatory signaling of the MPT pore [42], Rasola and collaborators proposed that CypD is phosphorylated by GSK3-β as a final step in a complex signaling cascade starting in the cytosol with ERK [43]. In this model, the authors provide evidence suggesting that in the unphosphorylated (active) state, ~1% of the GSK3-β pool is translocated to the mitochondrial matrix and is able to bind and phosphorylate human CypD potentially at S38, S39 or S123. Then, phospho-CypD would activate MPT pore resulting in tumor cell death. Regarding the putative phosphorylation of CypD, a sequence alignment between human and mouse CypD reveals that S38 and S39 are absent in mouse CypD, whereas S123 (S122 in mouse) is located in a remarkably conserved cluster of aminoacids in the periphery of the CsABD (Fig 3A). If current models suggesting MPT pore opening requires PPIase activity, then it makes sense that CypD phosphorylation (possibly at S122) increases enzyme activity. However, CypD presents 8 serines and 8 threonines, being all in the periphery of the enzyme (Fig. 3). Whether any of these residues is preferentially phosphorylated and CypD activity is consequently modified is still unknown.
According to Moll’s group, another possible regulatory factor where CypD can dock and potentially unleash pore opening is p53 [44]. Studies from this group support the notion that p53 translocates to the mitochondria upon oxidative insults to orchestrate MPT pore opening and activate necrosis. Accordingly, a robust p53-CypD complex could be detected following brain ischemia/reperfusion injury. The residues of p53 required for interaction were determined to encompass aminoacids 80-220. Binding was completely suppressed in the presence of CsA, implying that the CsABD docks to p53 on these aminoacids. However, this complex has been proposed to be irrelevant for Ca2+-induced MPT pore opening, thus narrowing down the potential situations under which p53 may induce cellular demise by interacting with CypD [45].
The last PTM reported for CypD is acetylation [46]. The balance between glycolysis and oxidative phosphorylation involves complex signaling and the acetylation-deacetylation pattern of many mitochondrial proteins is though to be a cornerstone of this regulation [47,48]. Changes in the acetylation profile of many mitochondrial proteins are evident following changes from glycolytic to aerobic metabolism [49]. These protein acetylation patterns are mainly regulated by deacetylases known as sirtuins. For instance, acetyl-CoA synthetase 2 is a target of mitochondrial sirtuin-3, and is activated once deacetylated (Hallows, 2006). CypD is not exempt from this regulation as previous studies demonstrate the binding and deacetylation enforced by sirtuin-3 as an adaptation mechanism following changes from a glycolytic to a respiratory environment [48]. Studies by Pastorino’s group suggest that CypD deacetylation mediated by sirtuin-3 occurs at lysine 145 (K166 in the unprocessed mouse CypD protein). A closer glance into the human CypD structure shown in Fig.3 reveals that K166 (K167 in human CypD) is located close to the catalytic W121 in the conserved short α-helical turn encompassing the CsABD [28]. Once deacetylated, the PPIase activity of CypD activity drops, potentially explaining why CypD deacetylation suppresses age-induced cardiac hypertrophy [46]. The results by Shulga et al. were validated using two CypD point mutants. In one case, a mutation that mimicked constitutive acetylation of CypD (K166Q) resulted in a sensitized MPT pore. The mutation K166R on the other hand, which mimics constitutive deacetylation of CypD, resulted in MPT pore inhibition [48,50]. Shulga and collaborators proposed that the main pathway involved in MPT pore desensitization -mediated by CypD deacetylation relied on the detachment of the protein from a putative binding site on the MPT regulator ANT.
3. How and where is CypD binding to modulate MPT pore opening?
To address this question, some groups have adopted bait-prey approaches. In early studies, Crompton’s group detected binding between the fusion protein GST-CypD and ~32-kDa proteins from heart mitochondrial membranes extracted with the zwitterionic detergent CHAPS [14]. These proteins reacted with antibodies against VDAC and ANT, which led to suggest that the MPT pore would form from the interaction between VDAC and ANT in a CypD-regulated way. Purified VDAC, ANT and GST-CypD reconstituted in fluorescein-loaded proteoliposomes were permeabilized by Ca2+ plus phosphate in a CsA-sensitive process. On the same year, Halestrap’s group performed similar “pulldown” experiments using GST-CypD and Triton-X100-solubilized inner mitochondrial membranes from liver [51]. After resolving the bound complexes, ANT was detected but not VDAC. Previously, Brustovetsky and Klingenberg demonstrated the reconstitution of a MPT pore-like activity, using ANT reconstituted in proteoliposomes [52]. These studies, plus the fact that selective ligands of ANT potently modulate the MPT pore led to the prevailing notion that the ANT plays a central role in the formation of the MPT pore [53]. Indeed, genetic elimination of ANT results in the desensitization of the MPT pore to Ca2+ (~3-fold Vs. ~2-fold upon CypD deletion) and traditional pore inducers such as diamide and t-butyl hydroperoxide [15]. This has led to a proposition that ANT probably affects the MPT pore through the modulation of the inner mitochondrial membrane surface potential (which is different from ΔΨ) [1,54].
Halestrap’s group recently proposed the possibility that ANT might be interacting closely with other components of the MPT pore [55]. One such component could be the mitochondrial phosphate carrier (PiC). Initial studies assessing the potential participation of this protein showed a nice correlation between PiC activity and MPT pore onset in addition to a CsA-sensitive CypD binding to this mitochondrial carrier [17,18]. Indeed, recent studies by our group have confirmed such binding and detected the requirement of residues 70-110 of CypD to bind PiC [20]. These residues form a subdomain encompassing the CsABD and the backface of the protein, which would explain why binding of CypD to the PiC is still detected (albeit decreased) in the presence of CsA. However, experiments where PiC was silenced in HeLa cells resulted in the detection of a canonical MPT pore activity [19]. Studies in cardiac-specific transgenic mice overexpressing or downregulating PiC resulted in no modulation of MPT pore readings as a function of PiC expression levels [20]. Nonetheless, cardiac-specific genetic deletion in mice results in a mild protection against Ca2+-induced MPT and reperfusion injury [21].
Previous evidence from Richelli’s group showed that CypD was able to bind to the lateral stalk of F1F0-ATP synthase and activate MPT pore opening [56]. By using a similar approach, Bernardi’s group recently showed that CypD binds the Oligomycin Sensitivity Conferring Protein (OSCP) subunit of ATP synthase potentially though electrostatic interactions [26]. General models of ATP synthases position OSCP in the F1 sector of the enzyme, closely interacting with α and β subunits from its docking site in the lateral stalk of the complex [57]. Giorgio et al. showed that dimeric (but not monomeric) F0F1-ATP synthase was able to form a multiple conductance channel (MCC) that closely resembled the electrophysiological behavior of the MPT pore [26]. However, divergences from a canonical MCC were found in terms of the lack of sensitivity of those preparations to CsA and Phenyl Arsine Oxide (PAO). In addition, the mechanism by which CypD triggers the (inner membrane) MPT pore through interaction with OSCP (in the matrix) remained unaddressed. Quite strikingly, siRNA-mediated depletion of OSCP (the alleged CypD target) resulted in a sensitized MPT pore to Ca2+. If OSCP were the actual CypD binding protein, it would be logical to assume that depletion of either CypD or OSCP would render the same phenotype (i.e. enhanced Ca2+-retention capacity), which apparently is not the case [26]. While this study did not ascribe the MCC-like activity to a single protein in the dimeric complex V, the authors suggested that the MPT pore could form at the membrane interphase between both dimers. This could explain the effect of chemically unrelated MPT pore effectors, potentially affecting the membrane such as low concentrations of mastoparan [58], fatty acids [59] and phospholipase A2 inhibitors [60].
Another line of evidence suggested a role of ATP synthase c-subunit as a critical component of the MPT pore [61]. Indeed, Bonora et al. showed that siRNA-mediated c subunit depletion protected HeLa cells from calcium overload and oxidative stress. While appealing, it is important to mention that disturbances in the membrane sector of ATP synthase can distort the tightly regulated morphology of the cristae membrane [62] and this can potentially affect MPT pore readings indirectly (For an editorial see [63]). Recent commentaries also pointed out that the antibody used by Bonora and colleagues detected a protein of ~15 kDa, whereas mature c-subunit migrates at 7 kDa, thus adding complexity to their results [55,64].
In line with a potential role of ATP synthase c-subunit as a core component of the MPT pore, recent evidence by Jonas group suggests that purified ATP-synthase c subunit presents MCC-like activity per se when reconstituted in liposomes with peak conductances up to ~1.5-2 nS [27]. Similar to the MPT pore, the channel formed by c-subunit presents negative rectification. This group reported that ATP (a potent MPT pore inhibitor) decreased channel conductance, albeit at much higher concentrations. Channel inhibition was also effectively accomplished following addition of an anti-c-subunit antibody. Strikingly, MCC activity was activated by Ca2+ and strongly attenuated with such anti-c-subunit antibody in sub mitochondrial vesicles strongly supporting the possibility that the c-subunit forms the actual channel of the MPT. Channel activity correlated with spatial clustering between c subunits in a CsA-dependent manner. With this evidence, Jonas’ group proposed a mechanism whereby the F0 sector “relaxation” and F1 sector partial “release” constitutes the fundamental structure of the MPT pore. Although this postulate is very provocative and would put an end to long lasting efforts to identify the identity of the MPT pore, there are still unaddressed questions concerning the possibility that the c-ring forms the MPT pore (see [55,64]). For instance, the authors successfully detected a c-subunit-dependent, Ca2+ and CypD-induced channel activity, being reversed by ATP synthase β subunit. However, there is still no evidence for a direct interaction between the c and β subunits [57].
Based on the available studies addressing potential mechanisms for CypD-dependent MPT pore, it is possible to infer an updated working model whereby CypD binds to PiC, ANT or ATP synthase to regulate pore opening (Fig. 4). In this model, Ca2+ and oxidative stress may activate MPT by inducing c-subunit loosening and increasing channel conductance, being facilitated by ANT in the “c” conformation. Conversely, blockade of the MPT pore by adenine nucleotides could target both ATP synthase and ANT to close the pore. This effect can also be recapitulated in the presence of bongkrekic acid, which blocks ANT in the “m” conformation.
Figure 4. Current proposed protein complexes influencing the MPT pore.

In this figure, ATP synthase (blue) is represented in the dimeric form and interacts with CypD (yellow) at the level of OSCP. CypD can also interact with ANT (red) or PiC (orange). Models were retrieved from the Protein Data Bank (except for the PiC, which was modeled previously [20]) and rendered using Pymol [65].
4. Concluding Remarks
Cyclophilin D is a matrix proline isomerase originally thought to exclusively facilitate nascent mitochondrial protein folding. However, its importance in the regulation of mitochondrial bioenergetics under normal and stress conditions is beginning to be understood. Posttranslational modifications or the addition of selective inhibitors can tune CypD’s PPIase activity and/or alter its binding properties, consequently affecting downstream targets such as ATP synthase, ANT or even PiC. This in turn would affect the probability of MPT pore opening. Given the disparate identity of MPT pore modulators, we do not discard the possibility that the MPT pore can form through the association of proposed regulatory factors. Studies aimed to unveil more molecular determinants, inhibitors and protein partners of CypD will allow more definite mechanistic insights into how this isomerase orchestrates MPT pore opening.
Acknowledgments
Funding This work was supported by the National Institutes of Health grant HL094404 (to C.P.B.). M.G.A. is currently supported by an American Heart Association Midwest Affiliate Postdoctoral Fellowship (13POST14060013).
Abbreviations
- ANT
adenine nucleotide translocase
- CsA
cyclosporin A
- CsABD
cyclosporin A binding domain
- CypA
cyclophilin A
- CypD
cyclophilin D
- GST
glutathione S-transferase
- GSNO
S-Nitrosoglutathione
- MCC
multiple conductance channel
- MPT
mitochondrial permeability transition
- OSCP
oligomycin sensitivity conferral protein
- PAO
phenyl arsine oxide
- PDB
protein data bank
- PiC
mitochondrial phosphate carrier
- PTM
posttranslational modification
- VDAC
voltage dependent anion channel
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, Di Lisa F, Forte MA. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- [2].Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. J. Biol. Chem. 1976;251:5069–5077. [PubMed] [Google Scholar]
- [3].Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria I The protective mechanisms. Arch. Biochem. Biophys. 1979;195:453–459. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- [4].Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria II Nature of the Ca2+ trigger site. Arch. Biochem. Biophys. 1979;195:460–467. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- [5].Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria III Transitional Ca2+ release. Arch. Biochem. Biophys. 1979;195:468–477. doi: 10.1016/0003-9861(79)90373-4. [DOI] [PubMed] [Google Scholar]
- [6].Baines CP. The cardiac mitochondrion: nexus of stress. Annu. Rev. Physiol. 2010;72:61–80. doi: 10.1146/annurev-physiol-021909-135929. [DOI] [PubMed] [Google Scholar]
- [7].Zoratti M, Szabo I, De Marchi U. Mitochondrial permeability transitions: how many doors to the house? Biochim. Biophys. Acta. 2005;1706:40–52. doi: 10.1016/j.bbabio.2004.10.006. [DOI] [PubMed] [Google Scholar]
- [8].Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 1988;255:357–360. [PMC free article] [PubMed] [Google Scholar]
- [9].Laupacis A, Keown PA, Ulan RA, McKenzie N, Stiller CR. Cyclosporin A: a powerful immunosuppressant. Can. Med. Assoc. J. 1982;126:1041–1046. [PMC free article] [PubMed] [Google Scholar]
- [10].Di Lisa F, Carpi A, Giorgio V, Bernardi P. The mitochondrial permeability transition pore and cyclophilin D in cardioprotection. Biochim. Biophys. Acta. 2011;1813:1316–1322. doi: 10.1016/j.bbamcr.2011.01.031. [DOI] [PubMed] [Google Scholar]
- [11].Fournier N, Ducet G, Crevat A. Action of cyclosporine on mitochondrial calcium fluxes. J. Bioenerg. Biomembr. 1987;19:297–303. doi: 10.1007/BF00762419. [DOI] [PubMed] [Google Scholar]
- [12].Tanveer A, Virji S, Andreeva L, Totty NF, Hsuan JJ, Ward JM, Crompton M. Involvement of cyclophilin D in the activation of a mitochondrial pore by Ca2+ and oxidant stress. Eur. J. Biochem. 1996;238:166–172. doi: 10.1111/j.1432-1033.1996.0166q.x. [DOI] [PubMed] [Google Scholar]
- [13].Connern CP, Halestrap AP. Purification and N-terminal sequencing of peptidyl-prolyl cis-trans-isomerase from rat liver mitochondrial matrix reveals the existence of a distinct mitochondrial cyclophilin. Biochem. J. 1992;284(Pt 2):381–385. doi: 10.1042/bj2840381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Crompton M, Virji S, Ward JM. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 1998;258:729–735. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- [15].Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Leung AW, Halestrap AP. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim. Biophys. Acta. 2008;1777:946–952. doi: 10.1016/j.bbabio.2008.03.009. [DOI] [PubMed] [Google Scholar]
- [18].Leung AW, Varanyuwatana P, Halestrap AP. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem. 2008;283:26312–26323. doi: 10.1074/jbc.M805235200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Varanyuwatana P, Halestrap AP. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion. 2012;12:120–125. doi: 10.1016/j.mito.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gutiérrez-Aguilar M, Douglas DL, Gibson AK, Domeier TL, Molkentin JD, Baines CP. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. J. Mol. Cell. Cardiol. 2014;72:316–325. doi: 10.1016/j.yjmcc.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kwong JQ, Davis J, Baines CP, Sargent MA, Karch J, Wang X, Huang T, Molkentin JD. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 2014;21:1209–1217. doi: 10.1038/cdd.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. U.S.A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- [24].Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J. Biol. Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- [25].Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- [26].Giorgio V, Von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabò I, Lippe G, Bernardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc. Natl. Acad. Sci. U.S.A. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park H-A, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA, Jonas EA. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc. Natl. Acad. Sci. U.S.A. 2014;111:10580–10585. doi: 10.1073/pnas.1401591111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Davis TL, Walker JR, Campagna-Slater V, Finerty PJ, Paramanathan R, Bernstein G, MacKenzie F, Tempel W, Ouyang H, Lee WH, Eisenmesser EZ, Dhe-Paganon S. Structural and biochemical characterization of the human cyclophilin family of peptidyl-prolyl isomerases. PLoS Biol. 2010;8:e1000439. doi: 10.1371/journal.pbio.1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Reidt U, Wahl MC, Fasshauer D, Horowitz DS, Lührmann R, Ficner R. Crystal structure of a complex between human spliceosomal cyclophilin H and a U4/U6 snRNP-60K peptide. J. Mol. Biol. 2003;331:45–56. doi: 10.1016/s0022-2836(03)00684-3. [DOI] [PubMed] [Google Scholar]
- [30].Xu C, Zhang J, Huang X, Sun J, Xu Y, Tang Y, Wu J, Shi Y, Huang Q, Zhang Q. Solution structure of human peptidyl prolyl isomerase-like protein 1 and insights into its interaction with SKIP. J. Biol. Chem. 2006;281:15900–15908. doi: 10.1074/jbc.M511155200. [DOI] [PubMed] [Google Scholar]
- [31].Howard BR, Vajdos FF, Li S, Sundquist WI, Hill CP. Structural insights into the catalytic mechanism of cyclophilin A. Nat. Struct. Biol. 2003;10:475–481. doi: 10.1038/nsb927. [DOI] [PubMed] [Google Scholar]
- [32].Zhao Y, Ke H. Crystal structure implies that cyclophilin predominantly catalyzes the trans to cis isomerization. Biochemistry. 1996;35:7356–7361. doi: 10.1021/bi9602775. [DOI] [PubMed] [Google Scholar]
- [33].Lin DT, Lechleiter JD. Mitochondrial targeted cyclophilin D protects cells from cell death by peptidyl prolyl isomerization. J. Biol. Chem. 2002;277:31134–31141. doi: 10.1074/jbc.M112035200. [DOI] [PubMed] [Google Scholar]
- [34].Kajitani K, Fujihashi M, Kobayashi Y, Shimizu S, Tsujimoto Y, Miki K. Crystal structure of human cyclophilin D in complex with its inhibitor, cyclosporin A at 096-A resolution. Proteins. 2008;70:1635–1639. doi: 10.1002/prot.21855. [DOI] [PubMed] [Google Scholar]
- [35].Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 1995;307(Pt 1):93–98. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Khaspekov L, Friberg H, Halestrap A, Viktorov I, Wieloch T. Cyclosporin A and its nonimmunosuppressive analogue N-Me-Val-4-cyclosporin A mitigate glucose/oxygen deprivation-induced damage to rat cultured hippocampal neurons, Eur. J. Neurosci. 1999;11:3194–3198. doi: 10.1046/j.1460-9568.1999.00743.x. [DOI] [PubMed] [Google Scholar]
- [37].Nicolli A, Basso E, Petronilli V, Wenger RM, Bernardi P. Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, and cyclosporin A-sensitive channel. J. Biol. Chem. 1996;271:2185–2192. doi: 10.1074/jbc.271.4.2185. [DOI] [PubMed] [Google Scholar]
- [38].Clarke SJ, McStay GP, Halestrap AP. Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J. Biol. Chem. 2002;277:34793–34799. doi: 10.1074/jbc.M202191200. [DOI] [PubMed] [Google Scholar]
- [39].Linard D, Kandlbinder A, Degand H, Morsomme P, Dietz KJ, Knoops B. Redox characterization of human cyclophilin D: identification of a new mammalian mitochondrial redox sensor? Arch. Biochem. Biophys. 2009;491:39–45. doi: 10.1016/j.abb.2009.09.002. [DOI] [PubMed] [Google Scholar]
- [40].Kohr MJ, Aponte AM, Sun J, Wang G, Murphy E, Gucek M, Steenbergen C. Characterization of potential S-nitrosylation sites in the myocardium. Am. J. Physiol. Heart. Circ. Physiol. 2011;300:H1327–35. doi: 10.1152/ajpheart.00997.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J. Biol. Chem. 2011;286:40184–40192. doi: 10.1074/jbc.M111.243469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rasola A, Sciacovelli M, Chiara F, Pantic B, Brusilow WS, Bernardi P. Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc. Natl. Acad. Sci. U.S.A. 2010;107:726–731. doi: 10.1073/pnas.0912742107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. p53 Opens the Mitochondrial Permeability Transition Pore to Trigger Necrosis. Cell. 2012;149:1536–1548. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Karch J, Molkentin JD. Is p53 the long-sought molecular trigger for cyclophilin D-regulated mitochondrial permeability transition pore formation and necrosis? Circ. Res. 2012;111:1258–1260. doi: 10.1161/CIRCRESAHA.112.280990. [DOI] [PubMed] [Google Scholar]
- [46].Hafner AV, Dai J, Gomes AP, Xiao CY, Palmeira CM, Rosenzweig A, Sinclair DA. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging (Albany NY) 2010;2:914–923. doi: 10.18632/aging.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lam MP, Lau E, Liem DA, Ping P. Cyclophilin D and acetylation: a new link in cardiac signaling. Circ. Res. 2013;113:1268–1269. doi: 10.1161/CIRCRESAHA.113.302687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Shulga N, Wilson-Smith R, Pastorino JG. Sirtuin-3 deacetylation of cyclophilin D induces dissociation of hexokinase II from the mitochondria. J. Cell Sci. 2010;123:894–902. doi: 10.1242/jcs.061846. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [49].Murphy E, Kohr M, Menazza S, Nguyen T, Evangelista A, Sun J, Steenbergen C. Signaling by S-nitrosylation in the heart. J. Mol. Cell. Cardiol. 2014;73:18–25. doi: 10.1016/j.yjmcc.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Shulga N, Pastorino JG. Ethanol sensitizes mitochondria to the permeability transition by inhibiting deacetylation of cyclophilin-D mediated by sirtuin-3. J. Cell Sci. 2010;123:4117–4127. doi: 10.1242/jcs.073502. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [51].Woodfield K, Ruck A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem. J. 1998;336(Pt 2):287–290. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Brustovetsky N, Klingenberg M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+ Biochemistry. 1996;35:8483–8488. doi: 10.1021/bi960833v. [DOI] [PubMed] [Google Scholar]
- [53].Halestrap AP, Woodfield KY, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J. Biol. Chem. 1997;272:3346–3354. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- [54].Rottenberg H, Marbach M. Regulation of Ca2+ transport in brain mitochondria II The mechanism of the adenine nucleotides enhancement of Ca2+ uptake and retention. Biochim. Biophys. Acta. 1990;1016:87–98. doi: 10.1016/0005-2728(90)90010-2. [DOI] [PubMed] [Google Scholar]
- [55].Halestrap AP, Richardson AP. The mitochondrial permeability transition: A current perspective on its identity and role in ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2014 doi: 10.1016/j.yjmcc.2014.08.018. DOI: 10.1016/j.yjmcc.2014.08.018. [DOI] [PubMed] [Google Scholar]
- [56].Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G. Cyclophilin D Modulates Mitochondrial F0F1-ATP Synthase by Interacting with the Lateral Stalk of the Complex. J. Biol. Chem. 2009;284:33982–33988. doi: 10.1074/jbc.M109.020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Walker JE. The ATP synthase: the understood, the uncertain and the unknown. Biochem. Soc. Trans. 2013;41:1–16. doi: 10.1042/BST20110773. [DOI] [PubMed] [Google Scholar]
- [58].Pfeiffer DR, Gudz TI, Novgorodov SA, Erdahl WL. The peptide mastoparan is a potent facilitator of the mitochondrial permeability transition. J. Biol. Chem. 1995;270:4923–4932. doi: 10.1074/jbc.270.9.4923. [DOI] [PubMed] [Google Scholar]
- [59].Bernardi P, Penzo D, Wojtczak L. Mitochondrial energy dissipation by fatty acids Mechanisms and implications for cell death. Vitam. Horm. 2002;65:97–126. doi: 10.1016/s0083-6729(02)65061-7. [DOI] [PubMed] [Google Scholar]
- [60].Broekemeier KM, Schmid PC, Schmid HH, Pfeiffer DR. Effects of phospholipase A2 inhibitors on ruthenium red-induced Ca2+ release from mitochondria. J. Biol. Chem. 1985;260:105–113. [PubMed] [Google Scholar]
- [61].Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, Wieckowski MR, Kroemer G, Galluzzi L, Pinton P. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle. 2013;12:674–683. doi: 10.4161/cc.23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Paumard P, Vaillier J, Coulary B, Schaeffer J, Soubannier V, Mueller DM, Brèthes D, di Rago J-P, Velours J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002;21:221–230. doi: 10.1093/emboj/21.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Gutierrez-Aguilar M, Baines CP. Mitochondrial ATP Synthase: Is the Molecular Engine of Life also an Efficient Death Machine. Bioenergetics. 2014;3:e119. [Google Scholar]
- [64].Halestrap AP. The C Ring of the F1F0 ATP Synthase Forms the Mitochondrial Permeability Transition Pore: A Critical Appraisal. Front. Oncol. 2014;4:234. doi: 10.3389/fonc.2014.00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].The PyMOL Molecular Graphics System. Version 1.5.0.4 Schrödinger, LLC; [Google Scholar]