

Abstract
Purpose
Congenital hypogonadotropic hypogonadism (CHH) and split hand/foot malformation (SHFM) are two rare genetic conditions. Here we report a clinical entity comprising CHH and SHFM.
Methods
We identified patients with CHH and SHFM through international collaboration. Probands and available family members underwent phenotyping and screening for FGFR1 mutations. The impact of identified mutations was assessed by sequence- and structure-based predictions, and/or functional assays.
Results
We identified 8 probands with CHH with (n=3, Kallmann Syndrome) or without anosmia (n=5) and SHFM, 7 of whom (88%) harbor FGFR1 mutations: one individual is homozygous for p.V429E; six individuals are heterozygous for p.G348R, p.G485R, p.Q594*, p.E670A, p.V688L, and p.L712P. All mutations were predicted to be loss-of-function by in silico analysis. Probands with FGFR1 mutations have severe GnRH deficiency (absent puberty and/or cryptorchidism and/or micropenis). SHFM in both hands and feet was only observed in the patient with the homozygous p.V429E mutation; V429 maps to the FRS2α binding domain of FGFR1, and functional studies of the p.V429E mutation demonstrated that it decreased recruitment and phosphorylation of FRS2α to FG FR 1 , thereby resulting in reduced MAPK signaling.
Conclusion
FGFR1 should be prioritized for genetic testing in patients with CHH and SHFM, because the likelihood of a mutation increases from 10% in the general CHH population to 88%.
Keywords: congenital hypogonadotropic hypogonadism, split hand/foot malformation, fibroblast growth factor receptor 1, FGF receptor substrate 2α
INTRODUCTION
Congenital hypogonadotropic hypogonadism (CHH [MIM 146110]) is a genetic disorder characterized by absent or incomplete pubertal development and infertility due to deficiency of gonadotropin-releasing hormone (GnRH) secretion or action. The co-occurrence of CHH with anosmia is termed Kallmann syndrome (KS [MIM 308700, 147950, 244200, 610628, 612370, 612702]). Anosmia in KS is usually linked to agenesis of the olfactory structures, which provide the anatomic path for the migration of GnRH neurons from the olfactory placode to the hypothalamic region during embryonic development.1 To date, mutations in >20 genes have been found to underlie CHH, acting either alone or in combination.2
Approximately 10-12% of CHH patients carry loss-of-function mutations in fibroblast growth factor receptor 1 (FGFR1), the first gene reported to be associated with both KS and normosmic CHH.3,4 CHH-associated FGFR1 mutations are typically heterozygous, and the disease is inherited as an autosomal dominant trait with variable expressivity. FGFR1 encodes a member of the FGFR subfamily of receptor tyrosine kinases. Upon binding a FGF ligand in the presence of heparan sulfate, FGFR1 dimerizes and its kinase domains are autophosphorylated. In turn, this activates intracellular pathways that culminate in diverse biological responses; activation of the phospholipase C gamma (PLCγ) pathway requires phosphorylation of FGFR1 tyrosine 766 (Y766), while activation of the Ras-MAPK and PI3-K pathways is mediated by recruitment of FGF receptor substrate 2α (FRS2α).5 FGFR1 is expressed in multiple tissues, including the brain and skeleton,6 among other functions, it is required for fate specification of GnRH neurons in the olfactory placode, as well as for GnRH neuron proliferation and migration to the hypothalamus.7 Alternative splicing of extracellular region-encoding exons of FGFR1 gives rise to the FGFR1b and FGFR1c isoforms; to date, the majority of CHH-associated mutations implicate FGFR1c as the major isoform relevant to GnRH neuron biology.3,4
CHH patients with loss-of-function FGFR1 mutations are enriched for additional skeletal phenotypes, such as cleft lip/palate, dental agenesis, mandibular hypoplasia, scoliosis, butterfly vertebrae, syndactyly, oligodactyly and clinodactyly.8,9 Recently, FGFR1 mutations (predicted to be loss-of-function) have been identified in patients with Hartsfield syndrome (MIM 615465),10 a rare disorder characterized by the association of holoprosencephaly, and split hand/foot malformation (SHFM, also called ectrodactyly), a severe malformation of the skeletal development with an absent or incomplete development of the central rays of hands, feet, or both.11 Notably, associated phenotypes including midline defect, multiple pituitary hormone deficiency and/or agenesis of the olfactory bulbs/tracts have been described in Hartsfield Syndrome patients.10,12 Herein, we report the association of CHH with SHFM, and show that the large majority of these SHFM-CHH patients carry loss-of-function FGFR1 mutations.
PATIENTS & METHODS
Patients
Via international collaboration (France, UK, Finland and United States), we identified 8 CHH patients with SHFM (7 males and 1 female). Diagnostic criteria for CHH included: (i) failure to initiate and/or complete spontaneous puberty by age 18 years; (ii) serum testosterone ≤3 nmol/L for men or estradiol ≤0.07 nmol/L for women, with low or normal levels of serum gonadotropins; (iii) otherwise normal pituitary function (absence of clinical and/or biochemical evidence of TSH, ACTH, GH deficiency, hyperprolactinemia, or diabetes insipidus), and (iv) normal magnetic resonance imaging (MRI) of the hypothalamic-pituitary region; or, in infants, (v) micropenis and/or cryptorchidism in the setting of low sex steroid and gonadotropin levels during the “mini-puberty”. 13 Assessment for spontaneous partial pubertal development was made based on clinical history, Tanner stage, and (in males) testicular size. Olfaction was assessed by self-report and/or formal smell testing (brief smell identification test, B-SIT or olfactometry). Skeletal phenotypes assessed included SHFM, cleft lip/palate, and dental agenesis. The institutional review board/ethics committee of the Massachusetts General Hospital, Hôpital Robert Debré, Helsinki University Central Hospital, and University College London Medical School approved the studies; all subjects or parents/legal guardians provided written informed consent.
Sequencing
Genomic DNA was obtained from peripheral blood samples using standard phenol-chloroform extraction. Mutation screening for FGFR1 (NM_023110.2) was performed as previously described.2 The coding exonic and proximal intronic (≥15 bp from splice sites) DNA sequences of FGFR1 were amplified by PCR and analyzed by direct sequencing. Sequence variations were found on both DNA strands and were confirmed in a separate PCR. Variants were considered pathogenic mutations if: (i) their allele frequency was <1% in the 1000 Genomes dataset and in Exome Variant Server (EVS), and (ii) the altered amino acid was predicted to be loss-of-function by structural modeling14 or by at least two prediction programs: PolyPhen-215, SIFT16, PMut17, Mutation Taster18 and Condel 19 for missense variants. All subjects were also screened for the presence of mutations in FGF8 (MIM 600483). Other CHH genes were selectively sequenced in some subjects (Supplementary data): KAL1 (MIM 300836), PROKR2 (MIM 607123), PROK2 (MIM 607002), TACR3 (MIM 162332), TAC3 (MIM 162330), GNRHR (MIM 138850), GNRH1 (MIM 152760), KISS1R (MIM 604161), KISS1 (MIM 603286), NSMF (MIM 60813), CHD7 (MIM 608892), and HS6ST1 (MIM 604846) (primers and PCR conditions are available upon request).
Structural modeling
To predict the functional consequences of the identified FGFR1 mutations, the following structures were used: (i) crystal structure of the extracellular ligand-binding domain of human FGFR1 in complex with human FGF2 (PDB ID: 1FQ9);20 (ii) crystal structure of the phosphorylated tyrosine kinase domain of human FGFR1 (PDB ID 3GQI);21 and (iii) nuclear magnetic resonance (NMR) solution structure of a 22 residue-long peptide derived from the juxtamembrane region of FGFR1 (residues 409-430) in complex with the FRS2α phosphotyrosine binding (PTB) domain (PDB ID: 1XR0).22 The structures were analyzed using program O and structural representations were prepared using PyMol.
Analysis of recruitment and phosphorylation of FRS2α by FGFR
Cell-based FRS2 phosphorylation assay
We first evaluated the effect of the V429E mutation on the ability of FGFR1c to phosphorylate FRS2 using cell-based assay. WT and V429E FGFR1c were cloned into the lentiviral vector FUCRW following standard protocols. BaF3 cells were maintained as described23 and transfected with FGFR1 pseudoviral stock in Hank's Balanced Salt Solution buffer. Stably transduced cells were treated with 1.5 nM of FGF1 for 10 min, rinsed in PBS and then lysed in RIPA buffer (Thermo Scientific). Cell extract (30 μg) was resolved by SDS-PAGE and analyzed by western blotting using anti FGFR1 (inhouse antibody raised in rabbit), anti-FRS2 (Abcam ab10425) and anti Phospho-FRS2-α (Tyr196) (Cell Signaling, #3864) antibodies.
Surface plasmon resonance (SPR) assay
The preparation of FGFR2 and FRS2 peptides and SPR spectroscopy analysis was performed following published protocol (for details see the supplementary materials).24
In vitro FRS2 phosphorylation assay
To study the impact of the V430E mutation on phosphorylation of FRS2α, FGFR2CDWT (2 μM) or FGFR2CDV430E were mixed with FRS2αPTB (40 μM) in a reaction buffer consisting of 25mM ATP, 50mM MgCl2 25mM Hepes (pH 7.5) and 150mM NaCl at ambient temperature. Reactions were quenched at different time points by the chelating the Mg2+ with equal moles of EDTA. Following tryptic digestion, the amount of a phospho-Y196-containing peptide derived from FRS2αPTB was quantified by Orbitrap mass spectrometry and expressed as a fraction of the total amount of Y196-containing tryptic peptide.
FGFR1 signaling reporter gene assays
The activation of downstream signaling pathways by wild type and mutated FGFR1 constructs was interrogated using two firefly luciferase-based reporter bioassays: the osteocalcin FGF response element (OCFRE) reporter, which reports activity of the MAPK pathway downstream of FRS2α signaling14 and a Nuclear Factor of Activated T-cells (NFAT) reporter (addgene plasmid 10959),25 which reports activity of the PLCγ /IP3/Ca2+ cascade independent of FRS2α.26 Transient transfection experiments in L6 myoblasts and luciferase assays were performed as previously described.14 Each experiment was performed in triplicate and repeated ≥3 times. The data were fitted with three-parameter sigmoidal curves using Prism5 (GraphPad Software Inc., San Diego, CA) and the dose-response curves of mutant receptors were compared to that of wild type FGFR1 using the Prism5 F-test function.
FGFR1 protein abundance, maturation, and cell surface expression assays
The FGFR1 V429E mutation was subcloned into the previously described FGFR1c expression construct myc-FGFR1WT, which incorporates a N-terminal myc-tag for antibody-mediated detection.14 The impact of the mutation on the total abundance, folding, and cell surface expression of the receptor were assayed in COS-7 cells as previously described;14 results of 3 experiments each performed in quadruplicate were compared by Mann-Whitney test.
RESULTS
Loss-of-function FGFR1 mutations are highly prevalent in patients with CHH and SHFM
We identified 8 CHH probands (7 males and 1 female) with SHFM, including 3 KS patients (Table 1, Supplementary data). Seven mutations in FGFR1 were identified in 7 male CHH probands (Table 1, Figure 1); thus 88% of CHH patients with SHFM harbor mutations in FGFR1. Among the probands with FGFR1 mutations, 4 also exhibit cleft palate; five cases are clearly familial (families 1-4, 7, Figure 1). The proband exhibiting the most severe SHFM phenotype (both hands and feet affected, with additional syndactylies) carries the homozygous missense mutation p.V429E.27 In contrast, 6 probands with SHFM (limited to either one foot or both feet) carry heterozygous mutations, 5 missense and one nonsense: p.G348R, p.G485R, p.Q594*, p.E670A, p.V688L, p.L712P (Table 1). Two mutations (p.G348R and p.E670A) have been previously reported 28,29
Table 1.
Clinical phenotypes of probands with CHH and SHFM.
| No. | Dx | nucleotide change | amino acid change | sex | puberty | OB on MRI | SHFM | Cleft lip/plate | other phenotypes | FSH (UI/l) | LH (UI/l) | T (nmol/l) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | KS | c.[1286T>A];[1286T>A] | p.[V429E]; [V429E] | M | absent | absent | both hands, both feet | - | cryptorchidism, absent septum pellucidum, hypoplastic anterior corpus callosum | <0.1 | <0.1 | <1 |
| 2 | KS | c.2062G>T | p.V688L | M | n/a | absent | left foot | + | micropenis, cryptorchidism | 0.4 | <0.1 | <1 |
| 3 | KS | c.2135T>C | p.L712P | M | absent | normal | both feet | - | micropenis | <0.1 | 2.3 | <1 |
| 4 | CHH | c.1453G>A | p.G485R | M | absent | ND | right foot | + | dental agenesis | 1.0 | 0.7 | <1 |
| 5 | CHH | c.1042G>A | p.G348R | M | absent | normal | both feet | + | micropenis, cryptorchidism, partial double teeth (canines) | 0.4 | 0.5 | <1 |
| 6 | CHH | c.1780C>T | p.Q594* | M | absent | ND | both feet | - | cryptorchidism | 0.3 | <0.1 | 2.4 |
| 7 | CHH | c.2009A>C | p.E670A | M | absent | ND | both feet | + | none | 0.7 | 0.9 | 1.8 |
| 8 | CHH | none | none | F | absent | ND | right hand, right foot | + | preauricular fistulas, hypoplastic labia majora | NA | NA | NA |
Dx: diagnosis; M: male; KS: Kallmann syndrome; CHH: congenital hypogonadotropic hypogonadism; n/a: not applicable, neonatal diagnosis of KS; OB: olfactory bulbs; MRI: magnetic resonance imaging; ND: not done; NA: not available
Figure 1. FGFR1 mutations underlie CHH with SHFM.

(A) Pedigrees of the 7 CHH and SHFM families with FGFR1 mutations; probands are denoted by arrows, SB: stillborn, OB: olfactory bulbs. A: homozygous mutation.
(B) Photographs and radiographs demonstrating severe skeletal anomalies of hands and feet among probands.
Previously reported mutations identified in CHH patients are distributed evenly between the extracellular and intracellular domains (Figure 2A). In contrast, the majority of the FGFR1 mutations (5/7) in the probands with CHH and SHFM affect amino acids located in the tyrosine kinase domain (Figure 2B). The nonsense mutation (p.Q594*) is expected to lead to synthesis of a truncated inactive receptor that lacks the part of the tyrosine kinase domain containing the catalytic site. For missense mutations, the affected residues are conserved across vertebrates (Figure 2B), and all mutations are predicted to be loss-of-function (Table S1, Figure S1, Supplementary data). p.E670A has been shown to impair FGFR1 downstream signaling as assessed by an in-vitro MAPK phosphorylation assay.29 p.V429E is the first reported mutation within the domain of FGFR1 that binds FRS2α, the docking protein that mediates MAPK pathway activation30 (Figure 3A, Supplementary data).
Figure 2. FGFR1 mutations identified in probands with CHH and SHFM.
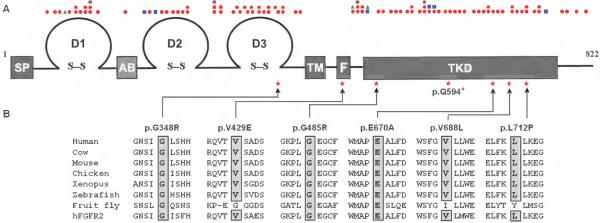
(A) Schematic of FGFR1 showing the locations of all published FGFR1 mutations associated with CHH (Kallmann syndrome or normosmic CHH, red circles), Hartsfield syndrome (blue squares), septo-optic-dysplasia (green triangles). SP: signal peptide, D1: immunoglobulin domain 1, AB: acid box domain, D2: immunoglobulin domain 2, D3: immunoglobulin domain 3, TM: transmembrane domain, F: FRS2α -binding domain, TKD: tyrosine kinase domain, stars: mutations described in this study.
(B) FGFR1 mutations identified in probands with CHH and SHFM, all the substituted residues of missense mutations are conserved across vertebrates (cow, mouse, chicken, xenopus, and zebrafish) and in human FGFR2 (hFGFR2).
Figure 3. The V429E substitution in FGFR1 impedes recruitment and phosphorylation of FRS2α and FGF2-induced MAPK signaling.

(A) Analysis of the impact of the V429E mutation based on the NMR structure of the FRS2 phosphotyrosine binding (PTB) domain in complex with the juxtamembrane (JM) region peptide of FGFR1. The FRS2 PTB domain and FGFR1 JM peptide are shown as purple and green ribbons respectively, and side chains of the V429 of FGFR1 and L62, M105, V112 of FRS2 are rendered in sticks. The molecular surfaces of L62, M105, and V112 of FRS2 PTB are also shown, to highlight their hydrophobic contacts with V429 of FGFR1.
(B) The V429E FGFR1c mutant fails to phosphorylate FRS2 in cell based assay. BaF3 were transfected with lentiviral vectors expressing WT or V429E FGFR1c and FRS2 phosphorylation was assessed upon FGF1 treatment by western blotting using anti phospho-FRS2-α specific antibodies . WT: wild type; EV: empty vector.
(C) Analysis of the impact of the V429E mutation on the ability of FGFR1 to recruit FRS2α . The assay was based on FGFR2 V430E, which is equivalent to FGFR1 V429E. Increasing concentrations of FGFR2CDWT and FGFR2CDV430E (carrying the equivalent mutation to FGFR1-V429E) ranging from 12.5 to 400 nM were passed over a CM5 chip onto which FRS2α had been immobilized. As representative of the full dataset, binding responses obtained for injections of 200 nM of FGFR2CDWT or FGFR2CDV430E are shown. The rising and falling parts of the wild type curve (blue) represent the association and dissociation phases, respectively, of FGFR2CDWT-FRS2α binding over time. At 200 nM, FGFR2CDWT exhibits maximal binding of 55 response units (blue), whereas FGFR2CDV430E shows negligible binding (red). According to a steady-state equilibrium analysis of the full data sets (not shown), FGFR2CDWT binds FRS2α with a KD of 320 nM, whereas the FGFR2CDV430E negligible binding to FRS2α.
(D) The V429E mutation reduces the ability of FGFR1 to phosphorylate FRS2 on Y196 in vitro. FGFR2WT and FGFR2V430E mutant kinases were allowed to phosphorylate FRS2 fragment PTB9-200 on Y196, a tyrosine phosphorylation site known to be required for Grb2 recruitment, at room temperature for 0, 1, 3, 5 10, 15, 20, 25, 30 minutes. Following tryptic digestion, samples were analyzed by Orbitrap mass spectrometry to quantify the phospho-Y196-containing tryptic peptide. FGFR2WT is shown in blue and FGFR2V430E is shown in red.
(E-F) The V429E mutation is loss-of-function in the OCFRE reporter assay (FRS2α dependent MAPK signaling) and not different from WT in the NFAT reporter assay (FRS2α independent PLCγ /IP3/Ca2+ signaling). Data shown represent the means ± S.E.M. of 3 experiments. FGFR1 WT is shown in blue, FGFR1V429E in red, empty vector in black. Relative to the maximal stimulation of WT (%), ** p<0.001, NS: not significant.
(G) Total abundance and maturation of recombinant FGFR1 proteins. COS-7 cells were transfected with FGFR1 constructs and the cell lysates were subjected to deglycosylation treatment followed by western blot analysis. PNGase-treated bands represent total protein abundance levels; 140kDa Endo H-treated bands represent the mature form while 100kDa Endo H-treated bands represent immature form of FGFR1. The experiment was performed three times, tested by Mann-Whitney test for statistic significance, no significant difference in overall expression and maturation index between WT and V429E. PNGase: Peptide N-Glycosidase F-treated; Endo H: endoglycosidase H-treated; WT: wild type; EV: empty vector.
(H) Cell surface abundance of the transiently transfected FGFR1 mutant in COS-7 cells. Cell surface abundance levels were measured by a radiolabeled antibody binding assay and plotted as a percentage of wild type levels. The abundance level of cell surface expressed FGFR1V429E was significantly higher than FGFR1wt. Values shown are the means ± SEM of 3 experiments each performed in quadruplicate. The difference between the mutant and the wild type receptor expression was compared by Mann-Whitney test, * p<0.05.
The FGFR1 -V429E substitution abolishes recruitment and phosphorylation of FRS2α by the FGF receptor
We chose to functionally verify our structural predictions on the p.V429E substitution as this is the first report of a mutation that would selectively impact a specific signaling pathway downstream of FGFR. A lentiviral expression system was used to express the wild type FGFR1c and its V429E variant in BaF3 cells, which lack endogenous FGFR expression. Cells were transfected with the wild type FGFR1c or the V429E FGFR1c mutant constructs, treated with FGF1, and FRS2α phosphorylation on tyrosine 196, one of the major Grb2 binding site of FRS2α , was assessed by western blotting with phosphospecific anitobodies. As shown in Figure 3B, wild type FGFR1c phosphorylated FRS2α on tyrosine 196 whereas the V429E mutant did not. For in vitro binding and phosphorylation experiments, we used the FGFR2 intracellular domain. This was necessary because the expression level of FGFR1 intracellular domain in E.coli is too low to allow for carrying out these experiments. In contrast, FGFR2 intracellular domain can be expressed abundantly and can be purified to high homogeneity using E.coli expression system. This approach was legitimate because FGFR1 and FGFR2 are structurally and functionally highly homologous. The FRS2α binding sites are highly conserved between these FGFRs and alternative splicing in both FGFRs, and the exclusion of the conserved Val-Thr motif eliminates FRS2α recruitm ent to the FG FR .31 Wild type FGFR2 intracellular domain (residues 401-821; FGFR2CDWT), the corresponding mutated fragment (FGFR2CDV430E), and the PTB domain of FRS2α (residues 11-140; FRS2αPTB) were expressed in E.coli as His-tagged proteins and purified to homogeneity. FRS2α was immobilized onto a CM5 chip sensor over and increasing concentrations of FGFR2CDWT or FGFR2CDV430E were flowed over the sensor chip. FGFR2CDWT bound FRS2αP with a KD of 320 nM, whereas the FGFR2CDV430E mutant failed to bind to FRS2αPTB (Figure 3C). We also examined the effect of the mutation on the ability of the mutant receptor to phosphorylate FRS2α using an in vitro kinase assay. Briefly, purified FRS2α protein was mixed with FGFR2CDWT or FGFR2CDV430E in a mole ratio of 20:1, and supplemented with a Mg-ATP mixture to start the phosphorylation reaction. Phosphorylation of FRS2α on tyrosine 196 as a function of time was monitored by mass spectrometry and was expressed as percentage of the phosphorylated substrate. As shown in Figure 3D, the V430E mutation diminishes the ability of FGFR2 to phosphorylate FRS2α. These data demonstrate that the FGFR2-V430E substitution diminishes the ability of FGFR2 to recruit and phosphorylate FRS2α in vitro. In contrast to cell-based data, the mutation did not abolish FRS2α phosphorylation in vitro. This was expected, because the high concentrations of FGFR kinase and FRS2α (several orders of magnitude greater than those in cell-based experiments) have resulted in recruitment-independent phosphorylation of FRS2α by the FGFR kinase. Collectively, these cell-based and in vitro data demonstrate that the FGFR1-V429E substitution diminishes the ability of FGFR1 to recruit and phosphorylate FRS2α.
The FGFR1 V429E substitution compromises FGFR1 MAPK signaling in vitro
We then studied the effects of the mutation on downstream signaling using the the OCFRE and NFAT reporter systems. Stimulation of wild type receptor with increasing doses of FGF2 resulted in a typical sigmoidal dose-response curve with ~5-fold maximal induction of reporter activity (Figure 3E). The V429E mutant displayed reduced inducibility: 56% of wild type at maximum activity (p <0.001, Figure 3E), suggesting compromised MAPK signaling. In contrast, V429E behaved similar to the wild type receptor in the NFAT reporter assay (Figure 3F), indicating normal PLCγ /IP3/Ca2+ signaling. These findings indicate that V429E represents a partial loss-of-function mutation that compromises the activation of FRS2α dependent MAPK signaling without affecting FRS2α independent PLCγ /IP3/Ca2+ signaling by FGFR1.
To exclude the possibility that the V429E mutation leads to loss of function by impairing FGFR1 receptor protein synthesis and/or maturation, we investigated whether it impacted FGFR1 total abundance, glycosylation, or cell surface expression. When the lysate of transiently transfected COS-7 cells was subjected to western blot analysis under reducing conditions, FGFR1 was detectable as two protein bands of 140 kDa and 120 kDa (Figure 3G). When the lysate was pretreated with Peptide N-Glycosidase F (PNGase), which removes all N-linked carbohydrate chains, FGFR1 was detectable as a single band of ~100 kDa, confirming that the two bands are differently N-glycosylated receptor pools. The total protein abundance was calculated as the ratios of PNGase-treated bands to ß-actin band densities; the value calculated for the V429E mutant was normalized to the WT receptor. Treatment with endoglycosidase H (Endo H), which removes only high mannose N-linked sugars, altered the mobility of only the 120 kDa FGFR1 band (Figure 3G), indicating that this pool represents the partially processed immature receptor, whereas the Endo H-resistant 140 kDa band represents the mature form of FGFR1. Receptor “maturation index” was estimated by calculating the ratio of the 140 kDa band to the total FGFR1 immunoreactivity of EndoH-treated samples. The total abundance (mean±SEM: 1.07±0.30, p>0.05) and the maturation index (1.04±0.05, p>0.05) of V429E were not different from wild type, suggesting that the mutation did not compromise receptor synthesis or maturation. The cell surface abundance of V429E was slightly elevated (by 20% relative to wild type, p<0.05) (Figure 3H), thus excluding reduced expression as cause for decreased signaling activity. Hence, the V429E mutation imparted loss of receptor function by selectively inhibiting the ability of the receptor to interact with its major downstream substrate FRS2α and to transduce respective intracellular signals.
Variable expressivity of phenotypes associated with FGFR1 mutations
In family 1, the proband with the homozygous mutation (p.V429E) exhibits severe reproductive, olfactory, and skeletal phenotypes (SHFM in hands and feet and syndactylies). The only detectable phenotype among the three heterozygous family members analyzed is hyposmia in the proband's sister. In family 2, the proband with KS, SHFM, and cleft lip and palate inherited the heterozygous mutation (p.V688L) from his affected mother who also exhibits KS and cleft lip and palate but not SHFM. In family 3, the proband with KS and SHFM inherited the heterozygous mutation (p.L712P) from his unaffected mother; his two sons (III-1 and III-2), who carry the mutation both have syndactyly and one has CHH. In family 4, the proband with CHH, SHFM, and cleft palate inherited his heterozygous mutation (p.G485R) from his unaffected mother. His brother, who carries the same mutation has CHH, was born with syndactyly and cleft palate. Finally, in family 7, the proband who carries the p.E670A mutation has CHH, SHFM, and cleft palate. DNA was not available from his parents. His affected brother with the identical FGFR1 mutation has KS and cleft palate. Thus, among the 5 familial cases, SHFM was found in the CHH probands and in three additional members in family 1 whose genotype and reproductive phenotype are unknown (two stillborn females and one male with neonatal death). On the other hand, CHH, anosmia/hyposmia, syndactyly, and cleft lip/palate were found in several family members carrying the FGFR1 mutation (Figure 1).
DISCUSSION
We show here that FGFR1 mutations are present in most cases of CHH with SHFM (88%). Several patients also exhibited anosmia (patients 1, 2, 3), cleft palate (patients 2, 4, 5, 7) or, more rarely, absent septum pellucidum and hypoplastic anterior corpus callosum (patient 1), phenotypes previously reported in association with FGFR1 mutations.3,4 FGFR1 mutations have been recently reported to underlie Hartsfield Syndrome, defined as the combination of holoprosencephaly and SHFM.10 A broad radiologic spectrum has been reported in holoprosencephaly, including absence of septum pellucidum and corpus callosum at the milder end.32 This phenotype was observed in patient 1 with CHH and SHFM. Further, patients with Hartsfield syndrome may have olfactory bulb agenesis and/or multiple pituitary hormone deficiency (including hypogonadotropic hypogonadism).10,12 Thus, our results show a substantial phenotypic and genetic overlap between CHH with SHFM on the one hand, and Hartsfield syndrome on the other. Indeed, FGFR1 is a pleiotropic gene involved not only in the GnRH neuron ontogeny and olfactory system development, but also implicated in forebrain development and embryonic limb morphogenesis. It is interesting to note that 7/8 probands reported here are males and 7/7 males were found to carry loss-of-function mutations in FGFR1. The sole CHH and SHFM patient without a FGFR1 mutation is a female with ectrodactyly of the right hand and foot, and cleft palate. A similar male predominance has been observed in Hartsfield Syndrome.10
In the consanguineous family 1, the disease is apparently inherited as a recessive trait, which is unusual for CHH associated with FGFR1 mutations.3 The proband carries a homozygous p.V429E mutation while 3 family members are heterozygous for the mutation and only one among them exhibits hyposmia, a phenotype which can be associated with CHH. This may be due to the fact that the p.V429E mutation causes only partial loss of function, such that two mutant alleles are needed to manifest the disease phenotype. In the other 6 families, FGFR1-associated CHH with SHFM was inherited as an autosomal dominant trait with incomplete penetrance. For example, among the informative pedigrees, the mutation was transmitted by unaffected mothers (families 3 and 4); additional mutation carriers without SHFM are present in families 2, 3, 4, and 7. However, although SHFM is classically described as an absence of the central ray, a broader range of limb extremity abnormalities has been described within this entity such as syndactyly.11 Accordingly, the apparent penetrance of FGFR1 mutations increases when the phenotype was considered as CHH associated to limb abnormality extremities and not only to SHFM (2 CHH patients and one prepubertal individual in families 3 and 4 harboring FGFR1 mutations exhibit syndactyly). Oligogenicity,2,27 and/or interaction with environmental factors33 may contribute to the incomplete penetrance and variable expressivity of CHH, SHFM with other limb extremity abnormalities and olfactory phenotypes.
Several lines of evidence support that the SHFM in these CHH probands is the consequence of the FGFR1 signaling defect. Fgfr1 restricts the number of cells in nascent limb buds and specifies digit placement and identity in developing limbs.34,35 Mice with inactivation of Fgfr1 in sonic hedgehog-expressing cells of developing limb buds lack the 3rd digit in all forelimbs and hindlimbs, which corresponds to the human SHFM phenotype.35 Additionally, mice with inactivation of Fgfr1 in limb mesenchyme immediately after limb bud initiation exhibit fused/missing 1st and 2nd digits similar to the phenotype of patients 1, 5 and 6.34 These data are consistent with a critical role for FGFR1 in limb development and SHFM pathogenesis. To date, the only gene associated with isolated (non-syndromic) SHFM in both mice and humans is TP63.36 Interestingly, the limb buds of Tp63−/− mice have markedly decreased expression of Fgf8 in the apical ectodermal ridge (AER) of limb buds;37 Fgf8 is required for proliferation of the underlying mesenchymal cells. Moreover, mice with double conditional knock out of Fgf8 and Fgf4 in AER cells exhibit aplasia of both proximal and distal limbs.38 Lastly, a locus for SHFM maps to a region that includes FGF8 (chromosome 10q24-q25).39 FGF8 is a potent ligand for FGFR1, and both FGF8 and FGFR1 loss-of-function mutations underlie CHH through defects in olfactory bulb or GnRH neuron development.40 Mutations in FGF8 are a rare cause of CHH (~1%), and none were found in the 8 CHH-SHFM probands reported here.
The most severe limb extremity phenotype, with median clefts in both hands and both feet as well as multiple syndactylies, was observed in the patient with a homozygous FGFR1 mutation (p.V429E). The p.V429E mutation is the first one identified in the binding domain for FRS2α; structural and functional studies show that the mutation selectively abolishes the interaction of the receptor with FRS2α, an adaptor critical for activation of the MAPK and PI3-K cascades.5 MAPK activation by FGF signaling is known to promote survival and neurite outgrowth in GnRH neurons.7 The p.V429E mutation suggests a role for FRS2α-mediated FGFR1 signaling not only for GnRH neuron ontogeny, but also for distal limb development in humans. The remaining 6 patients with heterozygous FGFR1 mutations exhibit milder SHFM phenotypes with a median cleft in either one foot (n=2) or both feet (n=4).
In conclusion, the association of CHH with SHFM is a clinical entity with a high frequency of FGFR1 mutations. A main limitation of this study is that it was not designed to assess the percentage of the general CHH population that presents SHFM; rather, the cases were assembled based on phenotypes that had come to the attention of the many collaborating physicians. To address the exact prevalence of CHH with SHFM, systematic phenotyping of defined CHH cohorts is necessary; which is beyond the scope of this study. Nevertheless, our findings have implications for clinic practice: Firstly, the high frequency of FGFR1 mutations in patients with CHH and SHFM compared to the general CHH population suggests that FGFR1 should be prioritized for genetic screening in CHH patients with SHFM. Secondly, as diagnosis of CHH in infancy can facilitate early treatment with gonadotropins/GnRH to promote both gonadal development and future fertility in adulthood,13 we propose that neonates with SHFM be assessed for CHH through evaluation at birth for micropenis and cryptorchidism in males; hormonal testing during the “minipuberty”; MRI of olfactory structures; and/or genetic screening of FGFR1. Lastly, further refining the clinical and genetic overlap between CHH, SHFM and Hartsfield syndrome is expected to facilitate a better understanding of their pathologic mechanisms, and to elaborate a rational algorithm for genetic diagnosis.
Supplementary Material
Figure S1 Structure-based prediction of the impact of FGFR1 mutations identified in probands with CHH and SHFM.
ACKNOWLEDGMENTS
We are grateful to all patients and families for their kind participation and to Prof. Kemal Topaloglu for clinical and genetic information. Funding: This work was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health [R01HD056264 to N.P., R01DE013686 to M.M.]; Swiss National Science Foundation grants [31003A, 135648 to N.P. and CRSII3141960 to N.P. & M.M.], Academy of Finland [TR & JT], INSERM, the DHU PROTECT and by COST Action [BM1105].
Footnotes
Supplementary information is available at the Genetics in Medicine website:
Supplementary methods: Preparation of FGFR2 and FRS2 peptides and SPR spectroscopy analysis.Case descriptions of CHH and SHFM patients and structural prediction of identified FGFR1 mutations.
REFERENCES
- 1.Tsai PS, Gill JC. Mechanisms of disease: Insights into X-linked and autosomal-dominant Kallmann syndrome. Nature clinical practice. Endocrinology & metabolism. 2006 Mar;2(3):160–171. doi: 10.1038/ncpendmet0119. [DOI] [PubMed] [Google Scholar]
- 2.Miraoui H, Dwyer AA, Sykiotis GP, et al. Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. American journal of human genetics. 2013 May 2;92(5):725–743. doi: 10.1016/j.ajhg.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dode C, Levilliers J, Dupont JM, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nature genetics. 2003 Apr;33(4):463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- 4.Pitteloud N, Acierno JS, Jr., Meysing A, et al. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proceedings of the National Academy of Sciences of the United States of America. 2006 Apr 18;103(16):6281–6286. doi: 10.1073/pnas.0600962103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine & growth factor reviews. 2005 Apr;16(2):139–149. doi: 10.1016/j.cytogfr.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Deng CX, Wynshaw-Boris A, Shen MM, Daugherty C, Ornitz DM, Leder P. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes & development. 1994 Dec 15;8(24):3045–3057. doi: 10.1101/gad.8.24.3045. [DOI] [PubMed] [Google Scholar]
- 7.Tsai PS, Moenter SM, Postigo HR, et al. Targeted expression of a dominant-negative fibroblast growth factor (FGF) receptor in gonadotropin-releasing hormone (GnRH) neurons reduces FGF responsiveness and the size of GnRH neuronal population. Molecular endocrinology. 2005 Jan;19(1):225–236. doi: 10.1210/me.2004-0330. [DOI] [PubMed] [Google Scholar]
- 8.Costa-Barbosa FA, Balasubramanian R, Keefe KW, et al. Prioritizing genetic testing in patients with Kallmann syndrome using clinical phenotypes. The Journal of clinical endocrinology and metabolism. 2013 May;98(5):E943–953. doi: 10.1210/jc.2012-4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jarzabek K, Wolczynski S, Lesniewicz R, Plessis G, Kottler ML. Evidence that FGFR1 loss-of-function mutations may cause variable skeletal malformations in patients with Kallmann syndrome. Advances in medical sciences. 2012;57(2):314–321. doi: 10.2478/v10039-012-0036-4. [DOI] [PubMed] [Google Scholar]
- 10.Simonis N, Migeotte I, Lambert N, et al. FGFR1 mutations cause Hartsfield syndrome, the unique association of holoprosencephaly and ectrodactyly. Journal of medical genetics. 2013 Sep;50(9):585–592. doi: 10.1136/jmedgenet-2013-101603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duijf PH, van Bokhoven H, Brunner HG. Pathogenesis of split-hand/split-foot malformation. Human molecular genetics. 2003 Apr 1;12:R51–60. doi: 10.1093/hmg/ddg090. Spec No 1. [DOI] [PubMed] [Google Scholar]
- 12.Vilain C, Mortier G, Van Vliet G, et al. Hartsfield holoprosencephaly-ectrodactyly syndrome in five male patients: further delineation and review. American journal of medical genetics. Part A. 2009 Jul;149A(7):1476–1481. doi: 10.1002/ajmg.a.32678. [DOI] [PubMed] [Google Scholar]
- 13.Bouvattier C, Maione L, Bouligand J, Dode C, Guiochon-Mantel A, Young J. Neonatal gonadotropin therapy in male congenital hypogonadotropic hypogonadism. Nature reviews. Endocrinology. 2012 Mar;8(3):172–182. doi: 10.1038/nrendo.2011.164. [DOI] [PubMed] [Google Scholar]
- 14.Raivio T, Sidis Y, Plummer L, et al. Impaired fibroblast growth factor receptor 1 signaling as a cause of normosmic idiopathic hypogonadotropic hypogonadism. The Journal of clinical endocrinology and metabolism. 2009 Nov;94(11):4380–4390. doi: 10.1210/jc.2009-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nature methods. 2010 Apr;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009;4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 17.Ferrer-Costa C, Orozco M, de la Cruz X. Sequence-based prediction of pathological mutations. Proteins. 2004 Dec 1;57(4):811–819. doi: 10.1002/prot.20252. [DOI] [PubMed] [Google Scholar]
- 18.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nature methods. 2010 Aug;7(8):575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez-Perez A, Lopez-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. American journal of human genetics. 2011 Apr 8;88(4):440–449. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schlessinger J, Plotnikov AN, Ibrahimi OA, et al. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Molecular cell. 2000 Sep;6(3):743–750. doi: 10.1016/s1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- 21.Bae JH, Lew ED, Yuzawa S, Tome F, Lax I, Schlessinger J. The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site. Cell. 2009 Aug 7;138(3):514–524. doi: 10.1016/j.cell.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhalluin C, Yan KS, Plotnikova O, et al. Structural basis of SNT PTB domain interactions with distinct neurotrophic receptors. Molecular cell. 2000 Oct;6(4):921–929. doi: 10.1016/s1097-2765(05)00087-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ornitz DM, Xu J, Colvin JS, et al. Receptor specificity of the fibroblast growth factor family. The Journal of biological chemistry. 1996 Jun 21;271(25):15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- 24.Ibrahimi OA, Zhang F, Eliseenkova AV, Itoh N, Linhardt RJ, Mohammadi M. Biochemical analysis of pathogenic ligand-dependent FGFR2 mutations suggests distinct pathophysiological mechanisms for craniofacial and limb abnormalities. Human molecular genetics. 2004 Oct 1;13(19):2313–2324. doi: 10.1093/hmg/ddh235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ichida M, Finkel T. Ras regulates NFAT3 activity in cardiac myocytes. The Journal of biological chemistry. 2001 Feb 2;276(5):3524–3530. doi: 10.1074/jbc.M004275200. [DOI] [PubMed] [Google Scholar]
- 26.Goetz R, Mohammadi M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nature reviews. Molecular cell biology. 2013 Mar;14(3):166–180. doi: 10.1038/nrm3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sykiotis GP, Plummer L, Hughes VA, et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proceedings of the National Academy of Sciences of the United States of America. 2010 Aug 24;107(34):15140–15144. doi: 10.1073/pnas.1009622107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bailleul-Forestier I, Gros C, Zenaty D, Bennaceur S, Leger J, de Roux N. Dental agenesis in Kallmann syndrome individuals with FGFR1 mutations. International journal of paediatric dentistry / the British Paedodontic Society [and] the International Association of Dentistry for Children. 2010 Jul;20(4):305–312. doi: 10.1111/j.1365-263X.2010.01056.x. [DOI] [PubMed] [Google Scholar]
- 29.Laitinen EM, Vaaralahti K, Tommiska J, et al. Incidence, phenotypic features and molecular genetics of Kallmann syndrome in Finland. Orphanet journal of rare diseases. 2011;6:41. doi: 10.1186/1750-1172-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burgar HR, Burns HD, Elsden JL, Lalioti MD, Heath JK. Association of the signaling adaptor FRS2 with fibroblast growth factor receptor 1 (Fgfr1) is mediated by alternative splicing of the juxtamembrane domain. The Journal of biological chemistry. 2002 Feb 8;277(6):4018–4023. doi: 10.1074/jbc.M107785200. [DOI] [PubMed] [Google Scholar]
- 31.Twigg SR, Burns HD, Oldridge M, Heath JK, Wilkie AO. Conserved use of a non-canonical 5′ splice site (/GA) in alternative splicing by fibroblast growth factor receptors 1, 2 and 3. Human molecular genetics. 1998 Apr;7(4):685–691. doi: 10.1093/hmg/7.4.685. [DOI] [PubMed] [Google Scholar]
- 32.Solomon BD, Pineda-Alvarez DE, Mercier S, Raam MS, Odent S, Muenke M. Holoprosencephaly flashcards: A summary for the clinician. American journal of medical genetics. Part C, Seminars in medical genetics. 2010 Feb 15;154C(1):3–7. doi: 10.1002/ajmg.c.30245. [DOI] [PubMed] [Google Scholar]
- 33.Raivio T, Falardeau J, Dwyer A, et al. Reversal of idiopathic hypogonadotropic hypogonadism. The New England journal of medicine. 2007 Aug 30;357(9):863–873. doi: 10.1056/NEJMoa066494. [DOI] [PubMed] [Google Scholar]
- 34.Li C, Xu X, Nelson DK, Williams T, Kuehn MR, Deng CX. FGFR1 function at the earliest staes of mouse limb development plays an indispensable role in subsequent autopod morphogenesis. Development. 2005 Nov;132(21):4755–4764. doi: 10.1242/dev.02065. [DOI] [PubMed] [Google Scholar]
- 35.Verheyden JM, Lewandoski M, Deng C, Harfe BD, Sun X. Conditional inactivation of Fgfr1 in mouse defines its role in limb bud establishment, outgrowth and digit patterning. Development. 2005 Oct;132(19):4235–4245. doi: 10.1242/dev.02001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Bokhoven H, Hamel BC, Bamshad M, et al. p63 Gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. American journal of human genetics. 2001 Sep;69(3):481–492. doi: 10.1086/323123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang A, Schweitzer R, Sun D, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999 Apr 22;398(6729):714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 38.Sun X, Mariani FV, Martin GR. Functions of FGF signalling from the apical ectodermal ridge in limb development. Nature. 2002 Aug 1;418(6897):501–508. doi: 10.1038/nature00902. [DOI] [PubMed] [Google Scholar]
- 39.Ozen RS, Baysal BE, Devlin B, et al. Fine mapping of the split-hand/split-foot locus (SHFM3) at 10q24: evidence for anticipation and segregation distortion. American journal of human genetics. 1999 Jun;64(6):1646–1654. doi: 10.1086/302403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Falardeau J, Chung WC, Beenken A, et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. The Journal of clinical investigation. 2008 Aug;118(8):2822–2831. doi: 10.1172/JCI34538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Structure-based prediction of the impact of FGFR1 mutations identified in probands with CHH and SHFM.