

Abstract
Phalaenopsis has a zygomorphic floral structure, including three outer tepals, two lateral inner tepals and a highly modified inner median tepal called labellum or lip; however, the regulation of its organ development remains unelucidated. We generated RNA-seq reads with the Illumina platform for floral organs of the Phalaenopsis wild-type and peloric mutant with a lip-like petal. A total of 43,552 contigs were obtained after de novo assembly. We used differentially expressed gene profiling to compare the transcriptional changes in floral organs for both the wild-type and peloric mutant. Pair-wise comparison of sepals, petals and labellum between peloric mutant and its wild-type revealed 1,838, 758 and 1,147 contigs, respectively, with significant differential expression. PhAGL6a (CUFF.17763), PhAGL6b (CUFF.17763.1), PhMADS1 (CUFF.36625.1), PhMADS4 (CUFF.25909) and PhMADS5 (CUFF.39479.1) were significantly upregulated in the lip-like petal of the peloric mutant. We used real-time PCR analysis of lip-like petals, lip-like sepals and the big lip of peloric mutants to confirm the five genes’ expression patterns. PhAGL6a, PhAGL6b and PhMADS4 were strongly expressed in the labellum and significantly upregulated in lip-like petals and lip-like sepals of peloric-mutant flowers. In addition, PhAGL6b was significantly downregulated in the labellum of the big lip mutant, with no change in expression of PhAGL6a. We provide a comprehensive transcript profile and functional analysis of Phalaenopsis floral organs. PhAGL6a PhAGL6b, and PhMADS4 might play crucial roles in the development of the labellum in Phalaenopsis. Our study provides new insights into how the orchid labellum differs and why the petal or sepal converts to a labellum in Phalaenopsis floral mutants.
Introduction
Orchids (Orchidaceae) represent one of the largest families of flowering plants, with more than 25,000 species [1]. Orchid production has become a worldwide important business in the floricultural industry. Potted Phalaenopsis is one of the most popular orchids in the trade. The Phalaenopsis genus belongs to the Orchidaceae, comprised of approximately 66 species [2], with distribution throughout tropical Asia from Taiwan and Sikkhim, in India, to Australia and the larger islands of the Pacific Ocean [2]. Phalaenopsis flowers have a zygomorphic floral structure, including three sepals (in the first floral whorl) and two petals as well as a highly modified inner median tepal called a labellum in the second floral whorl. In addition, Phalaenopsis flowers are highly evolved with a gynostemium or column because of the fusion of the male and female reproductive organs [3]. The use of tissue culture technology to massively produce elite Phalaenopsis orchid clones has been widely adopted by the orchid industry. However, unpredictable mutations or somaclonal variation may occur during tissue culture. Somaclonal variation, characterized by phenotypic changes of genetic or epigenetic origin [4], has been extensively studied in several plants. Such variation includes morphological traits such as flower color and morphologic features, leaf morphologic features and color, plant height, resistance to disease, improved quality and higher yield [5]. The labellum-like petal of the peloric mutant of Phalaenopsis is more common than in other somaclonal variants. Occasionally, a rare sepal peloric mutant has been observed. The orchid peloric mutant is thus valuable for investigating flower development at both morphological and molecular levels.
The genetic and molecular basis of floral organogenesis has been extensively studied in the model species Arabidopsis thaliana and Antirrhinum majus [6–9] and led to the evolving ABCDE model of five major classes of homeotic selector genes: A, B, C, D and E. Most of these key floral regulatory genes are the MADS-box gene family encoding MIKC-type MADS domain proteins that function as transcription factors (TFs) [10,11] A- and E-class genes control the development of sepals in the first whorl [12]. A-, B- and E-class genes work together to regulate petal formation in the second whorl, whereas B-, C- and E-class genes control stamen development in the third whorl. C- and E-class genes determine carpel development in the fourth whorl. D-class genes are involved in ovule development [7,13,14]. In Arabidopsis, the A-class genes include APETALA1 (AP1) and APETALA2 (AP2) [15,16]. The B-class genes are represented by APETALA3 (AP3) and PISTILLATA (PI) [17,18], the C-class gene AGAMOUS (AG) [19], the D-class gene SEEDSTICK/AGAMOUS-LIKE11 (STK/AGL11) [19] and the E-class genes SEPALLATAS (SEP1, SEP2, SEP3, and SEP4) [12,20].
The function of ABCDE model genes appear to be conserved across the angiosperms and provide detailed explanations for their floral morphologic features. However, study of the model has focused primarily on herbaceous plants and has not explained completely how diverse angiosperms evolved. The functions of many other expressed genes during floral development remain obscure [21,22]. The flower of orchid is similar to that of eudicots, with sepals and petals in the first and second whorls. However, during floral initiation in orchid, one of the petals develops into a labellum, which is a distinctive feature of a highly modified floral part for an unusual plant species for the study of flower development [23]. Despite its unique floral morphologic features, the molecular mechanism of floral development in orchid remains largely unclear, and more research is needed to identify genes involved in floral differentiation.
Previous studies of orchid floral development have depicted the involvement of certain MADS box genes, including members of the AP1, AP3/PI, DEF/GLO, AG, AGL6 and SEP subfamilies. Orchid A-class genes, such as ORAP11 and ORAP13 from Phalaenopsis [24], OMADS10 from Oncidium [25] and four MADS genes (DOMADS2, DthyrFL1, DthyrFL2 and DthyrFL3) from Dendrobium [26,27] have been identified. Four B-class DEF-like MADS-box genes expressed differently between the wild-type and peloric mutants with lip-like petals in Phalaenopsis [28,29]. In addition, OitaDEF-like genes exert a key function in the diversification of tepals and lip in Orchis italica [30]. A putative floral organ identity gene, OMADS3 (a paleoAP3 gene), isolated from Oncidium implied it to be an A-function gene regulating floral formation and initiation [31]. In addition, three paleoAP3 genes, OMADS5, OMADS3 and OMADS9, and one PISTILLATA gene, OMADS8, were characterized in Oncidium orchid [32]. In Habenaria, three DEF-like genes were identified, with HrGLO1 and HrGLO2 expressed in sepals, petals and columns but HrDEF expression detected only in petals and column [33]. C-class and D-class genes were identified from four orchid species: Phalaenopsis (PhalAG1, PhalAG2) [34], Dendrobium (DthyrAG1, DthyrAG2) [26], Dendrobium (DcOAG1, DcOAG2) [35] and AGAMOUS-like genes, denoted CeMADS1 and CeMADS2, from Cymbidium [36]. Orchid E-class and AGL-6 genes were identified from Oncidium and OMADS11 in the LOFSEP subclade, OMADS6 in the SEP3 subclade, and OMADS1 and OMADS7 in the AGL6 subclade [25,37]. However, this research is in line with current thoughts on how major evolutionary changes in the genetic basis of organ identity were established by gene duplication and the separation of functions. We have a long way to go to fully understand the role of MADS-box genes in orchid evolution.
Recently, next-generation deep-sequencing technology such as Solexa/Illumina RNA-seq and digital gene expression (DGE) have provided new approaches for studying global transcriptome profiling of species that lack reference genome information [38]. RNA-seq is widely used with model and non-model organisms to obtain massive sequence data for molecular marker development, gene discovery and transcriptome profiling [39–44]. So far, transcriptomic analyses have been performed with vegetative and reproductive tissues of Phalaenopsis [39,42], Oncidium leaf and floral buds [45], Cymbidium non-pseudobulb shoot and floral buds [46], Orchis italica floral buds [47] and Ophrys vegetative and reproductive tissues [48]. However, RNA-seq technology has not been used for transcriptomic analysis of floral-organ development in Phalaenopsis.
To better understand floral-organ development of Phalaenopsis orchid at the molecular level, we used RNA-seq technology to investigate the expression of a large number of genes, with emphasis on those differentially expressed in floral-organ development of the wild-type and peloric mutant of Phalaenopsis. To gain a comprehensive understanding of the Phalaenopsis floral development and related processes, we dissected sepal, petal and labellum of 0.2-cm wild-type and peloric mutant flower buds for RNA extraction and detailed transcriptome analysis. Differentially expressed transcripts and their expression patterns were analyzed, and several potential candidate transcripts were found to be regulator factors involved in floral development. We identified genes that are significantly differentially expressed in floral organs by comparing the wild-type and peloric mutant. Our study reveals the functional differentiation and coordination of floral organs and provides insights into possible regulatory networks underlying the development of floral buds in Phalaenopsis.
Materials and Methods
Plant material
Three distinct floral mutants in petal, sepal or labellum (lip) were used for this study. Wild-type (normal) and peloric petal mutant plants of the orchid hybrid Phalaenopsis Brother Spring Dancer ‘KHM190’ (Fig 1 and Fig A in S1 File) were obtained from I-Hsin Biotechnology (Chiayi, Taiwan). Wild-type Phalaenopsis aphrodite and its lip-like sepal mutant (peloric sepal) were from own collection. Wild-type flower of Phalaenopsis ‘NPU1458’ and its petal-like lip (big lip) mutant was from a breeding population. Wild-type and mutant plants were all grown in fan-and-pad greenhouse of National Pingtung University of Science and Technology (Pingtung, Taiwan) under natural day light and controlled temperature from 27 to 30°C. Flowering plants were maintained in a cooling greenhouse at 20/26°C (night/day) temperature. Virus-infected plants were determined by use of RT-PCR with virus-specific primers of Cymbidium mosaic virus (CymMV) and Odontoglossum ringspot virus (ORSV) and excluded. For all experiments, sepal, petal and labellum (lip) organs of 0.2-cm flower buds, which were collected from several clone plants derived from tissue culture to provide sufficient source of total RNAs, were collected (Fig A in S1 File), immersed in liquid nitrogen, and stored at—80°C.
Fig 1. Flowers of wild-type and peloric mutant of Phalaenopsis Brother Spring Dancer ‘KHM190’.
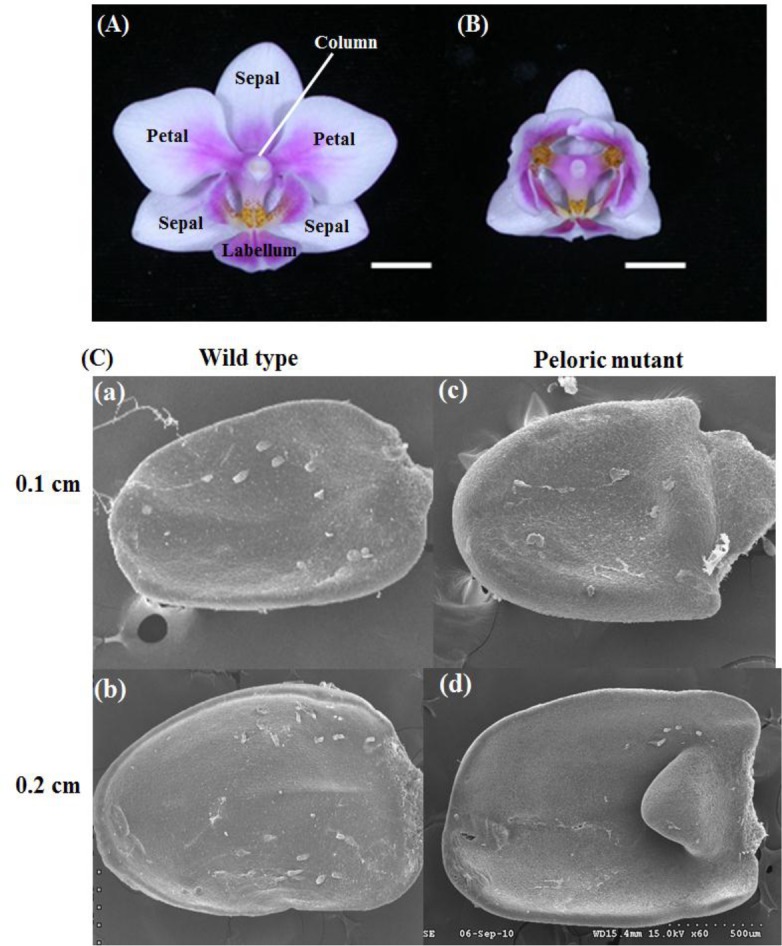
(A) Wild-type and (B) peloric mutant flower. Bar = 1 cm. (C) Scanning electron microscopy of petal of floral buds at early developmental stages of (a), (b) wild-type and (c), (d) peloric-mutant flower. Bar = 500 μm.
RNA extraction and deep sequencing
RNA was isolated from frozen orchid tissues by the TriSolution method (GeneMark, Taipei) [49]. RNA solution was treated with RNase-free DNase I (Promega, Taipei) to eliminate contaminating DNA. RNA quantity and quality were evaluated by the automated electrophoresis system (Experion, Bio-Rad). RNA samples with RNA quality indicator (RQI) >8 were sent to Yourgene Bioscience on dry ice (New Taipei City, Taiwan) for mRNA purification and cDNA construction. The cDNA library for transcriptome sequencing was constructed with use of the Illumina TruSeq RNA sample prep kit. An amount of 5 μg total RNA was directly fragmented after the oligo-T purification step. The first- and second-strand cDNA was synthesized from the fragmented RNA with random hexamer primers, then underwent end repair, A-tailing, adaptor ligation, size-selection of the range 320–420 bp (approximately insert size 200–300 bp) and then PCR amplification for 15 cycles. The products were loaded onto flow cell channels at 12 pM for pair-end 100 bp×2 sequencing with the Illumina HiSeq 2000 platform (Yourgene Bioscience, New Taipei City, Taiwan).
De novo assembly and analysis of Illumina reads
Before the transcriptome assembly, the clean reads were obtained from raw sequencing reads by removing adaptor sequences, reads with more than 5% unknown nucleotides, and low-quality reads (reads containing more than 50% bases with Q-value ≤ 20). For de novo assembly, we pooled all trimmed reads from samples and adopted Velvet (v1.2.07) [50] with distinct k-mer values (35, 45, 55, 65 and 75), followed by Oases (v0.2.06) [51]. The pair-read insert average size was set to 260 bases, with a standard deviation of 10%. Finally, the transcript datasets assembled at different k-mer values were merged by use of Oases with default settings (k-mer = 55).
Transcriptome annotation
Functional annotation of the contigs involved a local BLASTx search with our assembly against the NCBI Nr database (significant E-value threshold ≤ 1e-5). From the search results, to determine gene functions of the sequences, the Blast2GO program was used to obtain gene ontology (GO) annotation according to biological process cellular component and molecular function ontologies [52,53]. Pathway assignments were performed according to the KEGG pathway database [54] with BLASTx and E-value threshold 1e-5. Protein domain annotations involved use of RPS-BLAST 2.2.23 against the Pfam database (ver. 25.0) with the best hit and an E-value ≤ 1e-5 [55,56].
Identification of differentially expressed transcripts
To evaluate the expression of raw transcript, we first mapped trimmed reads to raw transcript sequence using gapped alignment mode of the program Bowtie 2.2.1.0 [57]. After alignment, we quantified raw transcript expression with the software package eXpress 1.3.0 [58]. The value of read counts from eXpress would be the input of DESeq [59], an R software package, was used to test for differential expression. Genes with differential expression of at least two-fold change at P ≤ 0.05 between normal and mutant floral organs were identified with the dispersion estimates obtained using blind method in DESeq package.
Real-time PCR analysis
Quantitative real-time RT-PCR and data analysis involved the ABI PRISM 7300 Sequence Detection System (Applied Biosystems) with SYBR Green PCR Master Mix (Applied Biosystems). Total RNA was isolated from Phalaenopsis sepal, petal and lip tissue. To remove contaminating DNA, RNA samples were treated with DNase I and used for first-strand cDNA synthesis by priming with oligo (dT)25 and catalyzed with Superscript II Reverse Transcriptase (Invitrogen) at 42°C for 1.5 h. The primers for the transcripts investigated were designed on the basis of open reading frame sequences for each gene with use of Primer Express (Applied Biosystems). The thermal cycling condition was 10 min at 95°C, and 40 cycles of 15 sec at 95°C and 1 min at 60°C. Before running real-time PCR, primer efficiency was evaluated by use of both gene-specific and internal-control Actin primers [49,60] (S1 Table) at 50-, 150- and 300-nM combinations as described [49]. We chose the 150-nM concentration for both the target and Actin genes as the most suitable combination. Each sample was amplified in triplicate. With the housekeeping gene Actin, the relative expression level of target genes was presented as 2-ΔCT by the ΔCT method (Applied Biosystems).
Results
Phalaenopsis floral morphogenesis in the wild-type and peloric mutant
The Phalaenopsis flower features bilateral symmetry with two whorls of tepals and a central gynostemium (Fig 1A). The three petal-like sepals include one at the top and two in the lower lateral positions. The flower has two lateral petals and a specialized, enlarged, flamboyant bottom petal, called a labellum. The gynostemium (column) is a reproductive organ with pistil and stigma organs fused together (Fig 1A). Flowers of peloric mutants are radially symmetrical with lip-like petals due to abnormal protrusion of upper cell layers and may lose their pollinia in severe cases (Fig 1B). Visible differences in the development of wild-type and peloric-mutant floral buds began at stage 2 (0.2-cm bud), with an asymmetric shape of petals. At this stage, the petals in the peloric mutant mimicked the labellum in shape, whereas its sepals were similar to those of the wild-type (Fig A in S1 File). Scanning electron microscopy revealed the stage-2 peloric mutant flower with an abnormal fin-like protrusion on both petals (Fig 1C).
Assembly of high-quality Phalaenopsis flower transcriptomes
To obtain an overview of the Phalaenopsis flower transcriptome profiles of stage-2 buds, when the peloric petal began to appear, we used RNA-seq for sequencing six cDNA preparations from sepal, petal and labellum tissues of both the wild-type and peloric mutant and generated 100-bp paired-end reads. Deep-sequencing of the six cDNA samples produced 36,697,424, 58,805,774, 41,084,182, 48,504,500, 78,458,762 and 65,091,470 clean paired-end reads for wild-type and peloric-mutant sepal, petal and labellum transcriptomes, respectively (Table 1). The mean read length was 90 bp. The raw data were submitted to and are available at NCBI and can be accessed in the Short Read Archive (NS: SRX396172; NP: SRX396784; NL: SRX396785; PS: SRX396786; PP: SRX396787; PL: SRX396788). All clean reads were assembled by Velvet and Oases and produced 752,203 assemblies (transcripts). We then chose the one with highest confidence score of a locus or if two isoforms of the same locus have the same confidence score, then the longer one was selected as unigenes. Finally, we obtained 43,552 contigs with a mean length of 1081 nt and a median length of 532 nt. The size distribution of the contigs is in Fig B in S1 File.
Table 1. Summary of Phalaenopsis floral-organ transcriptome assembly.
| Total no. of transcripts | NS | PS | NP | PP | NL | PL |
|---|---|---|---|---|---|---|
| Raw data | ||||||
| Total no. of reads | 39,281,522 | 63,691,838 | 43,899,068 | 51,106,016 | 81,707,498 | 67,469,926 |
| Total nucleotides (nt) | 3,967,433,722 | 6,432,875,638 | 4,433,805,868 | 5,161,707,616 | 8,252,457,298 | 6,814,462,526 |
| High-quality reads | 36,697,424 | 58,805,774 | 41,084,182 | 48,504,500 | 78,458,762 | 65,091,470 |
| Assembly * | ||||||
| Maximum CDS length (bp) | 4,816 | |||||
| Mean CDS length (bp) | 1,081 | |||||
| Total CDS length (bp) | 24,915,746 | |||||
| N50 size (bp) | 2,094 | |||||
| GC percentage | 43.36 |
NS, wild-type sepal; PS, peloric sepal; NP, wild-type petal; PP, peloric petal; NL, wild-type labellum; PL, peloric labellum (PL)
* All reads mixed.
Annotation of predicted proteins
We annotated, classified, and functionally mapped the 43,552 contig sequences based on BLASTx (cut-off E-value ≤ 1e-5) searches of four public protein databases: NCBI non-redundant (Nr) database, gene ontology (GO) database, Kyoto Encyclopedia of Genes and Genomes (KEGG) database, Enzyme Commission (EC) and Pfam (Fig 2; Fig 3 and S2 Table). Among 43,552 contig sequences, significant BLAST hits were found for 28,193 (64.7%) sequences, with no hit found for 15,359 (35.3%) sequences. From the Nr annotations, for the top hits, 44.4% of the annotated sequences (E-value 1e-5-1e-50) matched available plant sequences (Fig 2A). The species distribution based on Nr annotation is shown in Fig 2B, with the top matches being Vitis, Oryza, Zea, Populus and Ricinus.
Fig 2. Characterization of sequence homology of the Phalaenopsis assembled contigs against Nr databases.
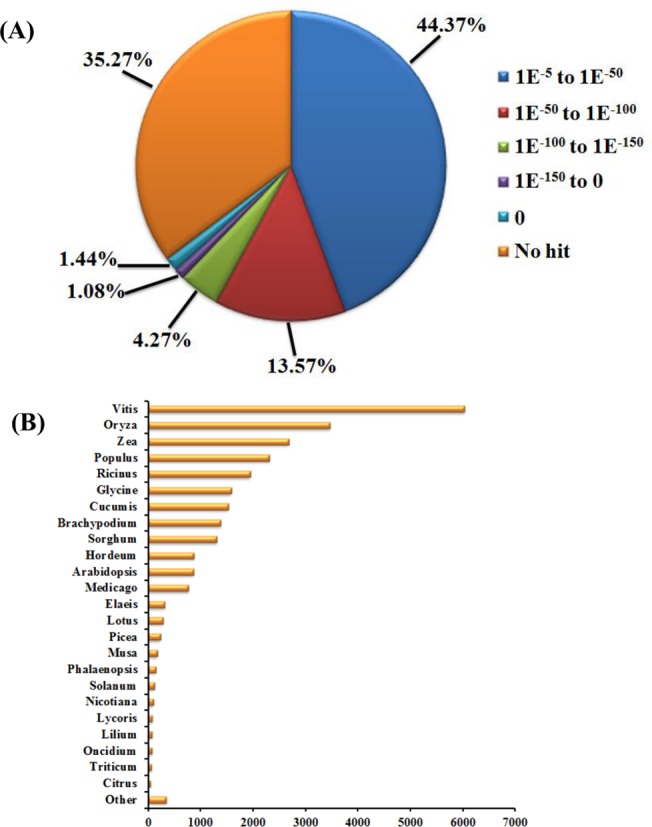
(A) E-value distribution of BLASTx hits for the assembled contigs with a cutoff of 1e-5 in the NCBI Nr database. (B) Species distribution of the 25 top BLASTx hits shown as number of contigs of the total homologous sequences with an E-value ≥ 1e-5.
Fig 3. Annotation of the Phalaenopsis transcriptome by gene ontology (GO), KEGG and Pfam classification.
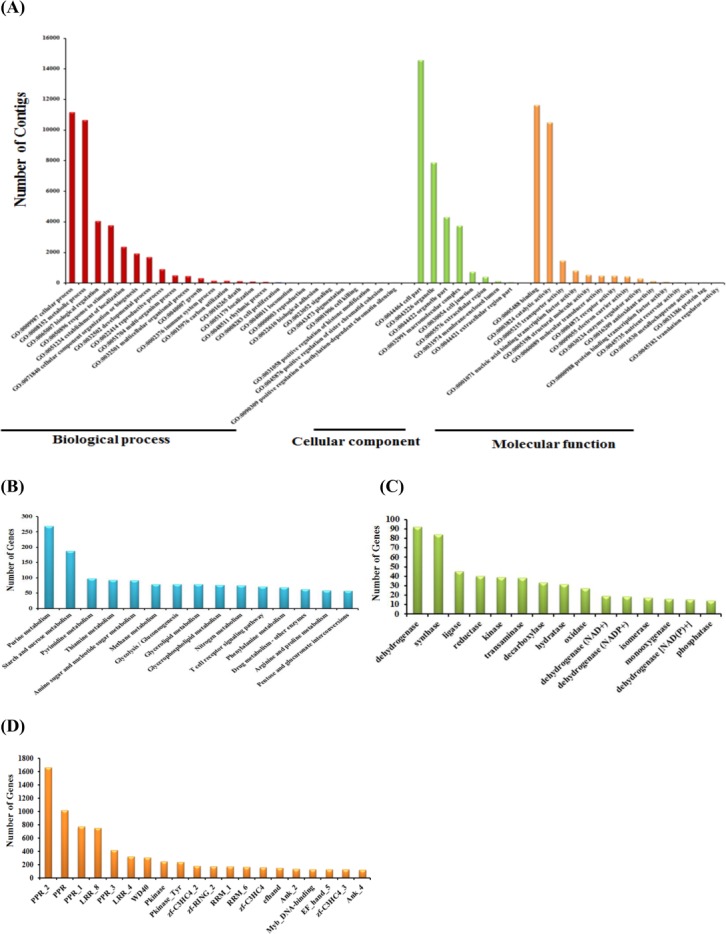
(A) GO classification summarized by three main categories: biological process, cellular component and molecular function. (B) Functional annotation of transcripts based on KEGG classification. (C) Functional characterization of transcripts for enzyme classes. (D) Pfam domains identified in translated Phalaenopsis transcripts.
Functional annotation and classification of Phalaenopsis floral transcriptome
GO assignments were used to classify the functions of the predicted Phalaenopsis flower-tissue transcripts. With Nr annotation, we used the Blast2GO 2.3.5 program to obtain GO annotations. In all, 4,609 GO annotations were assigned to 19,712 Phalaenopsis flower-tissue contigs, and the terms were summarized into three main GO categories and 49 GO functional groups (Fig 3A). We found 26 subsets within the Biological Process category, 8 within the Cellular Component category and 15 within the Molecular Function category (Fig 3A). Of these, 16,565 (84.0%) comprised the largest category of molecular function, followed by biological process (14,892, 75.6%) and cellular component (14,678, 74.5%) (S3 Table). Thus, the most abundant contigs were related to cellular and metabolic functions in the Biological Process category; cellular component, cell part and organelle functions in the Cellular Component category and molecular function, binding and catalytic activity in the Molecular Function category. Details of the gene annotation for significant hits of the three contig sets are in S3 Table.
To identify biological pathways activated in the flower tissues of Phalaenopsis, the assembled contigs were annotated with Enzyme Commission (EC) numbers from BLASTX alignments against the KEGG database (E-value ≤ 1e-5). A total of 3,724 transcripts were assigned to 139 KEGG pathways; of these 1,679 transcripts, EC numbers were also assigned (S4 Table). The top 15 KEGG pathways observed for Phalaenopsis flower tissue contigs are shown in Fig 3B. A large proportion of such contigs belonged to purine metabolism (map00230), starch and sucrose metabolism (map00500), pyrimidine metabolism (map00240), thiamine metabolism (map00730) and amino sugar and nucleotide sugar metabolism (map00520). The top 15 abundant enzyme classes for Phalaenopsis flower tissue contigs were dehydrogenase, synthase, ligase, reductase, kinase and transaminase (Fig 3C and S5 Table).
Identification of protein-coding domains
We obtained the conserved domain information for the transcriptome in the Pfam database [61] using RPS-BLAST, which scans a set of pre-calculated position-specific scoring matrices with a protein query. Comparison of the 43,552 contigs against the Pfam domain database with E-value cutoff 1e-5 and domain coverage >50% resulted in 9,607 contigs matching at least one protein domain model. The most abundant protein domains in Phalaenopsis flower buds were the pentatricopeptide repeat (PPR_2, pfam01535; PPR, pfam01535; PPR_1, pfam12854), followed by the leucine-rich repeat (LRR_8, pfam13855), pentatricopeptide repeat (PPR_3, pfam13812), leucine-rich repeat (LRR_4, pfam12799), and WD40 (WD40, pfam00400) domains (Fig 3D and S6 Table).
Global changes of transcriptome profile in wild-type and peloric-mutant flower organs
To identify differentially expressed genes (DEGs) between the wild-type and peloric-mutant flower tissues (sepal, petal and labellum) (Fig 1A and 1B), we determined relative expression levels from our RNA-seq data (43,552 contigs) using DEseq [46]. We found 1,838 distinct contig sequences between the peloric sepal (PS) and wild-type sepal (NS) libraries: 820 were upregulated and 1,018 downregulated in the peloric-mutant sepal (Fig 4). We found 758 distinct contig sequences between the peloric petal (PP) and wild-type petal (NP) libraries: 363 were upregulated and 395 downregulated in the peloric-mutant petal. We found 1,147 distinct contig sequences between the peloric labellum (PL) and wild-type labellum (NL) libraries: 854 were upregulated and 293 downregulated in the peloric-mutant labellum (Fig 4). The expression levels and annotation of these differentially expressed contig sequences are in S7 Table.
Fig 4. Changes in gene expression profiles between wild-type and peloric-mutant floral organs.
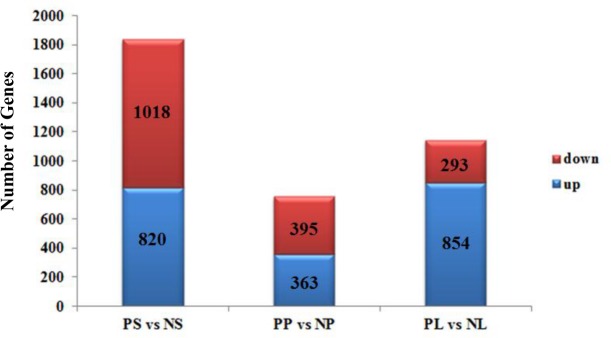
The number of up- and downregulated genes in peloric sepal (PS) and wild-type sepal (NS), peloric petal (PP) and wild-type petal (NP), and peloric labellum (PL) and wild-type labellum (NL). Six libraries were summarized.
Functional analysis of DEGs based on RNA-seq data
From the functional annotation of the Phalaenopsis flower tissue transcriptomes, the GO annotation of DEGs was obtained and underwent GO functional enrichment analysis, specifically the PS vs NS, PP vs NP and PL vs NL comparisons. GO annotation analysis involved groups of genes with greater than two-fold differential expression (log2 ratio ≥1 and log2 ratio ≤ -1, P< 0.05) in Phalaenopsis flower organs. Under PS vs NS, PP vs NP and PL vs NL, the DEGs were classified into 21, 18, and 20 categories of biological processes, respectively, 8 categories each on the basis of cellular components, and 13, 13, and 12 categories on the basis of molecular function (Fig C in S1 File).
Differential expression of TFs in flower organs
TFs bind DNA and target the assembly of protein complexes to regulate transcript levels of target gene expression [62]. In this study, we performed global TF classification for differentially expressed transcripts and found 878 that were differentially expressed between the wild-type and peloric mutant among Phalaenopsis floral organs (sepal, petal and labellum) (S8 Table), including members of the AP2/ERF, ARF, bHLH, homeobox, MADS, MYB and NAC families. This information is valuable in providing a deeper insight into the role of TFs during Phalalaenopsis wild-type and peloric-mutant floral-organ development. From DEseq analysis, most TF families from the most abundant transcripts were significantly downregulated and some were upregulated (S8 Table) in the peloric mutant. TFs with previously reported roles in Phalaenopsis floral-organ development included PeMADS4, which is among the most abundant MADS-box proteins expressed in the Phalaenopsis floral organs analyzed. The PeMADS4 transcript was detected in the wild-type Phalaenopsis labellum and ectopically expressed in the petal of the peloric mutant that transformed to a lip-like petal [29]. Another two candidate TFs (CUFF.17763 and CUFF.17763.1), upregulated in lip-like as compared with normal petals, encode a putative TF belonging to the MADS-box gene family that are homologous to AGL6-like genes (S8 Table). Some TF families, such as the AP2, homeobox, MYB and NAC family, will be further discussed below. A more in-depth analysis is necessary to determine how these gene families linked to molecular and cellular changes are activated during Phalaenopsis floral organ development.
RNA-seq expression validation by real-time PCR
The Illumina RNA-seq data were validated by real-time PCR analysis of selected genes with RNA isolated from floral organs (sepal, petal and labellum) of the 0.2-cm bud stage of both the wild-type and peloric mutant (Fig 1A and 1B). We selected 27 transcripts with differential expression patterns for real-time PCR analysis and performed a one-by-one comparison of each transcript by real-time PCR and RNA-seq. A total of 21 contigs showed differential expression in agreement with the RNA-seq data (Fig D in S1 File and S9 Table). Three transcripts showing upregulation in the RNA-seq results (CUFF.1482.1, CUFF.40149.1 and CUFF.29848.1) were slightly downregulated in the real-time PCR analysis, one transcript showing downregulation in the RNA-seq results (CUFF.39479.1) was slightly upregulated in the real-time PCR analysis and two transcripts showing downregulation in the RNA-seq results (CUFF. 19890.3 and CUFF. 29789.2) did no show significant difference in the real-time PCR analysis. These two methods yield completely opposite results that genome analyzer provides a holistic picture of all the isoforms of a gene into consideration, whereas the expression by real-time PCR is specific to the isoform of the gene into consideration owing to the use of gene specific primers. Overall, the real-time PCR results agreed well with the RNA-seq data.
PhAGL6 may be involved in labellum development of the Phalaenopsis flower
To reveal putative roles in labellum development of Phalaenopsis flower, we evaluated the association of the expression of five TFs (PhMADS1, PhMADS4, PhMADS5, PhAGL6a and PhAGL6b) in the floral organs (sepal, petal, labellum and gynostemium) and different floral morphologic features of Phalaenopsis orchid mutants (lip-like petal, lip-like sepal, and big lip) from our collection (Fig 5; Fig E in S1 File). We estimated the association of the expression of the five TFs and labellum development by real-time PCR. The PhMADS1 transcript was highly expressed in the gynostemium (Fig 6). The expression of PhMADS5 was significantly reduced in lip-like petals of peloric-mutant flowers (Fig 6A) but not in the lip-like sepal of peloric-mutant flowers (Fig 6B). The expression of three TFs (PhMADS4, PhAGL6a and PhAGL6b) was increased in the labellum of the wild-type and mutant (Fig 6). In contrast, PhMADS4 and PhAGL6b transcripts were significantly reduced in the labellum of the big-lip mutant (Fig 6C). Furthermore, the transcript levels of PhAGL6a and PhAGL6b were significantly increased in the lip-like petal and lip-like sepal of peloric- mutant flowers (Fig 6A and 6B). Thus, PhMADS4, PhAGL6a and PhAGL6b may play different roles in the development of the labellum in Phalaenopsis, especially PhAGL6b in peloric-mutant flowers and labellum development.
Fig 5. Phalaenopsis flower phenotypes of wild-type and peloric mutant.

(A) Wild-type flower of Phalaenopsis Brother Spring Dancer ‘KHM190’ and its lip-like petal mutant; (B) wild-type flower of Phalaenopsis aphrodite and its lip-like sepal mutant; (C) wild-type flower of Phalaenopsis ‘NPU1458’ and its big lip mutant. Bar = 1 cm.
Fig 6. Real-time PCR analysis of genes expressed in different floral mutants of Phalaenopsis orchid.
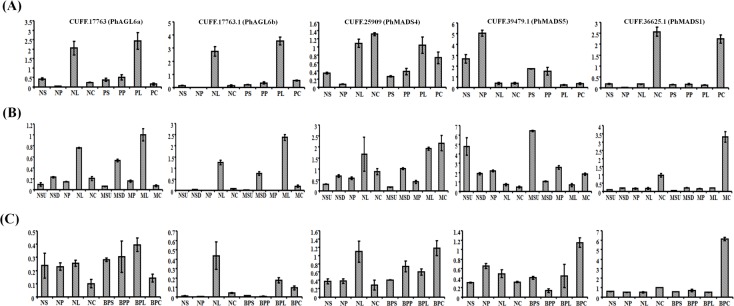
Total RNAs isolated from the sepal, petal, lip and column of mature flowers from Fig 5. The wild-type ([A] Phalaenopsis Brother Spring Dancer ‘KHM190’; [B] Phalaenopsis aphrodite and [C] Phalaenopsis ‘NPU1458’) and (N), lip-like petal mutant (P), lip-like sepal mutant (M) and big lip mutant (BP) were used as templates to detect the expression of CUFF.17763(PhAGL6a), CUFF.17763.1 (PhAGL6b), CUFF.25909 (PhMADS4), CUFF.39479.1 (PhMADS5) and CUFF.36625.1(PhMADS1). S, sepal (include upper and lateral sepals); SU, upper sepal; SD, lateral sepal; P, petal; L, lip; C, column.
Discussion
Construction of an informative floral-organ transcriptome for Phalaenopsis
Although the transcriptome of Phalaenopsis orchid has been reported previously with expressed sequence tags obtained through Sanger sequencing [49,63], three Roche 454 and Illumina platforms, and three Roche 454 and Illumina sequence datasets available in NCBI [40,43,45], an in-depth comparison of the orchid floral organ transcriptome to reveal developmental cues was lacking. In this study, we used the Illumina HiSeq 2000 sequencing platform to generate six transcriptome sequence datasets of different floral organs (sepal, petal and labellum) from the Phalaenopsis wild-type and peloric mutant. The assembled contigs of Phalaaenopsis floral organs were further analyzed to generate a functional characterization of the transcriptome and differential expression analysis. We assembled the contigs of the six libraries and obtained 43,552 transcripts; 64.7% of the contigs returned a significant BLAST result. The top five plant species with BLAST hits to annotated contigs were Vitis, Oryza, Zea, Populus and Ricinus (Fig 2B), for which the annotations of their genomes are comprehensive and largely accepted. Among 43,552 Phalaenopsis transcripts, 19,712 had GO annotations, 3,724 mapped to 139 pathways of KEGG, and 9,607 matched at least one protein domain with Pfam (Fig 3A, 3B and 3D). Thus, many unique processes and diverse pathways are involved in Phalaenopsis floral organ development.
Genes commonly expressed in floral organs
Transcript expression profiling is often compared among different developmental stages, different plant organs, or plants under different growth conditions [44,64–67]. In the present study, we identified many genes showing transcriptional changes in the Phalaenopsis floral organ by comparing the wild-type and peloric mutant. We identified 1,838 (PS vs. NS, 820 upregulated, 1,018 downregulated), 758 (PP vs. NP, 363 upregulated, 395 downregulated) and 1,147 (PL vs. NL, 854 upregulated, 293 downregulated) DEGs that were differentially expressed by greater than two-fold at P< 0.05 significance (Fig 4 and S7 Table). Most of these genes were involved in regulating flower development, such as floral meristem transformation or establishment of the floral meristem into different types of floral organs [13,68–70]. Our global transcript profiles provide a comprehensive high-resolution analysis of gene expression changes associated with Phalaenopsis floral-organ development.
From these DEGs, genes and TFs identified included floral homeotic MADS- box genes, MYB genes and NAC TFs (S8 Table) whose putative functions were linked to the morphogenesis of Phalaenopsis flower buds. By using the DEGs datasets, and computational and statistical analyses, this study led to the identification of genes that are likely involved in the control of key developmental processes during Phalaenopsis floral-organ development. Further functional characterization of the DEGs by GO functional analysis revealed an additional 42 (PS vs NS), 39 (PP vs NP) and 40 (PL vs NL) categories involved in biochemistry, metabolism, growth and regulation of biological processes (Fig C in S1 File). This DEG information will be valuable to elucidate floral organ development and to find novel floral-organ-related genes specific to Phalaenopsis orchids.
Transcriptional regulation in Phalaenopsis floral organs
Regulation of gene expression via TF binding is the primary mechanism by which dynamic complex processes of development and differentiation are controlled [71,72]. The specification of different types of floral organs is a key process regulated by the floral homeotic genes. These genes encode TFs that act in a combinatorial manner to regulate floral-organ developmental programs [6,73,74]. In the present study, we identified 878 TFs using global TF classification for the differentially expressed transcripts. Subsets of TF families were associated with functions in cell differentiation (bZIP, bHLH and MYB), meristem maintenance (homeobox, NAC and YABBY), floral-organ development (MADS and TCP) and other roles in hormone-mediated signalling by auxin (Aux/IAA, ARF), GA (GRAS and Dof) or ethylene (AP2/ ERF). The bHLH family contains genes regulating flower development, such as controlling floral-organ formation as well as the morphogenesis of sepals, petals, stamens and anthers in Arabidopsis [75–77], Eschscholzia californica [78] and rice [79]. The Phalaenopsis bHLH families showed complex expression profiles (S7 Table), which suggests their intricate roles in floral-organ development. Further investigation of bHLH TFs are required to verify floral-development regulation and interaction between these factors and other genes during early labellum and lip-like petal differentiation. The MYB transcription factors have conserved DNA binding domains and some have been known to regulate floral development [80–82]. Most of the Phalaenopsis MYB TFs in our RNA-seq data showed marked differential expression among the floral organs examined (S8 Table). Of the four MYB TFs, two (CUFF.26564.1 and CUFF.22705.1) were highly expressed in lip-like petals and the other two (CUFF.15031.1 and CUFF.15465.1) were upregulated in wild-type petals, which suggests different roles played by the four MYBs in petal organ development. The homeobox genes encode a group of transcriptional regulators that control meristem, floral and leaf maintenance and development [83–87]. In our RNA-seq data, one homeobox protein, KNOTTED-1-like 3 (CUFF.39041.1), was upregulated in lip-like petals. The roles of KNOTTED-1 genes such as HIRZINA and INVAGINATA in spur development have been reported in snapdragon (Antirrhinum majus) and Linaria vulgaris [83,88]. HIRZINA and INVAGINATA also induced sac-like outgrowths and distally dissected corolla tubes on flowers when constitutively expressed in transgenic tobacco [83]. Of note, KNOTTED-1-like 3 was ectopically expressed in Phalaenopsis lip-like petal, which suggests its probable function in inducing callosity structure and a sawtooth petal formation.
The MADS-box genes encode a family of TFs that are the best-studied floral TF family so far. Members of this family play prominent roles in floral organ specification [11,89]. In our study, the MADS-box TFs showed different expression profiles: four (CUFF.17763, CUFF.17763.1, CUFF.25909 and CUFF.36625.1) were upregulated and one (CUFF.39479.1) was downregulated in lip-like petals of the peloric mutant (S8 Table). In Phalaenopsis, PeMADS4 plays an important role in labellum development because its transcript was restricted to the labellum and lip-like petals of the peloric mutant [29]. Furthermore, the absence of OMADS5 expression is necessary for the formation of the large lips and the conversion of the sepal or petal into lips in Oncidium peloric mutants [32]. In our DEGs and real-time PCR analysis, we identified one transcript encoding PeMADS4 with higher expression in labellum and lip-like petals (Fig 6A), which is similar to findings by Tsai et al. (2004) [29]. Two members of the AGL6-like MADS-box subfamily were upregulated in the lip-like petal of peloric mutants and the labellum (Fig 6A, 6B and 6C). The AGL6-like genes define floral organ and meristem identity in Arabidopsis, rice, petunia and maize [64,90–93]. The AGL6-like genes from our Phalaenopsis floral transcriptomes might also play an important role in maintaining labellum development. Ectopic expression of AGL6-like genes in transgenic Phalaenopsis petals may reveal their function.
PhAGL6-like and PhMADS4 are critical candidate regulators of labellum development in Phalaenopsis
To reveal the regulation of five MADS genes (PhMADS1, PhMADS4, PhMADS5, PhAGL6a and PhAGL6b) in floral-organ development in Phalaenopsis, we examined their expression in different floral morphogens of Phalaenopsis orchid mutants (lip-like petal, lip-like sepal and big lip). The increased PhMADS4 transcript level in labellum and gynostemium of wild-type plants suggests its positive role in both labellum and gynostemium formation (Fig 6A, 6B and 6C). The PhMADS4 transcript level was 5.05-fold higher in lip-like petals of peloric mutant flowers than in wild-type petals (Fig 6A). In addition, the PhMADS4 transcript was also 1.48-fold increased in mutant lip-like sepal organ and 0.55-fold decreased in big-lip organs (Fig 6B and 6C). The results from these two different flower mutants suggest that PhMADS4 might be involved in labellum development and lip-like petal formation, as reported by Tsai et al. (2004) [29]. However, in the lip-like sepal of peloric mutants, PhMADS4 might play only a minor role in the conversion of the sepal into a labellum. The increased PhMADS4 transcript may be a secondary effect of sepal conversion into a lip-like structure. This assumption still requires further investigation. The expression of PhMADS5 was upregulated in sepals and petals but upregulated only in gynostemium of the big-lip mutant. PhMADS5 transcript level was slightly but significantly decreased (0.297-fold) in lip-like petals and decreased (0.56-fold) in lip-like sepals. This finding implies that PhMADS5 may be related to petal and sepal development. In all wild-types and peloric mutants examined, PhMADS1 transcript expression was stronger in the gynostemium than sepal, petal and labellum (Fig 6A, 6B and 6C), as reported by Chen et al. (2012) [94]. PhMADS1 is a C-function homeotic gene, which is associated with the formation of gynostemiums [94].
The expression of two AGL6-like TFs (PhAGL6a and PhAGL6b) was increased in the labellum of both the wild-type and lip-like petal mutant (Fig 6A). Furthermore, the expression of PhAGL6a and PhAGL6b was significantly increased in the lip-like petal of peloric mutant flowers. AGL6-like genes may be a positive regulator of labellum formation. Ectopic expression AGL6-like genes in petal or sepal may convert them into a lip-like structure. In contrast, downregulation of AGL6-like genes in the labellum may affect labellum development. As a further test of the significance of these assumptions, we examined the expression patterns of PhAGL6a and PhAGL6b in the lip-like petal, lip-like sepal and big lip of peloric mutant flowers. As expected, PhAGL6a and PhAGL6b were ectopically expressed in the lip-like petal and lip-like sepal organs in peloric mutants. In addition, the expression of PhAGL6b was reduced in the labellum of the big-lip mutant, with no change in expression of PhAGL6a. In summary, PhMADS4, PhAGL6a and PhAGL6b together play different roles in maintenance of labellum development. For petal and sepal mutants converted into a lip-like structure, PhMADS4, PhAGL6a and PhAGL6b are ectopically expressed in the peloric petal and sepal (Fig 7). Alternatively, PhMADS4 and PhAGL6b may be critical and involved in the labellum converting into a big lip (Fig 7).
Fig 7. Possible evolutionary relationships between PhAGL6a, PhAGL6b and PhMADS4 in the regulation of lip formation in Phalaenopsis orchid.

Conclusions
We examined global transcriptome landscapes from six floral organ tissues of the wild-type and peloric-mutant Phalaenopsis orchid and identified preferentially expressed genes within and among floral organ developmental tissues. By comparing genes showing floral-organ-preferential expression patterns between the wild-type and peloric mutant, we identified DEGs across a wide range of transcript abundances. Cumulative counts of contigs that mapped to predicted gene models enabled the identification of functionally interesting genes and gene families with altered expression in peloric mutants. Functional analysis and characterization of the differentially expressed genes provides new insight into peloric floral morphogenesis, in particular regarding the role of regulation for floral organ development. We provide a comprehensive list of genes that might be involved in labellum formation and petal conversion into a labellum process. Many TF genes, especially MADS-box genes were expressed in floral organs. We identified the MADS-box genes PhAGL6a, PhAGL6b and PhMADS4 as potential regulatory components of labellum development based on conversion of the petal or sepal mutant into a lip-like structure. Our study gives new insights into exploring PhAGL6a, PhAGL6b and PhMADS4 transgenic plants as a tool to investigate labellum development and petal or sepal conversion into a labellum in Phalaenopsis orchids. Our results provide a strong basis for future research into floral organ development in Phalaenopsis orchid.
Supporting Information
(DOC)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
We thank Yourgene Bioscience for assistance of technical works.
Data Availability
All RNA-seq files are available from the NCBI database (accession numbers: SRX396172, SRX396784,SRX396785, SRX396786, SRX396787, SRX396788).
Funding Statement
This work was supported by grants from the Council of Agriculture, Agriculture and Food Agency, Taiwan, with the grant numbers 103AS-9.1.1-FD-Z2 and 104AS-9.1.1-FD-Z2.
References
- 1. Pridgeon AM, Cribb PJ, Chase MW, Rasmussen FN (2005) Genera Orchidacearum: Epidendroideae (Part One). Oxford: Oxford University Press.696 p. [Google Scholar]
- 2. Christenson EA (2001) Phalaenopsis. Portland Oregon: Timber Press. 330 p. [Google Scholar]
- 3. Rudall PJ, Bateman RM (2002) Roles of synorganisation, zygomorphy and heterotopy in floral evolution: the gynostemium and labellum of orchids and other lilioid monocots. Biol Rev Camb Philos Soc 77: 403–441. [DOI] [PubMed] [Google Scholar]
- 4. Kaeppler SM, Kaeppler HF, Rhee Y (2000) Epigenetic aspects of somaclonal variation in plants. Plant Mol Biol 43: 179–188. [DOI] [PubMed] [Google Scholar]
- 5. Miguel C, Marum L (2011) An epigenetic view of plant cells cultured in vitro: somaclonal variation and beyond. J Exp Bot 62: 3713–3725. 10.1093/jxb/err155 [DOI] [PubMed] [Google Scholar]
- 6. Coen ES, Meyerowitz EM (1991) The war of the whorls: genetic interactions controlling flower development. Nature 353: 31–37. [DOI] [PubMed] [Google Scholar]
- 7. Theissen G (2001) Development of floral organ identity: stories from the MADS house. Curr Opin Plant Biol 4: 75–85. [DOI] [PubMed] [Google Scholar]
- 8. Theissen G, Saedler H (1995) MADS-box genes in plant ontogeny and phylogeny: Haeckel's 'biogenetic law' revisited. Curr Opin Genet Dev 5: 628–639. [DOI] [PubMed] [Google Scholar]
- 9. Weigel D, Meyerowitz EM (1994) The ABCs of floral homeotic genes. Cell 78: 203–209. [DOI] [PubMed] [Google Scholar]
- 10. Alvarez-Buylla ER, Pelaz S, Liljegren SJ, Gold SE, Burgeff C, Ditta GS, et al. (2000) An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc Natl Acad Sci U S A 97: 5328–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Theissen G, Becker A, Di Rosa A, Kanno A, Kim JT, Münster T, et al. (2000) A short history of MADS-box genes in plants. Plant Mol Biol 42: 115–149. [PubMed] [Google Scholar]
- 12. Ditta G, Pinyopich A, Robles P, Pelaz S, Yanofsky MF (2004) The SEP4 gene of Arabidopsis thaliana functions in floral organ and meristem identity. Curr Biol 14: 1935–1940. [DOI] [PubMed] [Google Scholar]
- 13. Krizek BA, Fletcher JC (2005) Molecular mechanisms of flower development: an armchair guide. Nat Rev Genet 6: 688–698. [DOI] [PubMed] [Google Scholar]
- 14. Theissen G, Melzer R (2007) Molecular mechanisms underlying origin and diversification of the angiosperm flower. Ann Bot 100: 603–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jofuku KD, den Boer BG, Van Montagu M, Okamuro JK (1994) Control of Arabidopsis flower and seed development by the homeotic gene APETALA2 . Plant Cell 6: 1211–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mandel MA, Yanofsky MF (1995) A gene triggering flower formation in Arabidopsis . Nature 377: 522–524. [DOI] [PubMed] [Google Scholar]
- 17. Goto K, Meyerowitz EM (1994) Function and regulation of the Arabidopsis floral homeotic gene PISTILLATA . Genes Dev 8: 1548–1560. [DOI] [PubMed] [Google Scholar]
- 18. Jack T, Brockman LL, Meyerowitz EM (1992) The homeotic gene APETALA3 of Arabidopsis thaliana encodes a MADS box and is expressed in petals and stamens. Cell 68: 683–697. [DOI] [PubMed] [Google Scholar]
- 19. Pinyopich A, Ditta GS, Savidge B, Liljegren SJ, Baumann E, Wisman E, et al. (2003) Assessing the redundancy of MADS-box genes during carpel and ovule development. Nature 424: 85–88. [DOI] [PubMed] [Google Scholar]
- 20. Pelaz S, Ditta GS, Baumann E, Wisman E, Yanofsky MF (2000) B and C floral organ identity functions require SEPALLATA MADS-box genes. Nature 405: 200–203. [DOI] [PubMed] [Google Scholar]
- 21. Ferrario S, Immink RG, Angenent GC (2004) Conservation and diversity in flower land. Curr Opin Plant Biol 7: 84–91. [DOI] [PubMed] [Google Scholar]
- 22. Kramer EM, Hall JC (2005) Evolutionary dynamics of genes controlling floral development. Curr Opin Plant Biol 8: 13–18. [DOI] [PubMed] [Google Scholar]
- 23. Cozzolino S, Widmer A (2005) Orchid diversity: an evolutionary consequence of deception? Trends Ecol Evol 20: 487–494. [DOI] [PubMed] [Google Scholar]
- 24. Chen D, Guo B, Hexige S, Zhang T, Shen D, Ming F (2007) SQUA-like genes in the orchid Phalaenopsis are expressed in both vegetative and reproductive tissues. Planta 226: 369–380. [DOI] [PubMed] [Google Scholar]
- 25. Chang YY, Chiu YF, Wu JW, Yang CH (2009) Four orchid (Oncidium Gower Ramsey) AP1/AGL9-like MADS box genes show novel expression patterns and cause different effects on floral transition and formation in Arabidopsis thaliana. Plant Cell Physiol 50: 1425–1438. 10.1093/pcp/pcp087 [DOI] [PubMed] [Google Scholar]
- 26. Skipper M, Johansen LB, Pedersen KB, Frederiksen S, Johansen BB (2006) Cloning and transcription analysis of an AGAMOUS- and SEEDSTICK ortholog in the orchid Dendrobium thyrsiflorum (Reichb. f.). Gene 366: 266–274. [DOI] [PubMed] [Google Scholar]
- 27. Yu H, Goh CJ (2000) Identification and characterization of three orchid MADS-box genes of the AP1/AGL9 subfamily during floral transition. Plant Physiol 123: 1325–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mondragon-Palomino M, Theissen G (2011) Conserved differential expression of paralogous DEFICIENS- and GLOBOSA-like MADS-box genes in the flowers of Orchidaceae: refining the 'orchid code'. Plant J 66: 1008–1019. 10.1111/j.1365-313X.2011.04560.x [DOI] [PubMed] [Google Scholar]
- 29. Tsai WC, Kuoh CS, Chuang MH, Chen WH, Chen HH (2004) Four DEF-like MADS box genes displayed distinct floral morphogenetic roles in Phalaenopsis orchid. Plant Cell Physiol 45: 831–844. [DOI] [PubMed] [Google Scholar]
- 30. Aceto S, Sica M, De Paolo S, D'Argenio V, Cantiello P, Salvatore F, et al. (2014) The analysis of the inflorescence miRNome of the orchid Orchis italica reveals a DEF-like MADS-box gene as a new miRNA target. PLoS One 9: e97839 10.1371/journal.pone.0097839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hsu HF, Yang CH (2002) An orchid (Oncidium Gower Ramsey) AP3-like MADS gene regulates floral formation and initiation. Plant Cell Physiol 43: 1198–1209. [DOI] [PubMed] [Google Scholar]
- 32. Chang YY, Kao NH, Li JY, Hsu WH, Liang YL, Wu JW, et al. (2010) Characterization of the possible roles for B class MADS box genes in regulation of perianth formation in orchid. Plant Physiol 152: 837–853. 10.1104/pp.109.147116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim SY, Yun PY, Fukuda T, Ochiai T, Yokoyama J, Toshiaki K, et al. (2007): Expression of a DEFICIENS-like gene correlates with the differentiation between sepal and petal in the orchid, Habenaria radiata (Orchidaceae). Plant Sci 172: 319–326. [Google Scholar]
- 34. Song IJ, Nakamura T, Fukuda T, Yokoyama J, Ito T, Ichikawa H, et al. (2006) Spatiotemporal expression of duplicate AGAMOUS orthologues during floral development in Phalaenopsis. Dev Genes Evol 216: 301–313. [DOI] [PubMed] [Google Scholar]
- 35. Xu Y, Teo LL, Zhou J, Kumar PP, Yu H (2006) Floral organ identity genes in the orchid Dendrobium crumenatum . Plant J 46: 54–68. [DOI] [PubMed] [Google Scholar]
- 36. Wang SY, Lee PF, Lee YI, Hsiao YY, Chen YY, Pan ZJ, et al. (2011) Duplicated C-class MADS-box genes reveal distinct roles in gynostemium development in Cymbidium ensifolium (Orchidaceae). Plant Cell Physiol 52: 563–577. 10.1093/pcp/pcr015 [DOI] [PubMed] [Google Scholar]
- 37. Hsu HF, Huang CH, Chou LT, Yang CH (2003) Ectopic expression of an orchid (Oncidium Gower Ramsey) AGL6-like gene promotes flowering by activating flowering time genes in Arabidopsis thaliana . Plant Cell Physiol 44: 783–794. [DOI] [PubMed] [Google Scholar]
- 38. Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10: 57–63. 10.1038/nrg2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brautigam A, Mullick T, Schliesky S, Weber AP (2011) Critical assessment of assembly strategies for non-model species mRNA-Seq data and application of next-generation sequencing to the comparison of C(3) and C(4) species. J Exp Bot 62: 3093–3102. 10.1093/jxb/err029 [DOI] [PubMed] [Google Scholar]
- 40. Hsiao YY, Chen YW, Huang SC, Pan ZJ, Fu CH, Chen W, et al. (2011) Gene discovery using next-generation pyrosequencing to develop ESTs for Phalaenopsis orchids. BMC Genomics 12: 360 10.1186/1471-2164-12-360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. O'Rourke JA, Yang SS, Miller SS, Bucciarelli B, Liu J, Rydeen A, et al. (2013) An RNA-Seq transcriptome analysis of orthophosphate-deficient white lupin reveals novel insights into phosphorus acclimation in plants. Plant Physiol 161: 705–724. 10.1104/pp.112.209254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Raju NL, Gnanesh BN, Lekha P, Jayashree B, Pande S, Hiremath PJ, et al. (2010) The first set of EST resource for gene discovery and marker development in pigeonpea (Cajanus cajan L.). BMC Plant Biol 10: 45 10.1186/1471-2229-10-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Su CL, Chao YT, Alex Chang YC, Chen WC, Chen CY, Lee AY, et al. (2011) De novo assembly of expressed transcripts and global analysis of the Phalaenopsis aphrodite transcriptome. Plant Cell Physiol 52: 1501–1514. 10.1093/pcp/pcr097 [DOI] [PubMed] [Google Scholar]
- 44. Uddenberg D, Reimegard J, Clapham D, Almqvist C, von Arnold S, Emanuelsson O, et al. (2013) Early cone setting in Picea abies acrocona is associated with increased transcriptional activity of a MADS box transcription factor. Plant Physiol 161: 813–823. 10.1104/pp.112.207746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chang YY, Chu YW, Chen CW, Leu WM, Hsu HF, Yang CH (2011) Characterization of Oncidium 'Gower Ramsey' transcriptomes using 454 GS-FLX pyrosequencing and their application to the identification of genes associated with flowering time. Plant Cell Physiol 52: 1532–1545. 10.1093/pcp/pcr101 [DOI] [PubMed] [Google Scholar]
- 46. Zhang J, Wu K, Zeng S, Teixeira da Silva JA, Zhao X, Tian CE, et al. (2013) Transcriptome analysis of Cymbidium sinense and its application to the identification of genes associated with floral development. BMC Genomics 14: 279 10.1186/1471-2164-14-279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. De Paolo S, Salvemini M, Gaudio L, Aceto S (2014) De novo transcriptome assembly from inflorescence of Orchis italica: analysis of coding and non-coding transcripts. PLoS One 9: e102155 10.1371/journal.pone.0102155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sedeek KE, Qi W, Schauer MA, Gupta AK, Poveda L, Xu S, et al. (2013) Transcriptome and proteome data reveal candidate genes for pollinator attraction in sexually deceptive orchids. PLoS One 8: e64621 10.1371/journal.pone.0064621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen YH, Tsai YJ, Huang JZ, Chen FC (2005) Transcription analysis of peloric mutants of Phalaenopsis orchids derived from tissue culture. Cell Res 15: 639–657. [DOI] [PubMed] [Google Scholar]
- 50. Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829. 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schulz MH, Zerbino DR, Vingron M, Birney E (2012) Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28: 1086–1092. 10.1093/bioinformatics/bts094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Conesa A, Gotz S (2008) Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics 2008: 619832 10.1155/2008/619832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen TW, Gan RC, Wu TH, Huang PJ, Lee CY, Chen YY, et al. (2012) FastAnnotator—an efficient transcript annotation web tool. BMC Genomics 13 Suppl 7: S9 10.1186/1471-2164-13-S7-S9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Finn RD, Mistry J, Schuster-Bockler B, Griffiths-Jones S, Hollich V, Lassmann T, et al. (2006) Pfam: clans, web tools and services. Nucleic Acids Res 34: D247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Roberts A, Pachter L (2013) Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods 10: 71–73. 10.1038/nmeth.2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yuan XY, Jiang SH, Wang MF, Ma J, Zhang XY, Cui B (2014) Evaluation of internal control for gene expression in Phalaenopsis by quantitative real-time PCR. Appl Biochem Biotechnol 173: 1431–1445. 10.1007/s12010-014-0951-x [DOI] [PubMed] [Google Scholar]
- 61. Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, et al. (2014) Pfam: the protein families database. Nucleic Acids Res 42: D222–230. 10.1093/nar/gkt1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wu S, Gallagher KL (2012) Transcription factors on the move. Curr Opin Plant Biol 15: 645–651. 10.1016/j.pbi.2012.09.010 [DOI] [PubMed] [Google Scholar]
- 63. Hsiao YY, Tsai WC, Kuoh CS, Huang TH, Wang HC, Wu TS, et al. (2006) Comparison of transcripts in Phalaenopsis bellina and Phalaenopsis equestris (Orchidaceae) flowers to deduce monoterpene biosynthesis pathway. BMC Plant Biol 6: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Li P, Ponnala L, Gandotra N, Wang L, Si Y, Tausta SL, et al. (2010) The developmental dynamics of the maize leaf transcriptome. Nat Genet 42: 1060–1067. 10.1038/ng.703 [DOI] [PubMed] [Google Scholar]
- 65. Matas AJ, Yeats TH, Buda GJ, Zheng Y, Chatterjee S, Tohge T, et al. (2011) Tissue- and cell-type specific transcriptome profiling of expanding tomato fruit provides insights into metabolic and regulatory specialization and cuticle formation. Plant Cell 23: 3893–3910. 10.1105/tpc.111.091173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sweetman C, Wong DC, Ford CM, Drew DP (2012) Transcriptome analysis at four developmental stages of grape berry (Vitis vinifera cv. Shiraz) provides insights into regulated and coordinated gene expression. BMC Genomics 13: 691 10.1186/1471-2164-13-691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang X, Liu D, Li A, Sun X, Zhang R, Wu L, et al. (2013) Transcriptome analysis of tomato flower pedicel tissues reveals abscission zone-specific modulation of key meristem activity genes. PLoS One 8: e55238 10.1371/journal.pone.0055238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jack T (2004) Molecular and genetic mechanisms of floral control. Plant Cell 16 Suppl: S1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lohmann JU, Weigel D (2002) Building beauty: the genetic control of floral patterning. Dev Cell 2: 135–142. [DOI] [PubMed] [Google Scholar]
- 70. Wellmer F, Alves-Ferreira M, Dubois A, Riechmann JL, Meyerowitz EM (2006) Genome-wide analysis of gene expression during early Arabidopsis flower development. PLoS Genet 2: e117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Riechmann JL, Ratcliffe OJ (2000) A genomic perspective on plant transcription factors. Curr Opin Plant Biol 3: 423–434. [DOI] [PubMed] [Google Scholar]
- 72. Zhang JZ (2003) Overexpression analysis of plant transcription factors. Curr Opin Plant Biol 6: 430–440. [DOI] [PubMed] [Google Scholar]
- 73. Bowman JL, Smyth DR, Meyerowitz EM (1991) Genetic interactions among floral homeotic genes of Arabidopsis . Development 112: 1–20. [DOI] [PubMed] [Google Scholar]
- 74. Honma T, Goto K (2001) Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 409: 525–529. [DOI] [PubMed] [Google Scholar]
- 75. Groszmann M, Bylstra Y, Lampugnani ER, Smyth DR (2010) Regulation of tissue-specific expression of SPATULA, a bHLH gene involved in carpel development, seedling germination, and lateral organ growth in Arabidopsis . J Exp Bot 61: 1495–1508. 10.1093/jxb/erq015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Szecsi J, Joly C, Bordji K, Varaud E, Cock JM, Dumas C, et al. (2006) BIGPETALp, a bHLH transcription factor is involved in the control of Arabidopsis petal size. EMBO J 25: 3912–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang W, Sun Y, Timofejeva L, Chen C, Grossniklaus U, Ma H (2006) Regulation of Arabidopsis tapetum development and function by DYSFUNCTIONAL TAPETUM1 (DYT1) encoding a putative bHLH transcription factor. Development 133: 3085–3095. [DOI] [PubMed] [Google Scholar]
- 78. Zahn LM, Ma X, Altman NS, Zhang Q, Wall PK, Tian D, et al. (2010) Comparative transcriptomics among floral organs of the basal eudicot Eschscholzia californica as reference for floral evolutionary developmental studies. Genome Biol 11: R101 10.1186/gb-2010-11-10-r101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Li N, Zhang DS, Liu HS, Yin CS, Li XX, Liang WQ, et al. (2006) The rice tapetum degeneration retardation gene is required for tapetum degradation and anther development. Plant Cell 18: 2999–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Corley SB, Carpenter R, Copsey L, Coen E (2005) Floral asymmetry involves an interplay between TCP and MYB transcription factors in Antirrhinum . Proc Natl Acad Sci U S A 102: 5068–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Galego L, Almeida J (2002) Role of DIVARICATA in the control of dorsoventral asymmetry in Antirrhinum flowers. Genes Dev 16: 880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Perez-Rodriguez M, Jaffe FW, Butelli E, Glover BJ, Martin C (2005) Development of three different cell types is associated with the activity of a specific MYB transcription factor in the ventral petal of Antirrhinum majus flowers. Development 132: 359–370. [DOI] [PubMed] [Google Scholar]
- 83. Box MS, Dodsworth S, Rudall PJ, Bateman RM, Glover BJ (2011) Characterization of Linaria KNOX genes suggests a role in petal-spur development. Plant J 68: 703–714. 10.1111/j.1365-313X.2011.04721.x [DOI] [PubMed] [Google Scholar]
- 84. Box MS, Dodsworth S, Rudall PJ, Bateman RM, Glover BJ (2012) Flower-specific KNOX phenotype in the orchid Dactylorhiza fuchsii. J Exp Bot 63: 4811–4819. 10.1093/jxb/ers152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chatterjee M, Bermudez-Lozano CL, Clancy MA, Davis TM, Folta KM (2011) A strawberry KNOX gene regulates leaf, flower and meristem architecture. PLoS One 6: e24752 10.1371/journal.pone.0024752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lenhard M, Jurgens G, Laux T (2002) The WUSCHEL and SHOOTMERISTEMLESS genes fulfil complementary roles in Arabidopsis shoot meristem regulation. Development 129: 3195–3206. [DOI] [PubMed] [Google Scholar]
- 87. Shani E, Burko Y, Ben-Yaakov L, Berger Y, Amsellem Z, Goldshmidt A, et al. (2009) Stage-specific regulation of Solanum lycopersicum leaf maturation by class 1 KNOTTED1-LIKE HOMEOBOX proteins. Plant Cell 21: 3078–3092. 10.1105/tpc.109.068148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Golz JF, Keck EJ, Hudson A (2002) Spontaneous mutations in KNOX genes give rise to a novel floral structure in Antirrhinum . Curr Biol 12: 515–522. [DOI] [PubMed] [Google Scholar]
- 89. Ng M, Yanofsky MF (2001) Function and evolution of the plant MADS-box gene family. Nat Rev Genet 2: 186–195. [DOI] [PubMed] [Google Scholar]
- 90. Duan Y, Xing Z, Diao Z, Xu W, Li S, Du X, et al. (2012) Characterization of Osmads6-5, a null allele, reveals that OsMADS6 is a critical regulator for early flower development in rice (Oryza sativa L.). Plant Mol Biol 80: 429–442. 10.1007/s11103-012-9958-2 [DOI] [PubMed] [Google Scholar]
- 91. Koo SC, Bracko O, Park MS, Schwab R, Chun HJ, Park KM, et al. (2010) Control of lateral organ development and flowering time by the Arabidopsis thaliana MADS-box Gene AGAMOUS-LIKE6 . Plant J 62: 807–816. 10.1111/j.1365-313X.2010.04192.x [DOI] [PubMed] [Google Scholar]
- 92. Rijpkema AS, Zethof J, Gerats T, Vandenbussche M (2009) The petunia AGL6 gene has a SEPALLATA-like function in floral patterning. Plant J 60: 1–9. 10.1111/j.1365-313X.2009.03917.x [DOI] [PubMed] [Google Scholar]
- 93. Thompson BE, Bartling L, Whipple C, Hall DH, Sakai H, Schmidt R, et al. (2009) bearded-ear encodes a MADS box transcription factor critical for maize floral development. Plant Cell 21: 2578–2590. 10.1105/tpc.109.067751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chen YY, Lee PF, Hsiao YY, Wu WL, Pan ZJ, Lee YI, et al. (2012) C- and D-class MADS-box genes from Phalaenopsis equestris (Orchidaceae) display functions in gynostemium and ovule development. Plant Cell Physiol 53: 1053–1067. 10.1093/pcp/pcs048 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
All RNA-seq files are available from the NCBI database (accession numbers: SRX396172, SRX396784,SRX396785, SRX396786, SRX396787, SRX396788).