

Abstract
The development of the adrenal cortex involves the formation and then subsequent regression of immature or fetal inner cell layers as the mature steroidogenic outer layers expand. However, controls over this remodeling, especially in the immature inner layer, are incompletely understood. Here we identify an inner cortical cell population that expresses thyroid hormone receptor-β1 (TRβ1), one of two receptor isoforms encoded by the Thrb gene. Using mice with a Thrbb1 reporter allele that expresses lacZ instead of TRβ1, β-galactosidase was detected in the inner cortex from early stages. Expression peaked at juvenile ages in an inner zone that included cells expressing 20-α-hydroxysteroid dehydrogenase, a marker of the transient, so-called X-zone in mice. The β-galactosidase-positive zone displayed sexually dimorphic regression in males after approximately 4 weeks of age but persisted in females into adulthood in either nulliparous or parous states. T3 treatment promoted hypertrophy of inner cortical cells, induced some markers of mature cortical cells, and, in males, delayed the regression of the TRβ1-positive zone, suggesting that TRβ1 could partly divert the differentiation fate and counteract male-specific regression of inner zone cells. TRβ1-deficient mice were resistant to these actions of T3, supporting a functional role for TRβ1 in the inner cortex.
During development, the adrenal cortex first forms as a fetal or immature cell layer that later regresses as it is replaced by mature cortical cell types. In rodent models, the mature cortex, which produces mineralocorticoids and glucocorticoids, is formed by inward migration of definitive cortical cells that are generated from outer capsular progenitor cells (1–3). Prior to regression, the fetal cortex in mice may produce corticosterone that aids fetal organ development (4) and in humans produces steroid hormones including dehydroepiandrosterone sulfate (5, 6). The need for orderly remodeling of cortical layers is suggested by human DAX1 mutations that result in failed regression of the fetal inner cortex, poor cortical differentiation, and deficiency of adrenal steroid hormones (7). In mice, Dax1 mutation results in retention of an immature cortex (8), enhanced steroidogenesis at young ages, and loss of adrenal steroidogenic activity at older ages (9). It has also been proposed that human adrenocortical carcinoma may derive from nonregressed fetal cortical cells (10).
The fetal cortex is generated from progenitor cells in the adrenal primordium (11), but the subsequent differentiation and then regression of the inner cortical layers is only partly understood. In juvenile mice, the inner cortex forms a transient zone of histologically dense cells, the so-called X-zone (12, 13) that expresses the steroid-metabolizing enzyme 20α-hydroxysteroid dehydrogenase (20αHSD) (14) but is otherwise poorly defined. This X-zone regresses in male mice after approximately 1 month but persists in adult females unless pregnancy occurs, which then induces regression (12). External factors such as gonadal factors and gonadotropins (15–19) influence the survival of this zone and thyroid extracts (20) can modify the histological appearance of the zone. Signaling factors such as activin and regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A) (17, 21) and transcription factors including GATA6 and steroidogenic factor 1 also regulate this process (18, 22).
To elucidate factors involved in adrenal differentiation, we have investigated thyroid hormone receptors (TRs). TRs act as ligand-regulated transcription factors and can mediate tissue remodeling, for example, in tail regression during amphibian metamorphosis (23) and intestinal maturation in mammals (24). The mammalian Thrb (Nr1a2) gene differentially expresses TRβ1 and TRβ2 receptor isoforms from independent promoters (25–30). Mutagenesis in mice has shown that TRβ2 contributes to photoreceptor differentiation (31) and control of the pituitary-thyroid axis (31, 32). Cell-specific functions of TRβ1 have not been defined directly by mutagenesis, although TRβ1 is expressed in a range of tissues (30, 33, 34). Here, using Thrb+/b1 (or +/b1) mice that express β-galactosidase instead of TRβ1 from one allele of the Thrb gene, we have identified a novel TRβ1-positive zone in the adrenal cortex that exhibits sexually dimorphic regression. T3 induces changes in morphology and expression of cortical markers in these cells, suggesting that TRβ1 modulates inner cortical responses at sensitive postnatal phases.
Materials and Methods
Mouse strains and treatment
For the developmental PCR analysis in Figure 1, wild-type C57BL/6J mice were used as the source of RNA samples. For other analyses, heterozygous male and female Thrb+/b1 (+/b1) or Thrb+/− (+/−) mice were crossed to generate control and mutant littermates. Mice were derived on a mixed C57BL/6J × 129/Sv background. The Thrbb1 allele, which carries lacZ targeted into a TRβ1-specific exon and leaves the TRβ2-specific exon intact (L. Ng and D. Forrest, manuscript in preparation), was further backcrossed for five generations and the Thrb+/− strain (35) for nine generations on a C57BL/6J background. Genotypes were determined by PCR as described (35). Mice were housed under a 12-hour light, 12-hour dark cycle. Mice were injected sc with 30 μL saline containing 0.5 μg L-T3 (Sigma-Aldrich) daily for 8–16 days, starting at the stated postnatal ages. Experiments followed the approved institutional protocols at the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health.
Figure 1.

Expression of TRβ1 in the adrenal inner cortex, A, X-gal staining (blue) showing expression in the adrenal inner cortex of lacZ in heterozygous Thrb+/b1 (+/b1) male and female mice. X-gal staining revealed sexually dimorphic regression after P20. B, Immunofluorescence showing the expression profile of 20αHSD (red, in inner cortical X-zone), tyrosine hydroxylase (Tyr. Hyd., green, in medulla), and DAPI (blue, all cell nuclei) in the adrenal. DAPI, 4′,6′-diamino-2-phenylindole; ic, inner cortex, m, medulla, oc, outer cortex. C and D, Relative levels of mRNA for TRβ1, TRα1, and 20αHSD in adrenal development detected by qPCR. The profiles of TRβ1 and 20αHSD were similar with a peak near weaning age, whereas TRα1 was expressed more constantly throughout development (means ± SD, n = 3 mice per age). E, embryonic day. E, RT-PCR showing predicted bands for wild-type TRβ1 (260 bases) and fusion TRβ1-lacZ (357 bases) mRNA in adrenal samples from +/+, +/b1, and b1/b1 mice. Scale bars, 50 μm.
Histology and histochemistry
Adrenal glands were fixed in 4% paraformaldehyde in PBS at 4°C for 4 hours, rinsed in PBS for 3 × 5 minutes, and then immersed in 30% sucrose at 4°C overnight. Samples were embedded in optimum cutting temperature compound (Sakura Finetek) for cryosectioning at 10 μm thickness or embedded in paraffin for microtome sectioning at a thickness 3 or 4 μm. Paraffin-embedded sections were stained with hematoxylin and eosin. Cryosections were stained using a 5-bromo-4-chloro-3-indolyl β-D-galactopyranoside (X-gal) staining kit (Sigma-Aldrich) with incubation for 2 hours to overnight at 37°C.
Immunohistochemistry
Paraffin-embedded sections were dewaxed and rehydrated in 100%, 95%, and then 70% ethanol then in PBS. Endogenous peroxidase activity was blocked using 3% H2O2 in PBS for 8 minutes followed by a rinse in PBS. Slides were pretreated in boiling 0.1 mM citric acid for 8 minutes in a microwave oven. After preincubating with blocking buffer (1.5% normal goat serum + 1.5% normal donkey serum in PBS with 0.1% Tween 20), sections were incubated with primary antibodies (Table 1). For double staining, two primary antibodies were coincubated at 4°C overnight followed by incubation with secondary antibodies at room temperature for 1 hour. For proliferating cell nuclear antigen (PCNA), tyrosine hydroxylase and the zona fasciculata marker aldoketoreductase 1B7 (AKR1B7), sections were further incubated with avidin-biotin-peroxidase complex for 10 minutes (avidin/biotin enzyme complex (ABC) kit; Vector Laboratory Inc) and then treated using the fluorescence TSA kit (PerkinElmer). Fluorescent images were obtained using a Nikon Eclipse 80i microscope with NIS-Elements AR software (Nikon Instruments) or a Leica DMI4000 confocal microscope with Leica LAS AF software. ImageJ (http://rsb.info.nih.gov/ij/) was used for adjusting the brightness, contrast, orientation, merging, and cell counting. Three sections that were 10 μm or farther apart were studied for each adrenal; the groups contained three or more adrenals from three or more mice.
Table 1.
Antibody Table
| Peptide/Protein Target | Name of Antibody | Manufacturer, Catalog Number | Species | Dilution |
|---|---|---|---|---|
| 20αHSD | Anti-20αHSD | Antibody Research, 591009 | Rabbit polyclonal | 1:500 |
| 3βHSD | Anti-3βHSD | TransGenic Inc, KO607 | Rabbit polyclonal | 1:500 |
| AKR1B7 | Anti-AKR1B7 | Santa Cruz, sc-27763 | Goat polyclonal | 1:200 |
| β-Galactosidase | Anti-β-galactosidase | Abcam, ab9361 | Chicken polyclonal | 1:1000 |
| BrdU | Anti-BrdU | Abcam, ab6326 | Rat monoclonal | 1:1000 |
| CYP11B2 | Anti-CYP11B2 | Gift from Dr C. Gomez-Sanchez | Rabbit polyclonal | 1:500 |
| PCNA | Anti-PCNA | Millipore, CBL407 | Mouse monoclonal | 1:1000 |
| Tyrosine hydroxylase | Anti-TH | Millipore, MAB318 | Mouse monoclonal | 1:1000 |
Abbreviation: BrdU, 5-bromo-2′-deoxyuridine.
PCR analysis of mRNA
Total RNA was isolated from whole adrenal glands using TRIzol reagent (Life Technologies). Samples contained both adrenals from a mouse up to postnatal day (P) 15 or single adrenals after P15. For quantitative PCR (qPCR), groups of three or more samples were analyzed independently. The RNA pellet was dissolved in 11 μL water, and then 8 μL was incubated with oligo-deoxythymidine primers and SuperScript III reverse transcriptase (Life Technologies). First-strand cDNA template was mixed with 250 nM of test gene primers with Power SYBR master mix in a StepOne Plus real-time PCR system (Life Technologies). Relative RNA amounts for test genes were calculated using relative standard curves with Actb as a control, as an average of three or more samples from three mice and expressed relative to levels at P8.
Primer pairs were as follows: Actb forward, 5′-TGC TGT CCC TGT ATG CCT CTG-3′, reverse, 5′-TTG ATG TCA CGC ACG ATT TCC-3′; Akr1c18 (20αHSD) forward, 5′-GAT AGG CCA GGC CAT TCT AAG C-3′, reverse, 5′-CAT TCC CTG GCT TCA GAG ACA C-3′; Thra (TRα1 isoform) forward, 5′-TCT GGC CCA AGC TGC TGA TG-3′, reverse, 5′-GGC CGC CTG AGG CTT TAG AC-3′; and Thrb (TRβ1 isoform) forward, 5′-GCT GGT AGG AAT GTC TGA AGC-3′, reverse, 5′-AGT CTG GAA AGT CTG GGC AC-3′. RT-PCR was used to detect TRβ1-lacZ fusion mRNA from the Thrbb1 allele and TRβ1-specific mRNA from the wild type Thrb allele (Figure 1E) using the primers as follows: common forward, 5′-GCT CGG CCG CAG CTC GCC AGC-3′, wild type reverse, 5′-CCT ACC AGC TTC CAG TCC TGG CC-3′, and lacZ reverse, 5′-CAT TCA GGC TGC GCA ACT GTT G-3′.
Results
A TRβ1-positive population of adrenal cortical cells
TRβ1 and TRβ2 isoforms possess unique N termini but common DNA binding and T3 binding domains, and each is expressed from its own promoter in the 5′ region of the Thrb gene (27, 28). To investigate the cellular expression of TRβ1 in the adrenal, we used Thrb+/b1 (or +/b1) mice that are heterozygous for the Thrbb1 allele, in which lacZ replaces the main TRβ1-specific coding exon of the endogenous Thrb gene. Expression of β-galactosidase was detected in the inner cortex. Expression began as early as embryonic day 14.5 (data not shown) and continued into the neonatal period in a diffuse inner cortical domain (Figure 1A). Signals were also detected in the immature medulla. Expression in the cortex peaked between P15 and P20, at which stage the inner cortical domain became more sharply defined. In males, β-galactosidase expression declined substantially by P28 and was undetectable by P35, whereas in females, the expression persisted into adulthood. No β-galactosidase signal was detected in adrenal sections from wild-type mice at any age examined (see also Figure 2).
Figure 2.
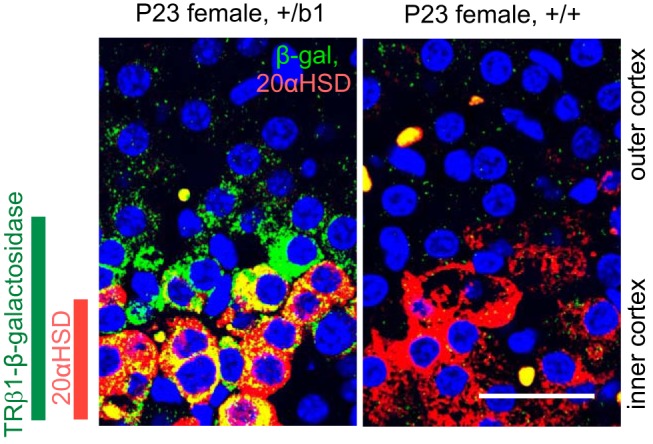
TRβ1-positive zone incorporates 20αHSD-positive zone in the adrenal inner cortex. Confocal image of double-immunofluorescence analysis for β-galactosidase (green) and 20αHSD (red) in adrenal sections of female mice. In +/b1 mice, the TRβ1-positive (β-galactosidase) cell zone is broader than and incorporates the 20αHSD-positive cell zone (orange or yellow, doubly positive cells). Specific β-galactosidase signal was not detected in +/+ control mice. DAPI (blue) marks all cell nuclei. Scale bars, 25 μm.
The expression pattern of β-galactosidase in the inner cortex resembled that of 20αHSD (encoded by Akr1c18), which marks the so-called X-zone in juvenile mice (14). We determined a fuller developmental comparison of TRβ1 and 20αHSD expression because the reported 20αHSD profile is incomplete for the prejuvenile phase (Figure 1B). As expected, 20αHSD immunofluorescence peaked in both males and females at approximately P20 and then displayed sexually dimorphic decline in males but not females (14). However, 20αHSD was not detected during the first postnatal week, unlike β-galactosidase, indicating that TRβ1 represented an earlier marker of the immature cortex.
The developmental profile of TRβ1 and 20αHSD mRNA in the adrenal was confirmed in wild-type mice by qPCR analysis (Figure 1, C and D). TRβ1 and 20αHSD mRNA levels peaked at approximately P21. Levels of both TRβ1 and 20αHSD mRNA declined in males but persisted in females after approximately P28. In contrast, the expression of TRα1 mRNA from the Thra gene was more even and lacked a peak in juvenile mice. TRβ1 expression in the adrenal was confirmed by gel electrophoresis of PCR products, which detected the predicted band sizes for wild-type TRβ1 and fusion TRβ1-lacZ mRNA in +/b1 mice (L. Ng, and D. Forrest, manuscript in preparation) (Figure 1E).
Double-immunofluorescence analysis in +/b1 mice demonstrated that 20αHSD-positive cells represented a subset of β-galactosidase-positive cells in the innermost cortical zone (Figure 2). All 20αHSD-positive cells examined were β-galactosidase positive. The outermost zone of this domain was β-galactosidase positive but 20αHSD negative, suggesting that TRβ1 marks a previously unrecognized inner cortical cell population of which the 20αHSD-positive x-zone is a subpopulation.
T3-induced hypertrophy of inner cortical cells
To test the T3 sensitivity of the inner cortex, T3 was given to mice daily from P14 to P28, encompassing the peak phase of TRβ1 expression. T3 but not saline treatment in female mice induced pronounced cellular hypertrophy in the inner cortex (Figure 3, A and B) and expansion of the β-galactosidase-positive zone (Figure 3, D and E). The mean area occupied by a given number of cells in the inner zone was almost twice as large in T3-treated mice as in saline-treated mice (P < .001), reflecting the increased cell size (Figure 3C). Zonal expansion did not reflect overt changes in cellular proliferation because T3 did not alter the staining pattern of PCNA, a proliferation marker, in the adrenal cortex. In the saline- or T3-treated mice, PCNA-positive cells were detected mainly in the outer zone. The small numbers of PCNA-positive cells in the inner cortex in +/b1 mice were negative for β-galactosidase (Figure 3, F and G) and 20αHSD (data not shown) under saline or T3 treatment. In males, T3 similarly induced hypertrophy of the inner cortex (see Figure 7).
Figure 3.
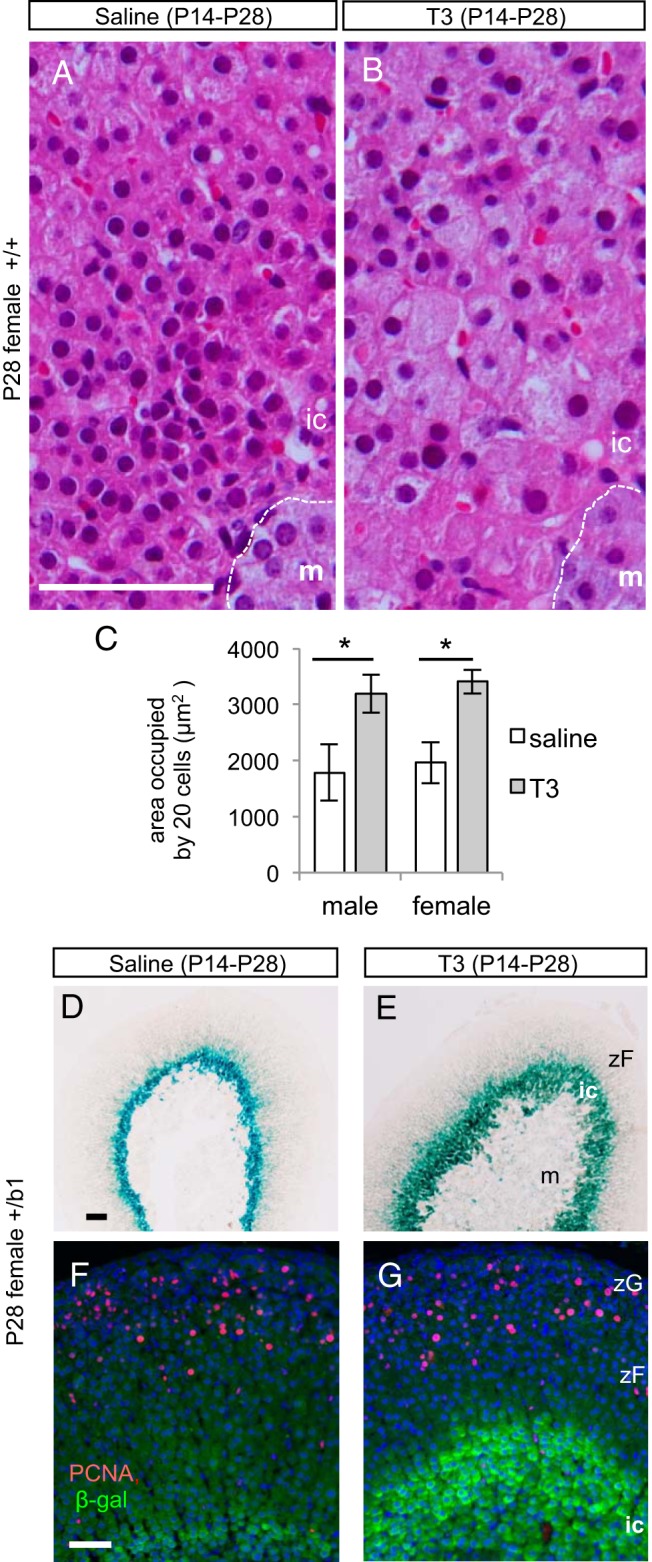
T3-induced hypertrophy of the TRβ1-positive inner cortical zone. A and B, Hematoxylin and eosin-stained paraffin sections, indicating that T3 but not saline treatment from P14 to P28 resulted in cellular hypertrophy in the inner cortex in +/+ female mice. C, Mean areas occupied by 20 cells in the inner cortex in histological sections reflect the increased cell size under T3 treatment. *, P < .001. Areas were measured in the same female as in panels A and B and in parallel groups of +/+ male mice. D and E, X-gal staining showing expansion of the inner cortex in response to T3 but not saline treatment. F and G, Immunofluorescence analysis showing that PCNA (red), an indicator of proliferating cells, was mainly in outer cortical zones in female mice at P28. PCNA showed very little colocalization with β-galactosidase (green) marker for TRβ1 in the inner cortex, regardless of saline or T3 treatment. In panels F and G, cell nuclei were stained by DAPI (blue). Scale bars, 50 μm. ic, inner cortex; m, medulla; zF, zona fasciculata, zG: zona glomerulosa.
Figure 7.

Requirement for Thrb gene for T3 action on the inner cortex. A–L, Male or female mice were treated with T3 or saline from P14 to P28. Immunofluorescence analysis showed that the T3-mediated expansion of the 20αHSD-positive zone (red) in +/+ mice (A–D) was impaired in Thrb−/− (E–H) and Thrbb1/b1 mice (I–L). M, Male mice treated with T3 or saline from P21 to P35. Hematoxylin and eosin-stained paraffin sections showed that T3-mediated hypertrophy in the inner cortex was impaired in Thrb−/− mice. Tyr. Hyd., tyrosine hydroxylase (for medulla, green). DAPI (blue) marks cell nuclei. Scale bars, 50 μm.
T3-induced changes in cortical differentiation markers
We tested whether T3 not only induced hypertrophy of the TRβ1-positive zone but also changed the differentiation status of these inner cortical cells. The inner zone expresses only a few known specific markers, such as 20αHSD and normally does not express markers of mature, outer cortical cell types. In both males and females under T3 treatment from P14 to P28, the expanded inner zone was 20αHSD-positive (Figure 4, A–D, only females shown), indicating that the hypertrophic cells retained an inner cortical cell identity. However, many cells, especially in the innermost zone next to the medulla, also coexpressed the outer cortical, zona fasciculata markers AKR1B7, which metabolizes aldehyde by-products of steroidogenesis (36) (Figure 4, A–D) and the steroidogenic enzyme 3-β-hydroxysteroid dehydrogenase (3βHSD) (Figure 4, E and F). This aberrant expression of AKR1B7 and 3βHSD suggested that T3 stimulated inner cortical cells to become partly like zona fasciculata cells.
Figure 4.
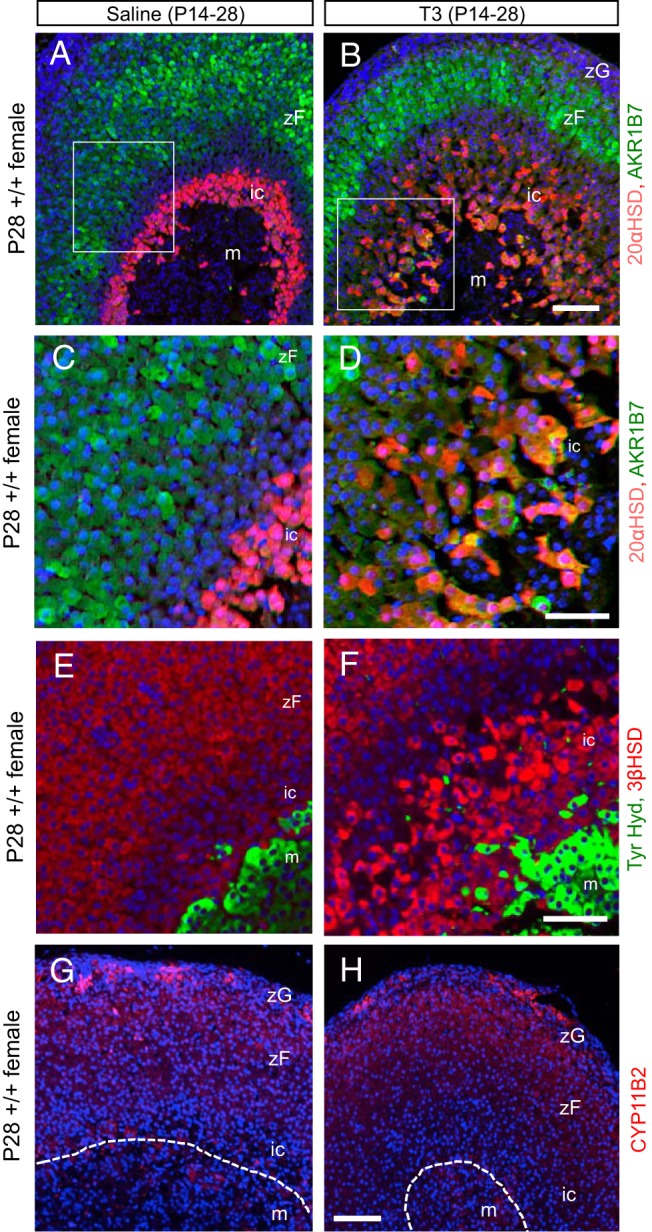
T3-induced changes in cortical marker expression in the inner cortex. A and B, Immunofluorescence analysis showing that T3 treatment expanded the 20αHSD-positive cell zone (red) and in the innermost cortical zone induced coexpression of the outer cortical, zona fasciculata marker AKR1B7 (green, or yellow for double positive). Mice were treated with T3 or saline from P14 to P28. C and D, Representative higher magnification of panels A and B showing cells positive for both 20αHSD and AKR1B7. E and F, Immunofluorescence analysis showing prominent expression of steroidogenic enzyme 3βHSD (red) in the expanded inner zone next to the medulla (green, tyrosine hydroxylase) after T3 treatment. G and H, Immunofluorescence analysis showing normal expression of CYP11B2 in the zona glomerulosa under saline or T3 treatment. In all panels, DAPI (blue) marks cell nuclei. Abbreviations are the same as in Figure 3. Scale bars for panels A, B, G, and H, 100 μm; for C–F, 50 μm.
Immunostaining indicated that the zona glomerulosa marker aldosterone synthase [cytochrome P450 family 11B polypeptide 2 (CYP11B2)] was unchanged by T3 and remained detectable only in the outermost cortical layer in saline- or T3-treated mice (Figure 4, G and H). The expression pattern of cytochrome P450 family 11A polypeptide 1 (CYP11A1), which is normally expressed in all cortical zones, was not altered by T3 and remained detectable in both the outer cortex and expanded inner cortex (data not shown). These findings suggested that T3 did not overtly change the differentiation status of outer cortical cells.
Developmental window of sensitivity to T3
To delineate the window of sensitivity of the inner cortex to T3 and to investigate whether T3 alters the progression of inner cortical development, T3 was given at different postnatal stages. Male mice were studied as this gave the opportunity to determine whether T3 also altered male-specific regression of the inner cortex after weaning. T3 given from P2 to P10 followed by analysis at P10 resulted in premature appearance of a 20αHSD-positive zone compared with saline-treated mice in which almost no 20αHSD-positive cells were present (Figure 5, A and B). T3 given from P14 to P28 expanded the 20αHSD-positive zone (Figure 5, C and D). T3 given from P21 to P35 expanded the 20αHSD-positive zone and notably prevented its regression compared with saline-treated mice in which a 20αHSD zone was no longer detectable (Figure 5, E and F). However, T3 given over a similar period from P14 to P30, followed by withdrawal of T3 treatment until analysis at P49 resulted in a loss of the 20αHSD-positive zone (Figure 5, G and H), suggesting that T3 treatment was continuously required to sustain the presence of the inner cortex. Furthermore, T3 treatment was unable to regenerate a previously regressed 20αHSD-positive zone in older male mice. Thus, T3 given from P35 to P49 failed to induce 20αHSD expression in the inner cortex (Figure 5, I and J).
Figure 5.

T3 influence on the appearance and regression of the 20αHSD-positive inner cortex. Periods of treatment of male mice with T3 or saline (postnatal days) and day of analysis are shown on the left. A and B, T3 resulted in premature appearance of a 20αHSD-positive zone (red) at P10. C and D, T3 stimulated expansion of the 20αHSD-positive zone at P28. E and F, T3 prolonged the presence of the 20αHSD-positive zone at P35. G and H, Removal of T3 treatment after P30 resulted in loss of 20αHSD-positive zone. I and J, T3 given after regression of the 20αHSD-positive zone did not recover a 20αHSD-positive zone. Tyr. Hyd., tyrosine hydroxylase (medulla marker, green). DAPI (blue) marks cell nuclei. Scale bars, 100 μm.
In summary, T3 accelerated formation of a 20αHSD-positive inner cortical zone at premature ages, induced hypertrophy of this zone at juvenile ages, and counteracted male-specific regression of this zone.
T3 sensitivity of the TRβ1-positive zone in adult females
In marked contrast to male mice, females retained a TRβ1-positive inner cortical zone into adulthood (Figure 1A), similar to the persistence of the 20αHSD-positive X-zone in adult female mice (14, 21). However, pregnancy is known to induce regression of the 20αHSD-positive zone in parous females (14). We therefore investigated the retention of the TRβ1-positive inner cortical zone and its T3 sensitivity in nulliparous and parous female mice at 6–8 months of age. In both states, the TRβ1-positive zone persisted and moreover expanded in response to T3 treatment (Figure 6). Thus, adult females retained a TRβ1-positive, T3-sensitive cortical zone, regardless of prior pregnancy. As expected in parous females, 20αHSD expression was largely lost (14) and in addition exhibited a minimal recovery in response to T3.
Figure 6.
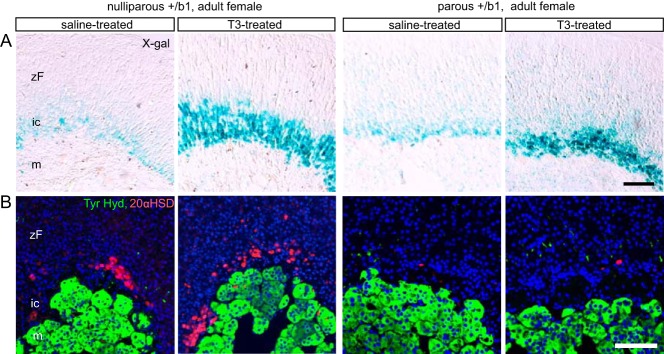
T3 sensitivity of the TRβ1-positive inner cortex in nulliparous or parous adult female mice. A, X-gal staining showing expansion of the TRβ1-positive inner cortex in response to T3 in both nulliparous and parous adult female mice (6 mo old, given T3 or saline for 2 wk before analysis). B, Immunofluorescence analysis showing substantial loss of 20αHSD expression after pregnancy (parous) and the inability of T3 to recover 20αHSD in parous mice. DAPI (blue) marks cell nuclei. Scale bars, 100 μm. ic, inner cortex; m, medulla; zF, zona fasciculata.
Requirement for TRβ1 for T3 action on the inner cortex
To demonstrate a functional requirement for TRβ1 for T3 action on the inner cortex, T3 was given to mice lacking all TRβ isoforms (Thrb−/−) or lacking specifically the TRβ1 isoform (Thrbb1/b1). In wild-type males and females, T3 treatment from P14 to P28 resulted in an expansion of the 20αHSD-positive zone, as expected (Figure 7, A–D) whereas Thrb−/− and Thrbb1/b1 mice of both genders showed little or no expansion of the 20αHSD-positive zone in response to T3 (Figure 7, E–L). Histological analysis demonstrated that Thrb−/− mice were also largely resistant to T3-induced hypertrophy of cells in the inner cortical zone (shown for males in Figure 7M). The results indicated that the TRβ1 isoform is required to mediate actions of T3.
Although TRβ1-deficient mice displayed impaired response to T3, these mice still formed a 20αHSD-positive zone (evident for females in Figure 7, G and K), suggesting that TRβ1 is not required for the gross development of an inner cortical zone.
Discussion
A TRβ1-positive zone of the adrenal inner cortex
TRβ1 displays a unique expression pattern in the adrenal cortex and to our knowledge is the first transcription factor found to be specific for the inner cortical zone. Our data indicate that TRβ1 marks a previously unrecognized cortical cell population and is an intrinsic mediator of T3 action in the inner cortex.
TRβ1-positive cells form a distinct inner zone of the immature cortex and then later display sexually dimorphic regression in males but not females (Figure 8). This zone incorporates but also differs in several ways from the so-called X-zone, described as a histologically dense inner cortical layer (12) that expresses 20αHSD (14). First, 20αHSD-positive cells represent a subset of the broader TRβ1-positive cell population. Second, TRβ1 appears earlier than 20αHSD in the cortex during embryogenesis, suggesting that TRβ1-positive cells are precursors of 20αHSD-positive cells. Third, the persistence of a TRβ1-positive zone in adult females after pregnancy contrasts with the pregnancy-induced loss of a histological X-zone (12) and loss of 20αHSD expression (14), suggesting that TRβ1 marks a novel cell population. Interestingly, it has been reported that thyroid gland extracts induce adrenocortical hypertrophy in species that lack a histologically identifiable X-zone, such as the guinea pig, rabbit, and cat (37). We speculate that in these species, a conserved TRβ1-positive cortical zone, rather than a histologically identified X-zone, may determine sensitivity to T3.
Figure 8.
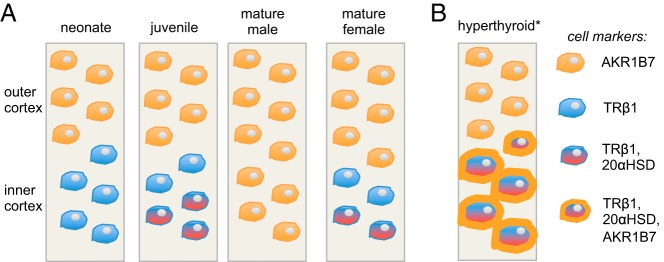
Summary of the sexually dimorphic TRβ1-positive cell zone during remodeling of the adrenal inner cortex. A, TRβ1-positive cells (blue) appear in the inner cortex prior to expression of 20αHSD. At juvenile stages, 20αHSD-positive X-zone cells appear as an inner subset of TRβ1-positive cells (red and blue). In males, TRβ1-positive cells regress at puberty, whereas in females, this cell population persists into adulthood. B, Hyperthyroidism induces cellular hypertrophy and expansion of the inner cortex. Many inner cortical cells also ectopically express the outer cortical marker AKR1B7 (orange outline). *, Response to T3 does not occur in mature male mice after regression of the TRβ1-positive inner cortical zone.
Our finding of a TRβ1-positive cortical zone is likely to explain early reports that thyroxine (38) or thyroid extracts (20) given to mice produce enlargement of a histologically defined cortical X-zone. Poor development of the X-zone also occurs in hyt hypothyroid female mice (39).
TRβ1 is also expressed at low levels in the adrenal medulla (Figure 1), in which its function is currently unclear, although T3 has been shown to influence neuronal differentiation of PC12 rat medullary chromaffin cells in culture (40).
During development, the immature cortex regresses as mature cortical cells generated from outer capsular cells migrate inward and differentiate into mineralocorticoid- and glucocorticoid-producing cells of the zona glomerulosa and zona fasciculata, respectively (1–3, 41). However, our data suggest that TRβ1 has the potential to divert the dead-end fate of the inner zone. Thus, in males, T3 counteracts age-dependent regression of this inner zone. Also, in this zone in both genders, T3 stimulates cytoplasmic expansion and expression of some steroidogenic enzyme markers of the outer cortex (AKR1B7 and 3βHSD). Thus, our findings suggest that inner cortical cells, rather than simply being eliminated, retain a degree of plasticity and may acquire some mature cortical markers. This concept may be consistent with proposals that X-zone cells in castrated male mice provide an alternative source of progenitor cells for restoration of the zona fasciculata (14, 42).
Possible role of the TRβ1-positive zone of the adrenal cortex
TRβ1 is unnecessary for the inner cortical zone to form (Figure 7), indicating that other factors initially direct the generation of this inner zone from the adrenal primordium (43, 44). Instead, we propose that TRβ1 may act more subtly at juvenile ages to modulate the differentiation or function of the inner cortex in response to T3. This proposal is in accord with the postnatal peak of TRβ1expression in the inner cortex. This specialized expression of TRβ1 in the inner cortex contrasts with the more widespread expression in the outer cortical layers of other transcription factors such as GATA6, steroidogenic factor 1, and dosage-sensitive sex reversal-adrenal hypoplasia congenita critical region on the X chromosome, gene 1 (DAX1) that are known to regulate gross development of the cortex (8, 18, 22, 45).
The TRβ1-positive cortical zone becomes most defined in mice at 2–3 weeks of age when there is a developmental peak of T4 and T3 levels in serum shortly before weaning (46, 47). This is a period when T3 promotes maturation of many systems such as the digestive (24) and sensory systems (48) in readiness for a fully autonomous lifestyle. Conceivably, the differentiation status of the residual inner cortical zone contributes in ways we do not yet understand to the maturation of stress responses. In mature females, the persistent TRβ1-positive zone may signify other unknown properties under different physiological conditions in adulthood.
The endocrine function of the transient, inner cortical zone in postnatal mice is enigmatic, although in the embryo, it may produce steroid hormones (4, 13) similar to the human fetal cortical zone (6). In juvenile mice, it is possible that these cells confer distinct functions in steroid hormone metabolism. 20αHSD has a progesterone-catabolizing activity in the ovary that influences parturition and can catabolize 11-deoxycorticosterone, but its role in adrenocortical physiology is unclear (14).
TRβ1 expression in the adrenal cortex suggests a direct link between T3 and the hypothalamic-pituitary-adrenal axis. Various correlations have been reported between thyroid hormone status and secretion of CRH, ACTH, or glucocorticoid. For example, in humans and animal models, hyperthyroidism may be associated with increased secretion (49–52) and hypothyroidism with decreased secretion of glucocorticoid (53, 54), possibly reflecting changes in adrenal function or sensitivity of the adrenal to ACTH. One explanation for these phenomena is that TRβ1 modifies the sensitivity of the adrenal cortex. An early proposal was that the X-zone in mice might act as a reserve tissue for increased demands for glucocorticoid (20). These proposals do not exclude a role for T3 at other levels of the hypothalamic-pituitary-adrenal axis. Thrb−/− mice did not display an overt defect in serum levels of corticosterone (C.J. Huang and D. Forrest, unpublished data), consistent with TRβ1 serving a relatively subtle role in adrenal function. The possibility that thyroid hormone and TRβ1 modulate responses of the hypothalamic-pituitary-adrenal axis to stress deserves further investigation.
The ability of Thrb to express two receptor isoforms in different tissues extends the versatility of the gene and provides a way of determining precise cellular responses. TRβ1 is expressed not only in the adrenal inner cortex but also in the pituitary, cochlea, liver, and other tissues (L. Ng and D. Forrest, manuscript in preparation). In human resistance to thyroid hormone, THRB mutations impair the function of both TRβ isoforms, and this may contribute to the varied tissue symptoms of this syndrome (55, 56). The study of adrenal function in these patients would be of interest in the light of the present findings. The possibility that the administration of thyroid hormone has clinical consequences on human adrenal function may also be considered. Of particular interest may be cases in which T4 is used to treat recently identified patients with mutations in the THRA gene, encoding TRα1 (57–59).
Acknowledgments
We thank Dr Celso Gomez-Sanchez (University of Mississippi Medical Center) for providing the CYP11B2 antibody and Dr Xuefeng Wu (National Institute of Diabetes and Digestive and Kidney Diseases) for assistance.
This work was supported by the intramural research program at the National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health.
Disclosure Summary: The authors have nothing to disclose.
For News & Views see page 1939
- AKR1B7
- aldoketoreductase 1B7
- CYP11B2
- cytochrome P450 family 11B polypeptide 2
- DAPI
- 4,6-Diamino-2-phenylindole
- DAX1
- dosage-sensitive sex reversal-adrenal hypoplasia congenita critical region on the X chromosome, gene 1
- 3βHSD
- 3-β-hydroxysteroid dehydrogenase
- 20αHSD
- 20α-hydroxysteroid dehydrogenase
- P
- postnatal day
- PCNA
- proliferating cell nuclear antigen
- qPCR
- quantitative PCR
- TR
- thyroid hormone receptor
- X-gal
- 5-bromo-4-chloro-3-indolyl β-D-galactopyranoside.
References
- 1. Wood MA, Acharya A, Finco I, et al. Fetal adrenal capsular cells serve as progenitor cells for steroidogenic and stromal adrenocortical cell lineages in M. musculus. Development. 2013;140:4522–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang CC, Miyagawa S, Matsumaru D, Parker KL, Yao HH. Progenitor cell expansion and organ size of mouse adrenal is regulated by sonic hedgehog. Endocrinology. 2010;151:1119–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. King P, Paul A, Laufer E. Shh signaling regulates adrenocortical development and identifies progenitors of steroidogenic lineages. Proc Natl Acad Sci USA. 2009;106:21185–21190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang CC, Shih MC, Hsu NC, Chien Y, Chung BC. Fetal glucocorticoid synthesis is required for development of fetal adrenal medulla and hypothalamus feedback suppression. Endocrinology. 2012;153:4749–4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ishimoto H, Jaffe RB. Development and function of the human fetal adrenal cortex: a key component in the feto-placental unit. Endocr Rev. 2011;32:317–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mesiano S, Jaffe RB. Developmental and functional biology of the primate fetal adrenal cortex. Endocr Rev. 1997;18:378–403. [DOI] [PubMed] [Google Scholar]
- 7. Muscatelli F, Strom TM, Walker AP, et al. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature. 1994;372:672–676. [DOI] [PubMed] [Google Scholar]
- 8. Yu RN, Ito M, Saunders TL, Camper SA, Jameson JL. Role of Ahch in gonadal development and gametogenesis. Nat Genet. 1998;20:353–357. [DOI] [PubMed] [Google Scholar]
- 9. Scheys JO, Heaton JH, Hammer GD. Evidence of adrenal failure in aging Dax1-deficient mice. Endocrinology. 2011;152:3430–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. James LA, Kelsey AM, Birch JM, Varley JM. Highly consistent genetic alterations in childhood adrenocortical tumours detected by comparative genomic hybridization. Br J Cancer. 1999;81:300–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zubair M, Ishihara S, Oka S, Okumura K, Morohashi K. Two-step regulation of Ad4BP/SF-1 gene transcription during fetal adrenal development: initiation by a Hox-Pbx1-Prep1 complex and maintenance via autoregulation by Ad4BP/SF-1. Mol Cell Biol. 2006;26:4111–4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miller EH. A transitory zone in the adrenal cortex which shows age and sex relationships. Am J Anat. 1927;40:251–293. [Google Scholar]
- 13. Sato T. The fine structure of the mouse adrenal X zone. Z Zellforsch Mikrosk Anat. 1968;87:315–329. [DOI] [PubMed] [Google Scholar]
- 14. Hershkovitz L, Beuschlein F, Klammer S, Krup M, Weinstein Y. Adrenal 20α-hydroxysteroid dehydrogenase in the mouse catabolizes progesterone and 11-deoxycorticosterone and is restricted to the X-zone. Endocrinology. 2007;148:976–988. [DOI] [PubMed] [Google Scholar]
- 15. Kananen K, Markkula M, Mikola M, Rainio EM, McNeilly A, Huhtaniemi I. Gonadectomy permits adrenocortical tumorigenesis in mice transgenic for the mouse inhibin α-subunit promoter/simian virus 40 T-antigen fusion gene: evidence for negative autoregulation of the inhibin α-subunit gene. Mol Endocrinol. 1996;10:1667–1677. [DOI] [PubMed] [Google Scholar]
- 16. Spencer SJ, Rabinovici J, Mesiano S, Goldsmith PC, Jaffe RB. Activin and inhibin in the human adrenal gland. Regulation and differential effects in fetal and adult cells. J Clin Invest. 1992;90:142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beuschlein F, Looyenga BD, Bleasdale SE, et al. Activin induces x-zone apoptosis that inhibits luteinizing hormone-dependent adrenocortical tumor formation in inhibin-deficient mice. Mol Cell Biol. 2003;23:3951–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pihlajoki M, Gretzinger E, Cochran R, et al. Conditional mutagenesis of Gata6 in SF1-positive cells causes gonadal-like differentiation in the adrenal cortex of mice. Endocrinology. 2013;154:1754–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deacon CF, Mosley W, Jones IC. The X zone of the mouse adrenal cortex of the Swiss albino strain. Gen Comp Endocrinol. 1986;61:87–99. [DOI] [PubMed] [Google Scholar]
- 20. Gersh I, Grollman A. The nature of the X-zone of the adrenal gland of the mouse. Anat Record. 1939;75:131–153. [Google Scholar]
- 21. Sahut-Barnola I, de Joussineau C, Val P, et al. Cushing's syndrome and fetal features resurgence in adrenal cortex-specific Prkar1a knockout mice. PLoS Genet. 2010;6:e1000980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee FY, Faivre EJ, Suzawa M, et al. Eliminating SF-1 (NR5A1) sumoylation in vivo results in ectopic hedgehog signaling and disruption of endocrine development. Dev Cell. 2011;21:315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ishizuya-Oka A, Hasebe T, Shi YB. Apoptosis in amphibian organs during metamorphosis. Apoptosis. 2010;15:350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sirakov M, Skah S, Nadjar J, Plateroti M. Thyroid hormone's action on progenitor/stem cell biology: new challenge for a classic hormone? Biochim Biophys Acta. 2013;1830:3917–3927. [DOI] [PubMed] [Google Scholar]
- 25. Weinberger C, Thompson CC, Ong ES, Lebo R, Gruol DJ, Evans RM. The c-erb-A gene encodes a thyroid hormone receptor. Nature. 1986;324:641–646. [DOI] [PubMed] [Google Scholar]
- 26. Hodin RA, Lazar MA, Wintman BI, et al. Identification of a thyroid-hormone receptor that is pituitary-specific. Science. 1989;244:76–79. [DOI] [PubMed] [Google Scholar]
- 27. Wood WM, Dowding JM, Haugen BR, Bright TM, Gordon DF, Ridgway EC. Structural and functional characterization of the genomic locus encoding the murine β2 thyroid hormone receptor. Mol Endocrinol. 1994;8:1605–1617. [DOI] [PubMed] [Google Scholar]
- 28. Jones I, Ng L, Liu H, Forrest D. An intron control region differentially regulates expression of thyroid hormone receptor β2 in the cochlea, pituitary, and cone photoreceptors. Mol Endocrinol. 2007;21:1108–1119. [DOI] [PubMed] [Google Scholar]
- 29. Frankton S, Harvey CB, Gleason LM, Fadel A, Williams GR. Multiple messenger ribonucleic acid variants regulate cell-specific expression of human thyroid hormone receptor β1. Mol Endocrinol. 2004;18:1631–1642. [DOI] [PubMed] [Google Scholar]
- 30. Hodin RA, Lazar MA, Chin WW. Differential and tissue-specific regulation of the multiple rat c-erbA messenger RNA species by thyroid hormone. J Clin Invest. 1990;85:101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ng L, Hurley JB, Dierks B, et al. A thyroid hormone receptor that is required for the development of green cone photoreceptors. Nat Genet. 2001;27:94–98. [DOI] [PubMed] [Google Scholar]
- 32. Abel ED, Boers ME, Pazos-Moura C, et al. Divergent roles for thyroid hormone receptor beta isoforms in the endocrine axis and auditory system. J Clin Invest. 1999;104:291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murray MB, Zilz ND, McCreary NL, MacDonald MJ, Towle HC. Isolation and characterization of rat cDNA clones for two distinct thyroid hormone receptors. J Biol Chem. 1988;263:12770–12777. [PubMed] [Google Scholar]
- 34. Wood WM, Ocran KW, Gordon DF, Ridgway EC. Isolation and characterization of mouse complementary DNAs encoding α and β thyroid hormone receptors from thyrotrope cells: the mouse pituitary-specific β2 isoform differs at the amino terminus from the corresponding species from rat pituitary tumor cells. Mol Endocrinol. 1991;5:1049–1061. [DOI] [PubMed] [Google Scholar]
- 35. Forrest D, Hanebuth E, Smeyne RJ, et al. Recessive resistance to thyroid hormone in mice lacking thyroid hormone receptor β: evidence for tissue-specific modulation of receptor function. EMBO J. 1996;15:3006–3015. [PMC free article] [PubMed] [Google Scholar]
- 36. Lambert-Langlais S, Pointud JC, Lefrancois-Martinez AM, et al. Aldo keto reductase 1B7 and prostaglandin F2α are regulators of adrenal endocrine functions. PLoS One. 2009;4:e7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Herring PT. The influence of the thyroids on the functions of the suprarenals. Endocrinology. 1920;4:577–599. [Google Scholar]
- 38. Preston MI. Effects of thyroxin injections on the suprarenal glands of the mouse. Endocrinology. 1928;12:323–334. [Google Scholar]
- 39. Shire JG, Beamer WG. Adrenal changes in genetically hypothyroid mice. J Endocrinol. 1984;102:277–280. [DOI] [PubMed] [Google Scholar]
- 40. Munoz A, Wrighton C, Seliger B, Bernal J, Beug H. Thyroid hormone receptor/c-erbA: control of commitment and differentiation in the neuronal/chromaffin progenitor line PC12. J Cell Biol. 1993;121:423–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zubair M, Parker KL, Morohashi K. Developmental links between the fetal and adult zones of the adrenal cortex revealed by lineage tracing. Mol Cell Biol. 2008;28:7030–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Freedman BD, Kempna PB, Carlone DL, et al. Adrenocortical zonation results from lineage conversion of differentiated zona glomerulosa cells. Dev Cell. 2013;26:666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim AC, Barlaskar FM, Heaton JH, et al. In search of adrenocortical stem and progenitor cells. Endocr Rev. 2009;30:241–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77:481–490. [DOI] [PubMed] [Google Scholar]
- 45. Kiiveri S, Liu J, Westerholm-Ormio M, et al. Differential expression of GATA-4 and GATA-6 in fetal and adult mouse and human adrenal tissue. Endocrinology. 2002;143:3136–3143. [DOI] [PubMed] [Google Scholar]
- 46. Campos-Barros A, Amma LL, Faris JS, Shailam R, Kelley MW, Forrest D. Type 2 iodothyronine deiodinase expression in the cochlea before the onset of hearing. Proc Natl Acad Sci USA. 2000;97:1287–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hadj-Sahraoui N, Seugnet I, Ghorbel MT, Demeneix B. Hypothyroidism prolongs mitotic activity in the post-natal mouse brain. Neurosci Lett. 2000;280:79–82. [DOI] [PubMed] [Google Scholar]
- 48. Ng L, Kelley MW, Forrest D. Making sense with thyroid hormone—the role of T(3) in auditory development. Nat Rev Endocrinol. 2013;9:296–307. [DOI] [PubMed] [Google Scholar]
- 49. Johnson EO, Kamilaris TC, Calogero AE, Gold PW, Chrousos GP. Experimentally induced hyperthyroidism is associated with activation of the rat hypothalamic-pituitary-adrenal axis. Eur J Endocrinol. 2005;153:177–185. [DOI] [PubMed] [Google Scholar]
- 50. Ramspott S, Hartmann K, Sauter-Louis C, Weber K, Wehner A. Adrenal function in cats with hyperthyroidism. J Feline Med Surg. 2012;14:262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lo MJ, Kau MM, Chen YH, et al. Acute effects of thyroid hormones on the production of adrenal cAMP and corticosterone in male rats. Am J Physiol Endocrinol Metab. 1998;274:E238–E245. [DOI] [PubMed] [Google Scholar]
- 52. Gallaghe TF, Fukushim DK, Weitzman ED, et al. Hyperthyroidism and cortisol secretion in man. J Clin Endocrinol Metab. 1972;34:919–927. [DOI] [PubMed] [Google Scholar]
- 53. Tohei A. Studies on the functional relationship between thyroid, adrenal and gonadal hormones. J Reprod Dev. 2004;50:9–20. [DOI] [PubMed] [Google Scholar]
- 54. Johnson EO, Calogero AE, Konstandi M, Kamilaris TC, La Vignera S, Chrousos GP. Effects of short- and long-duration hypothyroidism on hypothalamic-pituitary-adrenal axis function in rats: in vitro and in situ studies. Endocrine. 2012;42:684–693. [DOI] [PubMed] [Google Scholar]
- 55. Chatterjee V, Beck-Peccoz P. Resistance to thyroid hormone. In: deGroot L, Jameson J, eds. Endocrinology. 4th ed Philadelphia: WB Saunders Co; 2001:1609–1615. [Google Scholar]
- 56. Refetoff S, Weiss RE, Usala SJ. The syndromes of resistance to thyroid hormone. Endocr Rev. 1993;14:348–399. [DOI] [PubMed] [Google Scholar]
- 57. van Mullem AA, Chrysis D, Eythimiadou A, et al. Clinical phenotype of a new type of thyroid hormone resistance caused by a mutation of the TRα1 receptor: consequences of LT4 treatment. J Clin Endocrinol Metab. 2013;98:3029–3038. [DOI] [PubMed] [Google Scholar]
- 58. Moran C, Schoenmakers N, Agostini M, et al. An adult female with resistance to thyroid hormone mediated by defective thyroid hormone receptor α. J Clin Endocrinol Metab. 2013;98:4254–4261. [DOI] [PubMed] [Google Scholar]
- 59. Moran C, Agostini M, Visser WE, et al. Resistance to thyroid hormone caused by a mutation in thyroid hormone receptor (TR)α1 and TRα2: clinical, biochemical, and genetic analyses of three related patients. Lancet Diabetes Endocrinol. 2014;2:619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]