

Abstract
Estrogens dramatically dilate numerous vascular beds with the greatest response in the uterus. Endogenous hydrogen sulfide (H2S) is a potent vasodilator and proangiogenic second messenger, which is synthesized from L-cysteine by cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE). We hypothesized that estrogen replacement therapy (ERT) selectively stimulates H2S biosynthesis in uterine artery (UA) and other systemic arteries. Intact and endothelium-denuded UA, mesenteric artery (MA), and carotid artery (CA) were obtained from ovariectomized nonpregnant ewes (n = 5/group) receiving vehicle or estradiol-17β replacement therapy (ERT). Total RNA and protein were extracted for measuring CBS and CSE, and H2S production was determined by the methylene blue assay. Paraffin-embedded UA rings were used to localize CBS and CSE proteins by immunofluorescence microscopy. ERT significantly stimulated CBS mRNA and protein without altering CSE mRNA or protein in intact and denuded UA. Quantitative immunofluorescence microscopic analyses showed CBS and CSE protein localization in endothelium and smooth muscle and confirmed that ERT stimulated CBS but not CSE protein expression in UA endothelium and smooth muscle. ERT also stimulated CBS, but not CSE, mRNA and protein expression in intact and denuded MA but not CA in ovariectomized ewes. Concomitantly, ERT stimulated UA and MA but not CA H2S production. ERT-stimulated UA H2S production was completely blocked by a specific CBS but not CSE inhibitor. Thus, ERT selectively stimulates UA and MA but not CA H2S biosynthesis by specifically up-regulating CBS expression, implicating a role of H2S in estrogen-induced vasodilation and postmenopausal women's health.
Estrogens are potent vasodilators that cause blood flow to rise in selected organs throughout the body with the greatest response occurring in reproductive tissues, especially the uterus (1–4). In ovariectomized (OVX) nonpregnant ewes, daily estradiol-17β (E2β) treatment increases basal uterine blood flow (UBF) 30%–40% over 6–7 days. This increase in UBF occurs with increases in cardiac output and heart rate, whereas mean arterial pressure remains unchanged (2–4) and is associated with decreased responses to vasoconstrictors (5, 6). In addition, acute E2β exposure provokes an even more robust rise in UBF, up to 10-fold, within 90–120 minutes after a bolus iv injection of 1 μg/kg E2β (3, 4, 6–8). The vasodilatory effect of estrogens is of major physiological significance because: 1) circulating estrogen levels are significantly elevated during the follicular phase of the ovarian cycle and pregnancy to cause UBF to rise (9, 10); 2) during pregnancy rise in UBF provides all the support for fetal development and survival (2, 11, 12); and 3) insufficient rise in UBF during pregnancy results in intrauterine growth restriction (13), preeclampsia (14), and many other pregnancy disorders (15). This insufficient rise in UBF increases the risk of infant morbidity and mortality, is a significant contributor to maternal mortality, and increases susceptibility to cardiovascular and other diseases for both mother and neonate later in life (12, 13).
Enhanced nitric oxide (NO) production, via endothelial NO synthase (eNOS) in uterine artery (UA) endothelium, has been identified as a major contributor to the estrogen-induced uterine vasodilatation. Blockade of local UA NO production by L-NG-nitroarginine methyl ester (L-NAME) dose dependently inhibited estrogen-induced uterine vasodilatation in animals (16, 17). However, blockade of UA NO production by L-NAME only inhibits approximately 65% the E2β-induced UBF response in OVX nonpregnant (16, 17) and intact follicular phase (10) sheep. Thus, other vasodilators derived from UA endothelium and/or the smooth muscle, in addition to endothelium/NO, are likely to play a role in estrogen-induced uterine vasodilatation. To this end, prostacyclin is unlikely to play a key role as early studies have shown that estrogen-induced UBF in nonpregnant sheep is not affected by systemic infusion of indomethacin (17), supporting the notion that other components are involved.
Hydrogen sulfide (H2S) has long been known to be a toxic gas at high doses. However, because of the original discovery of its physiological action in the brain in 1996 (18), it has demonstrated that H2S possesses homologous biological and physiological functions to other “gasotransmitter” molecules, such as NO and carbon monoxide (19). H2S potently relaxes rat aortic vessels by activating KATP channels, which is confirmed by inhibition with the KATP channel blocker glibenclamide (20, 21). Akin to NO, exogenous and endogenous H2S promotes angiogenesis in vitro and in vivo through activation of KATP channels, protein kinase v-akt murine thymoma viral oncogene homolog 1 (Akt1) in endothelial cells (22–24), and interactions with NO signaling through eNOS activation in endothelial cells (24).
Endogenous H2S is primarily synthesized by cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) (20, 25, 26). Both enzymes produce H2S from L-cysteine: CBS via a β-replacement reaction with a variety of thiols and CSE by disulfide elimination followed by reaction with various thiols (24, 26). Because of the potent vasodilatory effects of H2S in many other vascular beds (24–27), it is highly likely that H2S also may have a critical role in the uterine circulation. A recent report has suggested that sc estrogen replacement therapy (ERT) (30 μg E2β · kg−1 · d−1) for 12 weeks in OVX rats resulted in enhanced endogenous H2S production via CSE up-regulation in the myocardium, which may provide a new avenue for the cardiovascular protective effects of estrogens (28). However, whether the H2S biosynthesis system exists and whether it is regulated by estrogen in the UA and systemic vasculatures have not been examined. Therefore, the objectives of the present study were to examine the effects of ERT on the expression of CSE and CBS expression and H2S production in UA and systemic mesenteric artery (MA) and carotid artery (CA) to test a hypothesis that ERT selectively stimulates H2S biosynthesis in UA and some systemic arteries.
Materials and Methods
Chemicals and antibodies
E2β, O-(carboxymethyl)hydroxylamine hemihydrochloride (CHH), and all other chemicals, unless specified, were from Sigma. β-Cyano-L-alanine (BCA) was from Cayman Chemical. Anti-β-actin monoclonal antibody was from Ambion. Anti-CBS monoclonal antibody and anti-CSE monoclonal antibody used were purchased from Santa Cruz Biotechnology, Inc. Anti-platelet endothelial cell adhesion molecule (PECAM1 or CD31) antibody was from Dako. Antibiotin antibody was from Cell Signaling Technology. Prolong Gold antifade reagent with 4′,6-diamidino-2-phenylindole (DAPI), Alexa Fluor 488 goat antimouse IgG, and Alexa Fluor 568 goat antimouse IgG were from Invitrogen.
Animal preparation and estrogen replacement treatment
Mixed Western breed ewes were OVX via midventral laparotomy as detailed previously (4, 17, 29). This particular animal use protocol was approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center. After at least 4–5 days of recovery, the ewes received vehicle (Veh) or ERT (n = 5/group) for 5–6 days. The ERT-treated ewes received a bolus injection of E2β (1 μg kg−1 · d−1, iv) over 1–2 minutes each morning, whereas UBF, mean arterial pressure, and heart rate were monitored continuously beginning 30 minutes before E2β injection and continuing for 120 minutes. Animals receiving bolus injection of ethanol and a saline flush of 0.7–0.9 mL, served as OVX/Veh controls, which alone produce no effect. This E2β treatment regime elicited a progressive increase in UBF that begins 30 minutes after treatment and reached its maximum at 90–120 minutes, increased mammary blood flow approximately 2-fold at 90–120 minutes, and resulted in plasma estrogen levels resembling those in women on ERT (6, 30), and those in sheep at the onset of parturition (9). E2β was dissolved in 95% ethanol and stored at 4°C at a stock of 1 mg/mL. The E2β dose and timed tissue collection were based on eNOS expression and hemodynamic responses as well as blood levels of E2β achieved, as detailed in the previous studies (3, 4, 6, 7, 11, 29, 31). The next morning after the last E2β infusion, the animals were killed with phenobarbital (10 mL of 100 mg/mL) to collect various intact artery tissues, including UA, MA, and CA. In addition, endothelium-denuded arteries were prepared from longitudinally opened artery segments whose endothelium was removed with a soft-tipped cotton swab. All samples were snap-frozen in liquid nitrogen and stored at −80°C until studied as described below.
RNA extraction, reverse transcription, and real-time quantitative polymerase chain reaction (qPCR)
Total RNAs were extracted from intact and endothelium-denuded artery segments using TRIzol reagent (Invitrogen) following the manufacturer's instructions. RNA was quantified by OD260/280. The first-strand cDNA was synthesized by reverse transcription with random primers and AMV Reverse Transcriptase (Promega) with 2-μg RNA as previously described (32). The cDNAs so derived were used for quantify CBS and CSE mRNAs by quantitative real-time PCR (run in triplicate) with gene-specific primers as listed in Table 1. Each real-time PCR reaction contained 7.5 μL of RT2 Real-Time PCR SYBR Green Master Mix (SABiosciences), 0.45 μL of forward and reverse primers (10μM), 3 μL of cDNA template in with 15-μL final volume in nuclease-free H2O. A StepOnePlus Real-Time PCR system (Life Technologies) was used with a 3-step thermal cycling program: initial denaturing at 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds, 55°C for 30 seconds, and 72°C for 30 seconds, and a melting curve program (95°C, 15 s; 60°C, 1 min, optics off; 60°C–95°C at 2°C/min, optics on). Comparative threshold cycle (CT) method (ΔΔCT method) was used to calculate relative CBS and CSE mRNA levels with the use of human ribosomal protein L19 as the internal reference control.
Table 1.
Primers Used for qPCR Analysis
| Gene | Sequence (5′→3′) | Amplicon |
|---|---|---|
| CSE | FWD, TTGTATGGATGATGTGTATGGAAGG | 141 bp |
| REV, CCAAACAAGCTTGGTTTCTGGTG | ||
| CBS | FWD, TGAGATTGTGAGGACGCCCAC | 177 bp |
| REV, TCACACTGCTGCAGGATCTC | ||
| L19 | FWD, AGACCCCAATGAGACCAATG | 129 bp |
| REV, GTGTTTTTCCGGCATCGAGC |
SDS-PAGE and Western blot analysis
Protein extracts were prepared from homogenates of artery segments in a nondenaturing buffer containing protease cocktail (32, 33). Protein concentrations were determined by using the Pierce BCA Protein Assay kit (Pierce Biotechnology). Proteins (10 μg/sample) were separated on 10% SDS-PAGE and transferred onto polyvinylidene fluoride membranes. The membranes were subjected to immunoblotting with mouse anti-CBS or anti-CSE monoclonal antibodies (0.4 μg/mL), followed by horseradish peroxidase-conjugated goat antimouse (0.01 μg/mL). Bound antibodies on membranes were visualized by using the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) and digital images were captured using a ChemiImager Imaging System (Alpha Innotech). The membranes were striped and reprobed with mouse anti-β-actin antibody (0.1 μg/mL) for normalizing sample loading. CBS and CSE proteins were quantified by NIH ImageJ software and presented as fold changes over controls.
Immunofluorescence microscopy and image analysis
The cellular specific expression of CBS and CSE proteins in sheep UA was determined by immunofluorescence microscopy with the use of archived UA samples from a sheep model of ERT as described in our previous studies (3, 34, 35). Tissue samples were collected from OVX ewes receiving Veh (OVX/Veh) or E2β treatment (OVX/ERT) as detailed previously (3, 6, 34). The animal use protocol was approved by the Institutional Animal Care and Use Committee at the University of Wisconsin-Madison. Briefly, paraffin-embedded UA rings from OVX nonpregnant ewes receiving E2β treatment (initial loading dose of 5 μg/kg, followed by 6 μg kg−1 · d−1 for 10 d) or Veh control were studied. Sections were baked at 55°C for 1 hour, deparaffinized in xylene, and rehydrated by passing through gradient ethanol. Sections were incubated in 0.05% trypsin to unmask antigens at room temperature for 30 minutes. Autofluorescence was quenched by washing the sections (3 × 20 min) with 300mM glycine in PBS; nonspecific binding was blocked by incubating with PBS containing 1% BSA, 0.125% saponin, and 1% gelatin at room temperature for 30 minutes. Sections were then incubated with anti-CD31 (5 μg/mL) overnight at 4°C. After 3 5-minute washes in PBS, the sections were incubated with Alexa Fluor 568-conjugated goat antimouse IgG (2 μg/mL) at room temperature for 1 hour. After 3 20-minute washes in PBS, sections were then incubated with 1 μg/mL of anti-CSE or anti-CBS antibodies at room temperature for 2 hours, followed by incubation with Alexa Fluor 488-conjugated goat antimouse IgG (2 μg/mL) at room temperature for 1 hour. After 3 20-minute PBS washes, the sections were mounted with Prolong Gold antifade reagent (Invitrogen) containing DAPI for labeling cell nuclei.
Samples were examined under a Leica fluorescence microscope (Leica Corp) and digital images were acquired using a charge-coupled device camera with the SimplePCI image analysis software (Hamamatsu Corp). The images were used to determine relative levels of CBS and CSE proteins by quantifying mean green fluorescence intensity by using NIH ImageJ software. For both OVX/Veh and OVX/ERT groups, CBS and CSE levels were averaged from data from 40 CD31-positive cells (endothelial cells) and 40 CD31-negative cells (smooth muscle cells) of each image, with 5–6 images per animal, and 3 animals per group. Cell bodies were outlined using the “region of interest” selection tool and “mean gray value” was recorded for a cell. The average mean gray value of cells from negative control without primary antibody accounted for autofluorescence and nonspecific background, which was subtracted from all counts generated from specific antibody-treated samples. CBS and CSE protein levels were presented as fold change in the average fluorescence intensity of OVX/Veh animals (Fig. 2).
Figure 2.
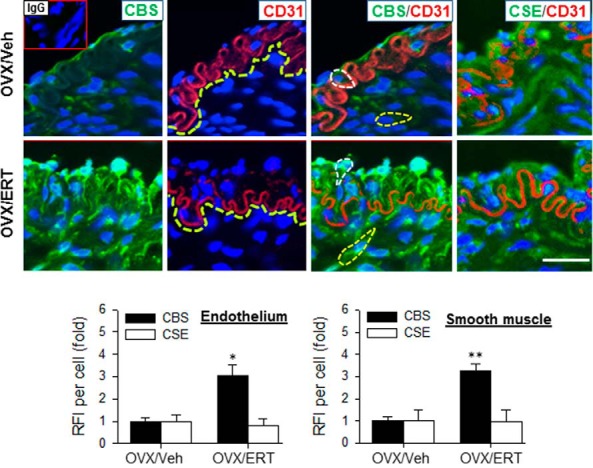
Immunofluorescence localization of CBS and CSE protein in UA from OVX ewes receiving Veh and E2β replacement therapy (ERT). Sections were labeled with specific primary antibodies against CBS (green) or CSE and endothelial cell marker CD31 (red) with nuclei stained with DAPI (blue). Negative control slides were treated similarly but without primary antibodies or with antimouse IgG; no positive staining was detected on these slides (inset of upper left image). Images were analyzed for relative fluorescence intensity (RFI). Representative outlines of cell bodies used for analysis are outlined (left panels) and border of CD31-positive endothelial cells was also indicated (center panels). Data (mean ± SEM) are from 3–5 different ewes/group. *, P < .05; **, P < .01. Scale bar, 50 μm.
Table 2.
Antibody Table (all used are listed)
| Peptide/Protein Target | Antigen Sequence (If Known) | Name of Antibody | Manufacturer, Catalog Number, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|
| CBS | Residues 101–400 of human CBS | CBS (A-2) | Santa Cruz Biotechnology, Inc; SC-133208 | Mouse monoclonal IgG2 | 1:500 (WB), 1:200 (IF) |
| Cystathionine γ-lyase | Residues 43–75 of human CTH | CTH (F-1) | Santa Cruz Biotechnology, Inc; SC-374249 | Mouse monoclonal IgG1 | 1:500 (WB), 1:200 (IF) |
| β-Actin | DDDIAALVIDNGSGK | β-Actin | Ambion; AM4302 | Mouse monoclonal IgG1 | 1:10 000 (WB) |
| Antimouse | HRP-conjugated antimouse | Fisher Scientific; PI32430 | Goat antimouse (H + L) | 1:1000 (WB) | |
| CD31 | CD31 | Dako; M0823 | Mouse monoclonal IgG1 | 1:40 (IF) | |
| Antimouse | Alexa Fluor 488 goat antimouse | Invitrogen; A11001 | Goat antimouse IgG (H + L) | 1:1000 (IF) | |
| Antimouse | Alexa Fluor 555 goat antimouse | Invitrogen; A11004 | Goat antimouse IgG (H + L) | 1:1000 (IF) |
Methylene blue assay for H2S production
Tissue H2S production was determined by the methylene blue assay as described previously (20, 36), with minor modifications. Briefly, segments of frozen endothelial denuded and intact artery tissue were homogenized in ice-cold 50mM potassium phosphate buffer, pH 8. The reaction mixture contained: 50mM potassium phosphate buffer pH 8.0, 10mM L-cysteine, and 2mM pyridoxal 5′-phosphate. Microtubes (2 mL) were used as the center wells; each contained 0.3 mL of 1% zinc acetate as trapping solution and a filter paper of 0.5 × 1.5 cm to increase the air/liquid contacting surface. The reaction was performed in 12-mL test tubes. The tubes containing the reaction mixture and center wells were flushed with N2 before being sealed with a double layer of Parafilm. The reaction was initiated by transferring the tubes from ice to a 37°C shaking water bath. After incubating at 37°C for 90 minutes, the reaction was stopped by adding 0.5 mL of 50% trichloroacetic acid. The tubes were sealed again and incubated at 37°C for another 60 minutes to ensure a complete trapping of the H2S released from the mixture. Subsequently, 0.05 mL of 20mM N,N-dimethyl-p-phenylenediamine sulfate in 7.2M HCl was added immediately after the addition of 0.05-mL 30mM FeCl3 in 1.2M HCl. The absorbance of the resulting solution at 670 nm was measured after 20 minutes. The H2S concentration was calculated based on a calibration curve generated from NaHS solutions. For CBS and CSE inhibition experiments, their respective inhibitor CHH or BCA, were added separately or in combination (final concentration, 2mM) to the reaction mixtures before initiating the methylene blue assay.
Statistical analysis
Data are presented as means ± SEM (n = 3–5 ewes/group) and analyzed by one- or two-way ANOVA, followed by Bonferroni test for multiple comparisons using Sigma Stat 3.5 (Systat Software, Inc). Student's paired t test was used for comparison of data between 2 groups. Significance was defined as P < .05, unless higher statistical power is indicated in the figure legends.
Results
Effects of ERT on UA CBS and CSE mRNA and protein expression
Because endogenous H2S is mainly synthesized by CBS and CSE (20, 26), we determined whether ERT stimulates ovine UA CBS and CSE expression. Real-time qPCR analysis showed that the mRNA levels of CBS in intact and denuded UA of OVX/ERT ewes were 5.04 ± 1.0- and 5.60 ± 0.9-fold greater (P < .01), respectively, than that of OVX/Veh ewes. CSE mRNA levels in intact and denuded UA from OVX/ERT and OVX/Veh ewes (Figure 1A) did not differ. Consistent with mRNA data, the levels of CBS protein determined by immunoblotting in intact and denuded UA of OVX/ERT ewes were 3.51 ± 0.5-fold (P < .01) and 4.67 ± 0.1-fold (P < .001) greater, respectively, than that of OVX/Veh ewes. CSE protein levels in both intact and denuded UA did not differ (Figure 1B).
Figure 1.
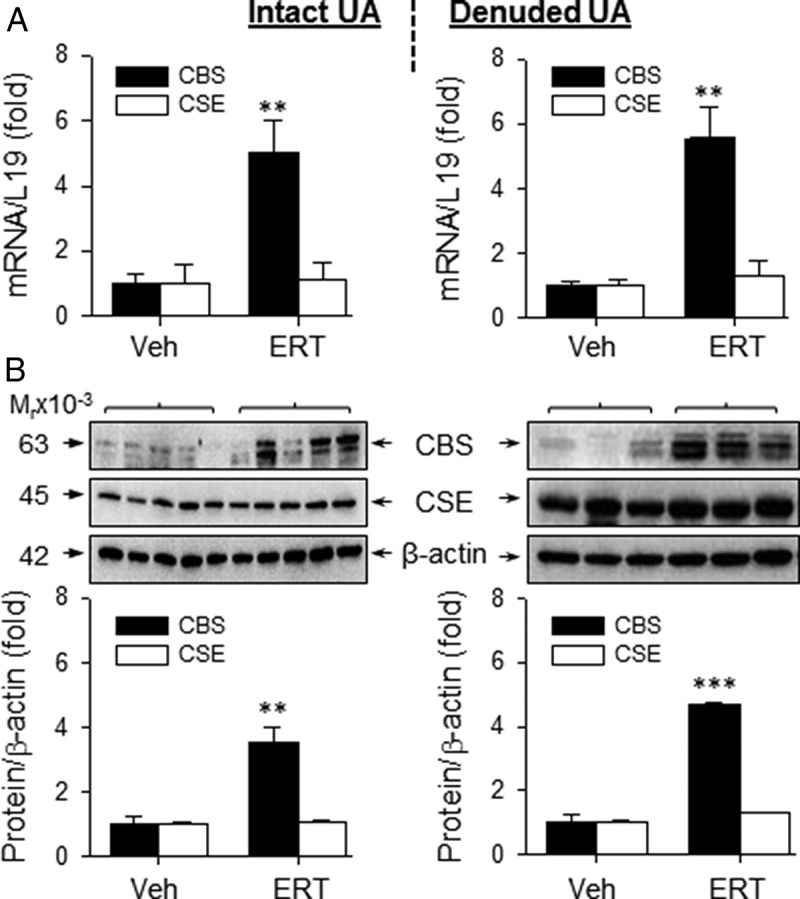
CBS and CSE mRNA and protein expression in UAs from OVX ewes receiving Veh and E2β replacement therapy (ERT) ewes. CBS and CSE mRNAs in intact (top, left) and denuded (top, right) UA. CBS and CSE protein analysis in intact (bottom, left) and denuded (bottom, right) UA. Data (mean ± SEM) are from 3–5 ewes/group. **, P < .01; ***, P < .001 compared with Veh.
Localization of CBS and CSE proteins in UA
We then determined the cell-specific expression of CBS and CSE proteins in UA by immunofluorescence microscopy. CBS and CSE proteins appeared to be coexpressed with CD31 in endothelial cells, but they did not coexist within the same subcellular location inside these cells in OVX ewes. In OVX/Veh ewes, CBS and CSE were associated with the apical surface and CD31 were mainly seen at the basement membrane. Weak green fluorescence labeling of CBS protein was observed in the endothelium (CD31-positive) and smooth muscle cells (CD31-negative) in OVX/Veh ewes. ERT treatment dramatically increased the green fluorescence intensity in both endothelium and smooth muscle; the average green immunofluorescence intensities in UA endothelial and smooth muscle cells of OVX/ERT ewes were 3.1 ± 0.5-fold (P < .05) and 3.3 ± 0.3-fold (P < .01) greater, respectively, than that of OVX/Veh ewes (Figure 2). Of note, ERT also altered the subcellular localization of CBS in endothelial cells from the apical surface to the basement membrane. Strong CSE immunofluorescence labeling was also observed in UA endothelial and smooth muscle cells in both OVX/Veh and OVX/ERT ewes, the green fluorescence intensities and subcellular localization did not differ between the groups.
Effects of ERT on CBS and CSE mRNA and protein expression in systemic arteries
In order to determine whether the effects of ERT on CBS and CSE expression were specific to UA, we evaluated the effects of ERT on H2S biosynthesis in “systemic” MA and CA from OVX/Veh and OVX/ERT ewes. The mRNA levels of CBS in intact and denuded MA of OVX/ERT ewes were 4.05 ± 1.2-fold (P < .01) and 5.37 ± 1.4-fold (P < .05) greater, respectively, than that of OVX/Veh ewes. The mRNA levels of CSE did not differ in either intact or denuded MA of OVX/ERT vs OVX/Veh ewes (Figure 3A). Similarly, the levels of CBS protein in intact and denuded MA of OVX/ERT ewes were 3.05 ± 0.3-fold (P < .01) and 6.15 ± 0.2-fold (P < .001) greater, respectively, than that of OVX/Veh ewes. The protein levels of CSE did not differ (P > .05) in either intact or denuded MA of OVX/ERT vs OVX/Veh ewes (Figure 3B). In contrast to both UA and MA, there were no differences in the mRNA or protein levels of CBS or CSE in either intact or denuded CA from OVX/ERT and OVX/Veh ewes (Figure 4). In comparison of these 3 vascular beds, ERT seemed to be more potent in stimulating CBS mRNA in UA vs MA, but less effective in stimulating CBS protein in UA than MA. These differences, however, did not reach statistical significance (Figure 5, A and B).
Figure 3.
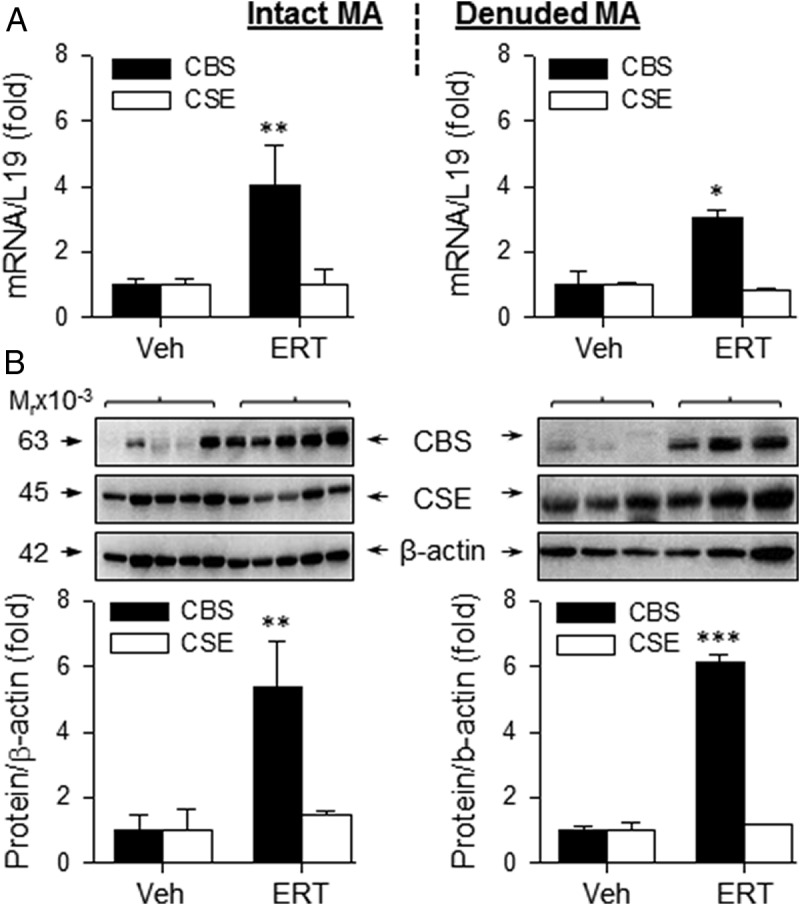
CBS and CSE mRNA and protein expression in MA from OVX ewes receiving Veh and E2β replacement therapy (ERT). Relative mRNA levels of CBS and CSE in intact (top, left) and denuded (top, right) MA. CBS and CSE proteins in intact (bottom, left) and denuded (bottom, right) MA. Data (mean ± SEM) are from 3–5 different ewes/group. *, P < .05; **, P < .01; ***, P < .001.
Figure 4.
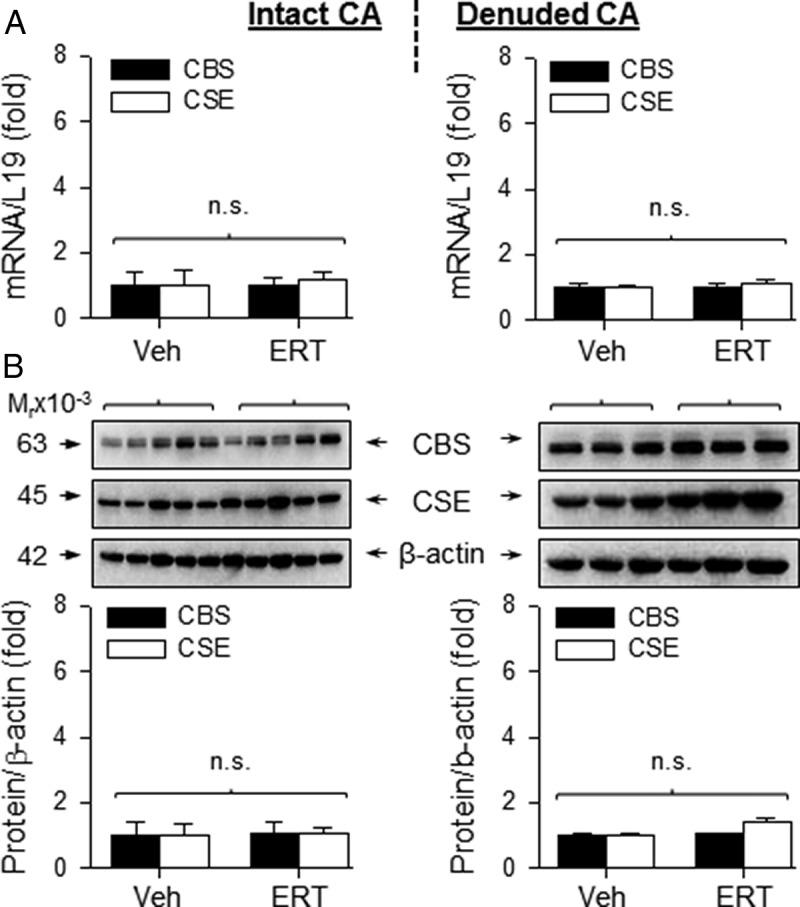
CBS and CSE mRNA and protein expression in CA from OVX ewes receiving Veh and E2β replacement therapy (ERT). Relative mRNA levels of CBS and CSE mRNAs in intact (top, left) and denuded (top, right) CA. CBS and CSE proteins from intact and denuded CA (bottom). ERT did not change CBS or CSE expression. Data (mean ± SEM) are from 3–5 different ewes/group; n.s., not statistically different.
Figure 5.
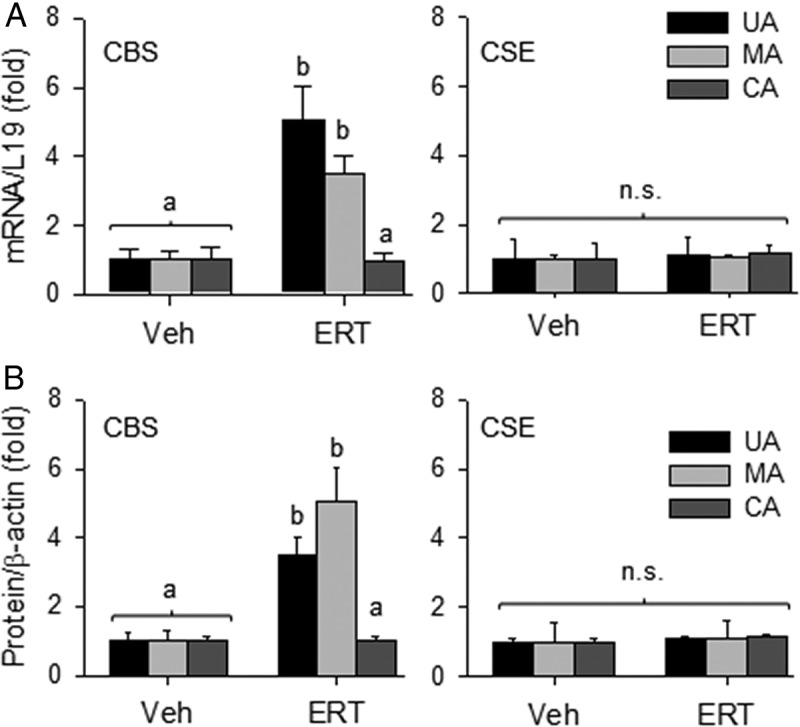
Comparisons of the effects of E2β replacement therapy (ERT) on CBS and CSE mRNA (A) and protein (B) expression in uterine and systemic arteries from OVX ewes. Data (mean ± SEM) were summed from 3–5 different ewes/group. Different letters differ significantly; n.s., no statistical difference.
Comparisons of the effects of ERT on H2S production in UA and systemic arteries
H2S production (nmol h−1 μg protein−1) by protein lysates of intact UA from OVX/ERT ewes (156 ± 9) was approximately 3-fold greater (P < .001) than that of OVX/Veh (54 ± 7) (Figure 6A) ewes. Treatment with the specific CBS inhibitor CHH, but not the specific CSE inhibitor BCA, inhibited H2S production in intact UA lysates from OVX/ERT ewes (P < .001), reducing H2S production to levels in OVX/Veh ewes. Coincubation with CHH and BCA also blocked the ERT-stimulated UA H2S production, but this did not differ from CHH alone (Figure 6A).
Figure 6.
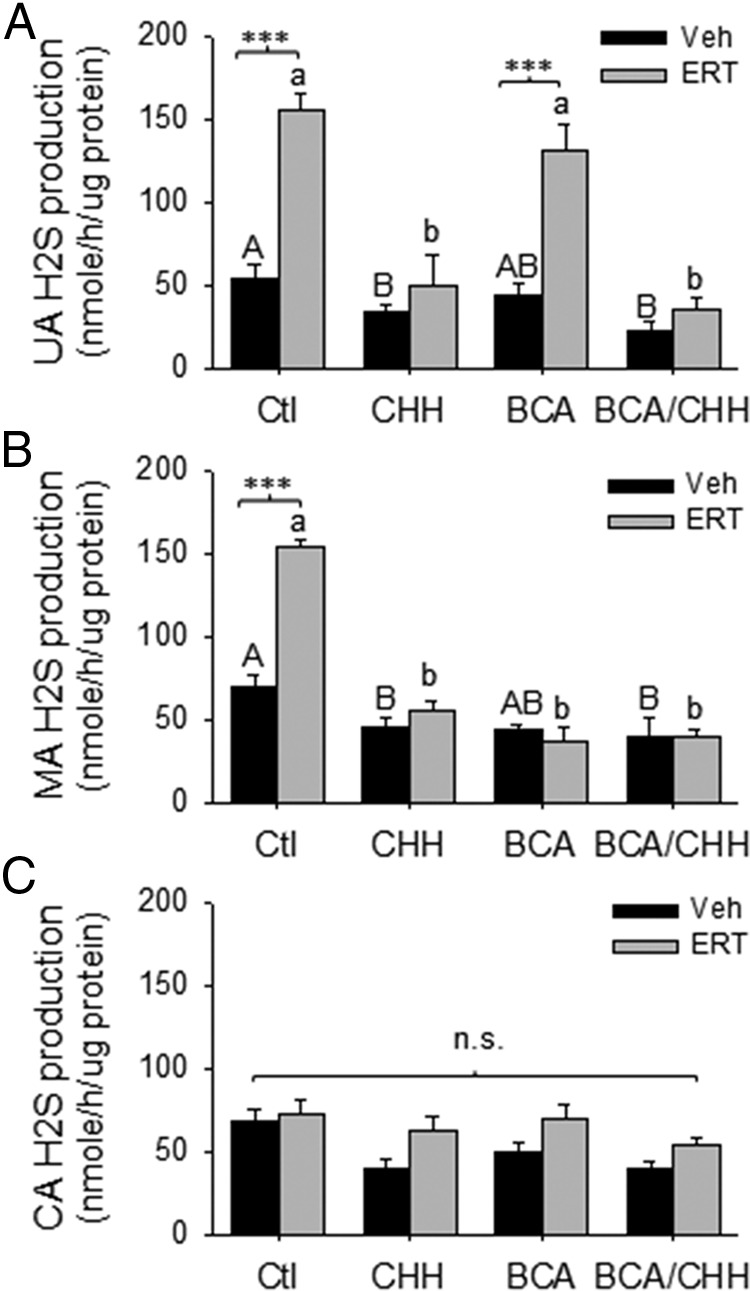
Effects of E2β replacement therapy (ERT) on H2S production in UA (A), MA (B), and CA (C) from OVX ewes. Proteins from OVX ewes were used to measure H2S production in the presence or absence of the specific inhibitors of CBS (CHH), CSE (BCA), or their combination. Data (mean ± SEM) are from 3 different ewes per group. ***, P < .001; bars with different letters differ significantly; n.s., not statistically different.
In the systemic arteries, H2S production by protein lysates of intact MA from OVX/ERT ewes (153 ± 4) was 2-fold greater (P < .001) compared with OVX/Veh ewes (70 ± 7) (Figure 6B). Pretreatment with either CHH or BCA alone and their combination inhibited ERT-stimulated H2S production in MA (Figure 6B). Consistent with no effects of ERT on CBS/CSE expression in CA, ERT also did not stimulate CA H2S production (Figure 6C).
Treatment with CHH or its combination with BCA, but not BCA alone, also reduced basal H2S production in lysates of UA and MA from OVX/Veh ewes (P < .05). However, basal CA H2S production was only inhibited by the combination of CHH and BCA.
Discussion
In the present study, we assessed the effects of ERT on H2S biosynthesis in UA and systemic arteries (MA and CA) using the well-characterized nonpregnant OVX sheep ERT models that have previously been used to assess the effects of estrogen on other vascular modulators, including eNOS (31, 34, 35, 37). We observed that: 1) CBS and CSE mRNA and protein are highly expressed in UA; 2) ERT stimulates CBS expression, but not CSE, reflected by increases in both mRNA and protein; 3) ERT stimulates CBS, but not CSE, expression in UA endothelium and smooth muscle by immunohistological analysis; 4) ERT also stimulates CBS, but not CSE mRNA and protein expression in the endothelium and smooth muscle of MA, but not CA; 5) ERT increases H2S synthesizing activity in UA and MA, but not CA; and 6) ERT-stimulated H2S production is sensitive to inhibition with the specific inhibitor of CBS (CHH), but not CSE (BCA), in the UA, whereas this is sensitive to both in the MA. These findings show that ERT stimulates UA H2S biosynthesis by selectively up-regulating CBS expression, possibly occurring mainly at the level of transcription in OVX nonpregnant sheep and there are selective regional effects of ERT in the systemic circulation consistent with the systemic vascular effects of E2β (3, 4).
The current study is the first to examine H2S biosynthesis in the UA and the effects of estrogens on the expression of the responsible enzymes. We initially hypothesized that CSE would be highly expressed in the UA endothelium and smooth muscle similarly to other vascular beds (26, 38). Indeed, we observed high basal CSE expression in the endothelium and smooth muscle of UA, MA, and CA. In contrast to previous studies showing that prolonged ERT (30 μg E2β · kg−1 · d−1 for 12 wk) stimulates CSE expression in the myocardium of OVX rats (28) and in human umbilical cord vein endothelial cells in vitro (39), our present data show that ERT did not stimulate CSE mRNA and protein expression in either the UA or the systemic arteries (ie, MA and CA) in the OVX sheep ERT model. However, our data are in agreement with a recent study showing that E2β does not stimulate CSE expression in mouse pulmonary artery smooth muscle cells in vitro (40).
In contrast, we have observed for the first time that CBS mRNA and protein expression are dramatically up-regulated in the UA and MA in OVX sheep receiving ERT. This is unexpected because CBS is considered the major enzyme for H2S biosynthesis in tissues/cells of the central nervous system (26). Nonetheless, there are recent studies describing the presence of CBS in the vasculature. For example, CBS has been identified in the cerebral arteries of rats (41) and pulmonary arteries of mice with decreased expression in a rat model of pulmonary hypertension (42). Although others examined the stimulatory effects of E2β on H2S production and CSE expression in the vasculature, the effects of estrogen on CBS expression were not evaluated (28, 39, 40).
Because both CBS and CSE proteins are expressed in UA endothelium and smooth muscle, both may contribute to basal H2S synthesis. However, basal and ERT-stimulated H2S production is only sensitive to the CBS inhibitor CHH. There was little or no contribution by CSE because the combination of CHH with the CSE inhibitor BCA did not further inhibit basal or ERT-stimulated H2S biosynthesis. In keeping with the findings that ERT only stimulates CBS, but not CSE expression in UA endothelium and smooth muscle, these findings show that CBS is the major enzymatic pathway for H2S biosynthesis in UA in response to estrogen treatment. Moreover, estrogen stimulation of UA H2S biosynthesis seems to be mainly regulated at the level of gene transcription because ERT increases the levels of both CBS mRNA and protein expression. In addition, ERT treatment seems to also alter the subcellular localization of CBS protein in UA endothelial cells from apical surface to basement membrane, which may contribute to enzyme activation.
Currently, there is a large body of evidence suggesting that local UA endothelial production of NO via increased expression and activation of endothelial eNOS contributes to the dramatic increases in UBF after exogenous or during endogenous estrogen stimulation (10, 16, 17). However, the E2β-stimulated rises in UBF in OVX and intact follicular phase nonpregnant sheep is only inhibited approximately 65% after local NOS inhibition with L-NAME (16, 17, 43). Thus, additional mechanisms appear to be involved in or contribute to E2β-stimulated rises in UBF. In keeping with the potent vasodilatory (20, 21) and antigenic properties (24) of H2S, increased H2S production may function as a novel UA vasodilator, contributing to estrogen-induced uterine vasodilation, reflecting its role as the third described member of the gasotransmitter family after NO and carbon monoxide in the uterine circulation. Indeed, our current findings showing estrogen up-regulation of CBS expression and enhanced H2S production in UA endothelium and smooth muscle suggests that the CBS/H2S system has the potential of a novel pathway contributing to estrogen-induced vasodilation, although a functional role of a CBS/H2S pathway in estrogen-induced uterine vasodilatation requires further investigation.
Previous studies using experimental long-term ERT animal models have shown that estrogen stimulates sustained vasodilation in the uterine and various systemic vascular beds, including MA in OVX sheep (1, 3). ERT also improves circulation and hyporeactivity of MA by alleviating oxidative stress in a rat portal hypertension model (44). Furthermore, ERT improves MA relaxation and decreases wall thickness associated with aging (45) and attenuates phenylephrine-induced MA contraction in OVX rats (46). These effects of estrogens are important in the regulation of blood pressure, blood flow, vascular tone, vascular remodeling, and stiffness in the UA and some systemic vascular beds (3, 47–49). Previous studies also have shown that estrogens promote vascular function by stimulating the expression of various genes such as NOS, vascular endothelial growth factor, and prostacyclin synthase in different blood vessels (29, 31, 34, 35, 50, 51). Blockade of the eNOS/NO pathway by L-NAME significantly reduced estrogen-induced MA relaxation in rats; however, it does not completely block the E2β-induced relaxation response (52). These studies demonstrate a similar role for eNOS/NO in estrogen-induced vasodilatation in UA (16, 17, 43, 53) and MA (52). Our current study also shows that ERT increases MA expression of CBS, but not CSE, mRNA and protein expression in OVX ewes. Thus, the CBS/H2S pathway may contribute to MA relaxation in response to estrogens as with the UA; however, unlike the UA, CSE/H2S may also have a role in the MA because ERT-stimulated UA H2S production was only inhibited by CHH and ERT-stimulated MA production was sensitive to both CHH and BCA. In contrast, ERT only stimulated CBS but not CSE mRNA/protein in both UA and MA. This discrepancy between H2S production and CSE expression in these 2 vascular beds in OVX sheep receiving ERT are currently unclear. The contribution of CSE to ERT-stimulated MA H2S production without increased protein expression is likely related to its activation via posttranslational modification(s) such as interaction with Ca2+/calmodulin (27). In addition, previous studies have shown a role of CSE/H2S in dilating resistant MA in rats (21, 54). Regardless, this discrepancy suggests different regulatory mechanisms responsible for the expression and activation of CSE protein, and possibly the functional role(s) of CSE/H2S, in different vascular beds.
Contrasting to the significantly elevated CBS, but not CSE expression in UA and MA endothelium and smooth muscle, and increased UA H2S production in OVX/ERT ewes, ERT did not alter CA endothelial and smooth muscle CBS and CSE expression or H2S production, even though basal CBS/CSE expression levels are high in the CA. Although the mechanisms underlying vascular bed-specific expression of CBS and CSE by ERT are elusive, this may be, at least in part, associated with regulation of tissue/cell-specific expression and/or activation of specific estrogen receptors (ie, ERα and ERβ) (37). Nonetheless, our current study shows that estrogens selectively stimulate H2S biosynthesis in the uterine and other systemic vascular beds. These findings suggest that differential expression and regulation of the H2S biosynthesis system in different vascular beds have important implications to comprehend the understanding of the cardiovascular protective effects of estrogens. In contrast, ERT has been shown to not significantly affect cerebral blood flow or cognitive function in postmenopausal women (55), and to be associated with a decreased common CA intima-media thickness and arterial stiffness, which are thought to be predictors of cardiovascular diseases in postmenopausal women (56). However, other studies have shown ERT to promote cerebral blood flow and cognitive function in women over 55 years of age (57) and to reduce age-associated increases in common CA stiffness (58). In rats, estrogens stimulate vasorelaxation of UA and MA but not CA (59). However, in the OVX sheep model of in which we gave identical ERT prolonged exposure of E2β (5-μg/kg bolus, followed by 6 μg kg−1 · d−1 for 10 d), prolonged but not acute, increased blood flow to many nonreproductive organs, including the brain, were noted (3). However, H2S dilates cerebral microvessels, such as cerebral pial arterioles of the newborn pig (26). Therefore, further assessment of the role of ERT on CA and cerebral blood flow may be required to delineate these discrepancies.
Although our current study does not provide the mechanism by which estrogens selectively stimulate endothelial and smooth muscle CBS expression and H2S production in different vascular beds, it clearly demonstrates that ERT selectively stimulates CBS mRNA and protein in UA and MA endothelium and smooth muscle, but not CA, suggesting that CBS is a novel tissue-specific estrogen-responsive gene in vascular endothelium and smooth muscle. The rat and human CBS promoter is more than 4500 bp and consists of 5 alternative noncoding exons and multiple putative binding sites for various transcription factors, including ER, specificity protein 1, activator protein (AP), -1, and AP-3 (60, 61). The mechanism by which estrogens stimulates CBS mRNA and protein expression in the endothelium and smooth muscle of UA and MA is most likely related to ER-dependent transcription because these cells express both estrogen receptors ERα and ERβ (32, 37). Alternatively, ligand-bound ERs interact or “cross talk” with other transcription factors such as Sp1 and AP-1 (62), which may also have a role in estrogen stimulation of CBS expression.
Altogether, our current data have demonstrated that prolonged estrogen replacement treatment selectively stimulate H2S biosynthesis in the UA, and some (MA) but not other (CA) systemic vascular beds by up-regulating CBS expression via transcription in nonpregnant OVX sheep. Due to the potent vasodilatory effects of H2S, we further postulate that increased H2S via enhanced CBS expression plays a critical role in estrogen-induced uterine vasodilation. In keeping with the important physiological significance of estrogens in regulating uterine blood flow during the follicular phase of the ovarian cycle and pregnancy and in postmenopausal women's health, further investigations are warranted to establish a role of CBS-CSE/H2S in estrogen-induced uterine and systemic vascular dilation.
Acknowledgments
This work was supported by National Institutes of Health Grants RO1 HL70562 and R21 HL98746 (D.-b.C.), RO1 HL49210 (R.R.M.), and R01 HD08783 (C.R.R.) and the American Heart Association Grant SDG13910006 (H.H.Z.). T.J.L. was an American Heart Association Predoctoral Fellow (AHA 14PRE18570033).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AP
- activator protein
- BCA
- β-cyano-L-alanine
- CA
- carotid artery
- CBS
- cystathionine β-synthase
- CD31
- cell adhesion molecule 31
- CHH
- O-(carboxymethyl)hydroxylamine hemihydrochloride
- CSE
- cystathionine γ-lyase
- DAPI
- 4′,6-diamidino-2-phenylindole
- E2β
- estradiol-17β
- eNOS
- endothelial NO synthase
- ER
- estrogen receptor
- ERT
- estrogen replacement therapy
- H2S
- hydrogen sulfide
- L-NAME
- L-NG-nitroarginine methyl ester
- MA
- mesenteric artery
- NO
- nitric oxide
- OVX
- ovariectomized
- qPCR
- quantitative polymerase chain reaction
- UA
- uterine artery
- UBF
- uterine blood flow
- Veh
- vehicle.
References
- 1. Rosenfeld CR, Killam AP, Battaglia FC, Makowski EL, Meschia G. Effect of estradiol-17, on the magnitude and distribution of uterine blood flow in nonpregnant, oophorectomized ewes. Pediatr Res. 1973;7:139–148. [DOI] [PubMed] [Google Scholar]
- 2. Rosenfeld CR. Distribution of cardiac output in ovine pregnancy. Am J Physiol. 1977;232:H231–H235. [DOI] [PubMed] [Google Scholar]
- 3. Magness RR, Phernetton TM, Zheng J. Systemic and uterine blood flow distribution during prolonged infusion of 17β-estradiol. Am J Physiol. 1998;275:H731–H743. [DOI] [PubMed] [Google Scholar]
- 4. Magness RR, Rosenfeld CR. Local and systemic estradiol-17 β: effects on uterine and systemic vasodilation. Am J Physiol. 1989;256:E536–E542. [DOI] [PubMed] [Google Scholar]
- 5. Naden RP, Rosenfeld CR. Systemic and uterine responsiveness to angiotensin II and norepinephrine in estrogen-treated nonpregnant sheep. Am J Obstet Gynecol. 1985;153:417–425. [DOI] [PubMed] [Google Scholar]
- 6. Magness RR, Parker CR, Jr, Rosenfeld CR. Systemic and uterine responses to chronic infusion of estradiol-17 β. Am J Physiol. 1993;265:E690–E698. [DOI] [PubMed] [Google Scholar]
- 7. Killam AP, Rosenfeld CR, Battaglia FC, Makowski EL, Meschia G. Effect of estrogens on the uterine blood flow of oophorectomized ewes. Am J Obstet Gynecol. 1973;115:1045–1052. [DOI] [PubMed] [Google Scholar]
- 8. Magness RR, Phernetton TM, Gibson TC, Chen DB. Uterine blood flow responses to ICI 182 780 in ovariectomized oestradiol-17β-treated, intact follicular and pregnant sheep. J Physiol. 2005;565:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Magness RR, Rosenfeld CR, Carr BR. Protein kinase C in uterine and systemic arteries during ovarian cycle and pregnancy. Am J Physiol. 1991;260:E464–E470. [DOI] [PubMed] [Google Scholar]
- 10. Gibson TC, Phernetton TM, Wiltbank MC, Magness RR. Development and use of an ovarian synchronization model to study the effects of endogenous estrogen and nitric oxide on uterine blood flow during ovarian cycles in sheep. Biol Reprod. 2004;70:1886–1894. [DOI] [PubMed] [Google Scholar]
- 11. Magness RR, Rosenfeld CR. The role of steroid hormones in the control of uterine blood flow. In: Rosenfeld CR, ed. The Uterine Circulation. Ithaca, NY: Perinatology Press; 1989:239–271. [Google Scholar]
- 12. Lang U, Baker RS, Braems G, Zygmunt M, Kunzel W, Clark KE. Uterine blood flow–a determinant of fetal growth. Eur J Obstet Gynecol Reprod Biol. 2003;110(suppl 1):S55–S61. [DOI] [PubMed] [Google Scholar]
- 13. Osol G, Moore LG. Maternal uterine vascular remodeling during pregnancy. Microcirculation. 2014;21:38–47. [DOI] [PubMed] [Google Scholar]
- 14. Khalil RA, Granger JP. Vascular mechanisms of increased arterial pressure in preeclampsia: lessons from animal models. Am J Physiol Regul Integr Comp Physiol. 2002;283:R29–R45. [DOI] [PubMed] [Google Scholar]
- 15. Jobe SO, Tyler CT, Magness RR. Aberrant synthesis, metabolism, and plasma accumulation of circulating estrogens and estrogen metabolites in preeclampsia implications for vascular dysfunction. Hypertension. 2013;61:480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Van Buren GA, Yang DS, Clark KE. Estrogen-induced uterine vasodilatation is antagonized by L-nitroarginine methyl ester, an inhibitor of nitric oxide synthesis. Am J Obstet Gynecol. 1992;167:828–833. [DOI] [PubMed] [Google Scholar]
- 17. Rosenfeld CR, Cox BE, Roy T, Magness RR. Nitric oxide contributes to estrogen-induced vasodilation of the ovine uterine circulation. J Clin Invest. 1996;98:2158–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Abe K, Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. 1996;16:1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mustafa AK, Gadalla MM, Snyder SH. Signaling by gasotransmitters. Sci Signal. 2009;2:re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20:6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheng Y, Ndisang JF, Tang G, Cao K, Wang R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol. 2004;287:H2316–H2323. [DOI] [PubMed] [Google Scholar]
- 22. Yang G, Cao K, Wu L, Wang R. Cystathionine γ-lyase overexpression inhibits cell proliferation via a H2S-dependent modulation of ERK1/2 phosphorylation and p21Cip/WAK-1. J Biol Chem. 2004;279:49199–49205. [DOI] [PubMed] [Google Scholar]
- 23. Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T, Zhu YC. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc Res. 2007;76:29–40. [DOI] [PubMed] [Google Scholar]
- 24. Papapetropoulos A, Pyriochou A, Altaany Z, et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci USA. 2009;106:21972–21977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang G, Wu L, Jiang B, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science. 2008;322:587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leffler CW, Parfenova H, Basuroy S, Jaggar JH, Umstot ES, Fedinec AL. Hydrogen sulfide and cerebral microvascular tone in newborn pigs. Am J Physiol Heart Circ Physiol. 2011;300:H440–H447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Szabó C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. [DOI] [PubMed] [Google Scholar]
- 28. Zhu X, Tang Z, Cong B, et al. Estrogens increase cystathionine-γ-lyase expression and decrease inflammation and oxidative stress in the myocardium of ovariectomized rats. Menopause. 2013;20:1084–1091. [DOI] [PubMed] [Google Scholar]
- 29. Rosenfeld CR, Chen C, Roy T, Liu X. Estrogen selectively up-regulates eNOS and nNOS in reproductive arteries by transcriptional mechanisms. J Soc Gynecol Investig. 2003;10:205–215. [DOI] [PubMed] [Google Scholar]
- 30. Schwartz J, Freeman R, Frishman W. Clinical pharmacology of estrogens: cardiovascular actions and cardioprotective benefits of replacement therapy in postmenopausal women. J Clin Pharmacol. 1995;35:314–329. [DOI] [PubMed] [Google Scholar]
- 31. Vagnoni KE, Shaw CE, Phernetton TM, Meglin BM, Bird IM, Magness RR. Endothelial vasodilator production by uterine and systemic arteries. III. Ovarian and estrogen effects on NO synthase. Am J Physiol. 1998;275:H1845–H1856. [DOI] [PubMed] [Google Scholar]
- 32. Liao WX, Magness RR, Chen DB. Expression of estrogen receptors-α and -β in the pregnant ovine uterine artery endothelial cells in vivo and in vitro. Biol Reprod. 2005;72:530–537. [DOI] [PubMed] [Google Scholar]
- 33. Chen DB, Bird IM, Zheng J, Magness RR. Membrane estrogen receptor-dependent extracellular signal-regulated kinase pathway mediates acute activation of endothelial nitric oxide synthase by estrogen in uterine artery endothelial cells. Endocrinology. 2004;145:113–125. [DOI] [PubMed] [Google Scholar]
- 34. Rupnow HL, Phernetton TM, Shaw CE, Modrick ML, Bird IM, Magness RR. Endothelial vasodilator production by uterine and systemic arteries. VII. Estrogen and progesterone effects on eNOS. Am J Physiol Heart Circ Physiol. 2001;280:H1699–H1705. [DOI] [PubMed] [Google Scholar]
- 35. Chen DB, Jia S, King AG, et al. Global protein expression profiling underlines reciprocal regulation of caveolin 1 and endothelial nitric oxide synthase expression in ovariectomized sheep uterine artery by estrogen/progesterone replacement therapy. Biol Reprod. 2006;74:832–838. [DOI] [PubMed] [Google Scholar]
- 36. Webb GD, Lim LH, Oh VM, et al. Contractile and vasorelaxant effects of hydrogen sulfide and its biosynthesis in the human internal mammary artery. J Pharmacol Exp Ther. 2008;324:876–882. [DOI] [PubMed] [Google Scholar]
- 37. Byers MJ, Zangl A, Phernetton TM, Lopez G, Chen DB, Magness RR. Endothelial vasodilator production by ovine uterine and systemic arteries: ovarian steroid and pregnancy control of ERα and ERβ levels. J Physiol. 2005;565:85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang G, Pei Y, Teng H, Cao Q, Wang R. Specificity protein-1 as a critical regulator of human cystathionine γ-lyase in smooth muscle cells. J Biol Chem. 2011;286:26450–26460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou K, Gao Q, Zheng S, et al. 17β-estradiol induces vasorelaxation by stimulating endothelial hydrogen sulfide release. Mol Hum Reprod. 2013;19:169–176. [DOI] [PubMed] [Google Scholar]
- 40. Li H, Mani S, Cao W, et al. Interaction of hydrogen sulfide and estrogen on the proliferation of vascular smooth muscle cells. PLoS One. 2012;7:e41614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chertok VM, Kotsyuba AE. Distribution of H2S synthesis enzymes in the walls of cerebral arteries in rats. Bulletin of Experimental Biology and Medicine. 2012;154:104–107. [DOI] [PubMed] [Google Scholar]
- 42. Luo L, Liu D, Tang C, et al. Sulfur dioxide upregulates the inhibited endogenous hydrogen sulfide pathway in rats with pulmonary hypertension induced by high pulmonary blood flow. Biochem Biophys Res Commun. 2013;433:519–525. [DOI] [PubMed] [Google Scholar]
- 43. Nelson SH, Steinsland OS, Suresh MS, Lee NM. Pregnancy augments nitric oxide-dependent dilator response to acetylcholine in the human uterine artery. Hum Reprod. 1998;13:1361–1367. [DOI] [PubMed] [Google Scholar]
- 44. Zhang B, Zhang CG, Zhou QB, Chen W, Wu ZY. Estrogen improves the hyperdynamic circulation and hyporeactivity of mesenteric arteries by alleviating oxidative stress in partial portal vein ligated rats. World J Gastroenterol. 2013;19:6863–6868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Y, Stewart KG, Davidge ST. Estrogen replacement reduces age-associated remodeling in rat mesenteric arteries. Hypertension. 2000;36:970–974. [DOI] [PubMed] [Google Scholar]
- 46. Keung W, Man RY. Circulating sex hormones modulate vascular contractions and acute response to 17β-estradiol in rat mesenteric arteries. Pharmacology. 2011;88:55–64. [DOI] [PubMed] [Google Scholar]
- 47. Rosenfeld CR, Roy T. Large conductance Ca2+-activated and voltage-activated K+ channels contribute to the rise and maintenance of estrogen-induced uterine vasodilation and maintenance of blood pressure. Endocrinology. 2012;153:6012–6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Scott PA, Tremblay A, Brochu M, St-Louis J. Vasorelaxant action of 17 -estradiol in rat uterine arteries: role of nitric oxide synthases and estrogen receptors. Am J Physiol Heart Circ Physiol. 2007;293:H3713–H3719. [DOI] [PubMed] [Google Scholar]
- 49. Xiao D, Huang X, Yang S, Zhang L. Direct chronic effect of steroid hormones in attenuating uterine arterial myogenic tone: role of protein kinase c/extracellular signal-regulated kinase 1/2. Hypertension. 2009;54:352–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Magness RR, Shideman CR, Habermehl DA, Sullivan JA, Bird IM. Endothelial vasodilator production by uterine and systemic arteries. V. Effects of ovariectomy, the ovarian cycle, and pregnancy on prostacyclin synthase expression. Prostaglandins Other Lipid Mediat. 2000;60:103–118. [DOI] [PubMed] [Google Scholar]
- 51. Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med. 1999;340:1801–1811. [DOI] [PubMed] [Google Scholar]
- 52. Otter D, Austin C. Effects of 17β-oestradiol on rat isolated coronary and mesenteric artery tone: involvement of nitric oxide. J Pharm Pharmacol. 1998;50:531–538. [DOI] [PubMed] [Google Scholar]
- 53. Weiner CP, Lizasoain I, Baylis SA, Knowles RG, Charles IG, Moncada S. Induction of calcium-dependent nitric oxide synthases by sex hormones. Proc Natl Acad Sci USA. 1994;91:5212–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jackson-Weaver O, Osmond JM, Riddle MA, et al. Hydrogen sulfide dilates rat mesenteric arteries by activating endothelial large-conductance Ca(2)(+)-activated K(+) channels and smooth muscle Ca(2)(+) sparks. Am J Physiol Heart Circ Physiol. 304:H1446–H1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Guvenal T, Durna A, Erden O, Guvenal F, Cetin M, Cetin A. Effects of different postmenopausal hormone therapy regimens on cerebral blood flow and cognitive functions. Adv Ther. 2009;26:805–811. [DOI] [PubMed] [Google Scholar]
- 56. Gompel A, Boutouyrie P, Joannides R, et al. Association of menopause and hormone replacement therapy with large artery remodeling. Fertil Steril. 2011;96:1445–1450. [DOI] [PubMed] [Google Scholar]
- 57. Resnick SM, Maki PM, Golski S, Kraut MA, Zonderman AB. Effects of estrogen replacement therapy on PET cerebral blood flow and neuropsychological performance. Horm Behav. 1998;34:171–182. [DOI] [PubMed] [Google Scholar]
- 58. Nagai Y, Earley CJ, Kemper MK, Bacal CS, Metter EJ. Influence of age and postmenopausal estrogen replacement therapy on carotid arterial stiffness in women. Cardiovasc Res. 1999;41:307–311. [DOI] [PubMed] [Google Scholar]
- 59. Hilgers RH, Oparil S, Wouters W, Coelingh Bennink HJ. Vasorelaxing effects of estetrol in rat arteries. J Endocrinol. 2012;215:97–106. [DOI] [PubMed] [Google Scholar]
- 60. Kraus JP, Oliveriusová J, Sokolová J, et al. The human cystathionine β-synthase (CBS) gene: complete sequence, alternative splicing, and polymorphisms. Genomics. 1998;52:312–324. [DOI] [PubMed] [Google Scholar]
- 61. Bao L, Vlcek C, Paces V, Kraus JP. Identification and tissue distribution of human cystathionine β-synthase mRNA isoforms. Arch Biochem Biophys. 1998;350:95–103. [DOI] [PubMed] [Google Scholar]
- 62. Schultz JR, Petz LN, Nardulli AM. Cell- and ligand-specific regulation of promoters containing activator protein-1 and Sp1 sites by estrogen receptors α and β. J Biol Chem. 2005;280:347–354. [DOI] [PubMed] [Google Scholar]