

Abstract
Aldosterone is a steroid hormone important in the regulation of blood pressure. Aberrant production of aldosterone results in the development and progression of diseases including hypertension and congestive heart failure; therefore, a complete understanding of aldosterone production is important for developing more effective treatments. Angiotensin II (AngII) regulates steroidogenesis, in part through its ability to increase intracellular calcium levels. Calcium can activate calpains, proteases classified as typical or atypical based on the presence or absence of penta-EF-hands, which are involved in various cellular responses. We hypothesized that calpain, in particular calpain-10, is activated by AngII in adrenal glomerulosa cells and underlies aldosterone production. Our studies showed that pan-calpain inhibitors reduced AngII-induced aldosterone production in 2 adrenal glomerulosa cell models, primary bovine zona glomerulosa and human adrenocortical carcinoma (HAC15) cells, as well as CYP11B2 expression in the HAC15 cells. Although AngII induced calpain activation in these cells, typical calpain inhibitors had no effect on AngII-elicited aldosterone production, suggesting a lack of involvement of classical calpains in this process. However, an inhibitor of the atypical calpain, calpain-10, decreased AngII-induced aldosterone production. Consistent with this result, small interfering RNA (siRNA)-mediated knockdown of calpain-10 inhibited aldosterone production and CYP11B2 expression, whereas adenovirus-mediated overexpression of calpain-10 resulted in increased AngII-induced aldosterone production. Our results indicate that AngII-induced activation of calpain-10 in glomerulosa cells underlies aldosterone production and identify calpain-10 or its downstream pathways as potential targets for the development of drug therapies for the treatment of hypertension.
Aldosterone, a mineralocorticoid hormone responsible for regulating fluid and electrolyte balance, is involved in blood pressure control. Excessive production of aldosterone results in the development and progression of hypertension, and increases the risk of cardiac fibrosis, congestive heart failure, and renal failure and stroke, all of which can lead to premature death and disability. The addition of mineralocorticoid receptor antagonists to standard therapies has been shown to reduce morbidity and mortality rates in chronic heart failure and acute myocardial infarction patients, suggesting the involvement of aldosterone in cardiovascular disease (1).
Aldosterone biosynthesis occurs in the zona glomerulosa (ZG) of the adrenal cortex upon stimulation of the ZG cells with angiotensin II (AngII), increased extracellular potassium (K+) levels or ACTH. The main secretagogues, AngII and elevated extracellular K+ levels, activate signal transduction pathways that increase cytosolic Ca2+ levels and underlie aldosterone production (2). The initial rate-limiting step in steroidogenesis requires steroidogenic acute regulatory protein (StAR) protein, which mediates translocation of cholesterol from the outer to the inner mitochondrial membrane, at which site the side-chain cleavage enzyme complex that initiates steroidogenesis is located (3). The final stages of aldosterone biosynthesis occur by the action of the aldosterone synthase enzyme (encoded by CYP11B2), which is the late rate-limiting step in aldosterone production (4). AngII increases the expression of both StAR and CYP11B2 (4), as do increased K+ levels (5).
Calpains are intracellular Ca2+-dependent cysteine proteases that are active at neutral pH (6). Calpains can be classified based on penta-EF-hand structures, which allow for the distinction between classical/typical and nonclassical/atypical calpains: nonclassical calpains have a loosely defined T-domain instead of the penta-EF-hands found in the classical calpains (7). These classes can be further subdivided into ubiquitous and tissue-specific calpains (reviewed in Refs. 7, 8), and to date, 16 calpain genes have been identified in mammals. Several mechanisms are thought to regulate cellular calpain activity including autolysis, phosphorylation, interactions with phospholipids, activator proteins or the small calpain subunit and inhibition by calpastatin, an endogenous calpain inhibitor (9). The physiological roles of calpains include effects on cytoskeletal remodeling, signal transduction, gene expression, cell cycle, apoptosis and long-term potentiation. Aberrant increases in intracellular Ca2+ lead to hyper-activation of calpains, which is associated with various pathologies that can be categorized as either genetic diseases or Ca2+ homeostasis-linked diseases. Calpain pathologies with a genetic background include limb girdle muscular dystrophy type 2A, gastric cancer and type 2 diabetes (T2D), whereas calpain pathologies that are linked to aberrant Ca2+ homeostasis include neurodegenerative disorders, cataract formation, atrial fibrillation, myocardial infarction, and hypertension. Due to the involvement of calpains in multiple pathologies, calpains are now targeted for the development of therapeutic treatments.
Calpain-10 is the most extensively studied atypical calpain and has been identified as a T2D susceptibility gene as well as an important mediator of insulin secretion (10). Calpain-10 is ubiquitously expressed in human and animal tissues and has been detected in the cytosol, nucleus and mitochondria of cultured cells (11). Human calpain-10 has up to 8 different variants as a result of alternative splicing, with calpain-10a being the most abundant. Calpain-10 has been associated with renal cell death, ryanodine-induced apoptosis, pancreatic β-cell exocytosis, glucose transporter type 4 vesicle translocation, cataractogenesis, and T2D. Moreover, mitochondrial calpain-10 has been shown to play a role in the regulation of the mitochondrial electron transport chain, and calpain-10 overexpression leads to mitochondrial dysfunction (12). Calpain-10 is also required for cell viability, and a decrease in calpain-10 levels is observed in aging kidneys of rats, mice, and humans, associated with a decrease in renal function (13).
Calpains have been shown to contribute to the development of AngII-induced cardiovascular remodeling (14), and inhibition of calpain activity prevents endothelial dysfunction, myocardial and vascular hypertrophy and tissue fibrosis in AngII-induced hypertension (15, 16). However, to date no study has addressed the role of calpain in agonist-induced aldosterone production in adrenal glomerulosa cells. Because key regulators of aldosterone production increase cytosolic Ca2+ levels (2, 17, 18), and there is a Ca2+ requirement for activation of calpains (6), it seems possible that AngII could increase calpain activity in these cells. We hypothesized that calpain, and in particular calpain-10, is activated by AngII in adrenal glomerulosa cells and underlies increased aldosterone production. In this study, we are the first to identify the involvement of calpains in agonist-induced aldosterone production.
Materials and Methods
Materials
Calpain inhibitors and the fluorescent substrate were purchased from EMD Millipore. Primary antibodies were purchased from the next suppliers, anticalpain-10 antibody was purchased from Abcam, anti-CYP11B2 antibody was a generous gift from Dr Celso Gomez-Sanchez (University of Mississippi, Jackson, MS), and the anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody was purchased from Cell Signaling Technology, Inc. All secondary antibodies were purchased from LI-COR Biosciences, and antibody information is provided in Supplemental Table 1. A detailed description of all reagents and methods used in this study is included in Supplemental Materials.
Methods
Culture of human adrenocortical carcinoma cells
Human adrenocortical carcinoma cells (HAC15 cells) were cultured as previously described (19, 20). Before experiments the HAC15 cells were incubated overnight in low-serum medium (containing 0.1% Cosmic Calf serum). Cells were then treated with pan-calpain inhibitors or typical calpain inhibitors in the presence and absence of the indicated agonist; all treatments contained 0.1% dimethyl sulfoxide (DMSO). Supernatants were collected and frozen for subsequent measurement of aldosterone production. Cells were processed for the techniques described below, with greater detail provided in Supplemental Methods.
Culture of primary bovine adrenal ZG cells
Bovine adrenal ZG (bovine ZG) cells were isolated as previously described (21, 22). For experimental treatments cells were pretreated with pan-calpain inhibitors, typical calpain inhibitors or vehicle (0.1% DMSO) in equilibrated bicarbonate-buffered Kreb's Ringer containing 2.5mM sodium acetate (KRB+) followed by treatment with equilibrated KRB+ in the presence or absence of 10nM AngII or with or without 15mM K+ (for which KCl was isoosmotically substituted for NaCl). Supernatants were collected and frozen for subsequent measurement of aldosterone production.
Measurement of aldosterone or cortisol production
Upon treatment with the appropriate agents, cells were collected and solubilized in 0.3M NaOH to determine protein content using the Bio-Rad protein assay (Bio-Rad Laboratories) with BSA as standard. Aldosterone content of the supernatants was assayed using a solid-phase RIA kit (Siemens) or a cortisol enzyme immunoassay kit (Oxford Biomedical Research).
RNA extraction and cDNA synthesis
Total RNA was extracted from cells using the PerfectPure RNA Cultured Cell kit according to the protocols of the manufacturer (5 Prime). Purity, quantification and integrity of the RNA were determined using a Nanodrop instrument (Thermo Scientific). Total RNA was reverse transcribed using Iscript cDNA synthesis kits according to the manufacturer's instructions.
Quantitative real-time PCR (qRT-PCR) analysis
qRT-PCR amplifications were performed using an ABI Step-One Plus Fast Real-Time PCR system (Applied Biosystems by Life Technologies) according to the reaction parameters recommended by the manufacturer. All primers for the amplification of target sequences were purchased from Applied Biosystems and are listed in Supplemental Table 2. Relative gene expression was calculated by the δ-δ cycle threshold (Ct) (ΔΔCt) method, and the results are expressed as the fold difference in gene expression normalized to the endogenous housekeeping gene (cyclophilin A) and relative to untreated samples.
Semiquantitative RT-PCR analysis
For semiquantitative RT-PCR analysis, primers were designed using Oligoperfect Designer (Life Technologies) and purchased from Integrated DNA Technologies (Supplemental Table 3). RT-PCR of cDNA was performed using RED Extract-N-AMP PCR reaction mix according to the manufacturer's protocol. The amplified products were resolved on a 1% Tris-acetate-EDTA agarose gel and visualized using a Syngene UV transilluminator.
Western blot analysis
After treatment cells were harvested and lysed. The proteins were separated with 10% SDS-PAGE and probed with anticalpain-10 (1:1000 dilution), anti-aldosterone synthase (encoded by CYP11B2) (1:100 dilution) and anti-GAPDH (1:5000 dilution) primary antibodies at 4°C. Protein bands were visualized by IRDye-conjugated secondary antibodies with the Odyssey-SA imaging system (LI-COR Biosciences). Fluorescent bands were quantified using the Odyssey-SA software, and data were normalized to GAPDH levels.
In vitro calpain activity assay
Calpain activity was measured using the fluorogenic substrate, [4-((4-(dimethylamino) phenyl) azo) benzoic acid, succinimidyl ester]-threonine-proline-leucine-lysine∼serine-proline-proline-proline-serine-proline-arginine-[5-((2-aminoethyl)amino) naphthalene-1-sulfonic acid], or (DABCYL)-TPLK∼SPPPSPR-(EDANS) (Calbiochem by EMD Millipore) (23). Fluorescence was measured using a Tecan Spectrafluor Plus fluorescent reader (Tecan, Mannedorf, Switzerland) with 320-nm excitation and 480-nm emission wavelengths at 30°C for 30 minutes. The initial velocity of the rate of increase in fluorescence was determined by calculating the slope after subtracting the background fluorescence.
Small interfering RNA (siRNA) silencing in HAC15 cells
We generated pooled siRNA by combining 2 mission predesigned siRNAs (Sigma-Aldrich) that target calpain-10, SASI_Hs01_00149015 (RefSeqID NM_023083) and SASI_Hs01_00088088 (RefSeqID NM_023085). Cultured HAC15 cells were transfected with pooled calpain-10 siRNA and control siRNA with green fluorescent protein (GFP) by nucleofection (Lonza Group Ltd) as described in detail in Supplemental Methods. The efficiency of transfection was monitored by microscopically monitoring the percentage of GFP-positive cells upon transfection with control siRNA.
Adenoviral amplification and infection of HAC15 cells
Calpain-10 recombinant adenovirus purchased from Signagen was infected into Ad293 cells (Agilent Technologies). After purification as described previously (see reference 35 below), HAC15 cells were infected with the calpain-10- or GFP-expressing adenoviral constructs at multiplicities of infection (MOIs) of 25, 50, and 100. The efficiency of infection was monitored by microscopically determining the percentage of GFP-positive cells, after 6 hours of infection.
Statistical analysis
All experiments were performed independently and repeated a minimum of 3 times in duplicate or triplicate. The values were statistically analyzed by one way-ANOVA, with a Student-Newmann-Keuls post hoc test, using Prism software (Graph Pad Software, Inc), with statistical significance assigned at P < .05.
Results
The pan-calpain inhibitors, calpeptin and MDL-28170 (MDL), inhibit agonist-induced aldosterone production in primary bovine ZG (bovine ZG) cells
To determine whether calpain plays a role in agonist-induced aldosterone production in adrenal glomerulosa cells, we investigated the effect of pan-calpain inhibitors in primary cultures of bovine ZG cells. Bovine ZG cells were stimulated with medium containing 15mM K+ or 10nM AngII, secretagogues known to stimulate aldosterone production in these cells (21). The calpain inhibitor calpeptin reduced aldosterone production in AngII- and K+-treated cells (Figure 1A). Cells pretreated with calpeptin exhibited an approximately 50% inhibition (AngII) and 80% inhibition (K+) of agonist-induced aldosterone secretion. To ensure that the inhibitory response was not a result of nonspecific cytotoxicity, glomerulosa cells were treated with 22R-hydroxycholesterol (22ROH) for 1 hour. Water-soluble 22ROH is a cholesterol analog that bypasses normal signaling pathways regulating the StAR-mediated rate-limiting step of cholesterol shuttling from the outer to the inner mitochondrial membrane where steroidogenesis is initiated. Therefore, a comparison of steroid synthesis occurring upon exposure to 22ROH in the presence or absence of calpeptin provides an indication of possible nonspecific effects on adrenal glomerulosa cell viability/health or steroidogenic enzyme activity. Calpeptin had no significant effect on 22ROH-mediated aldosterone production (Figure 1A). Thus, the ability of calpeptin to inhibit agonist-induced aldosterone production in bovine ZG cells was not due to nonspecific cytotoxicity.
Figure 1.

The pan-calpain inhibitors, calpeptin and MDL, inhibited agonist-induced aldosterone production in primary bovine ZG cells. Primary bovine ZG cells were pretreated with KRB+ for 1 hour, then immediately treated with or without 10nM AngII or an elevated extracellular potassium concentration (15mM, with KCl substituted iso-osmotically for NaCl) in the presence and absence of 25μM calpeptin (A) or 30μM MDL (B) (or 0.1% DMSO vehicle) for 1 hour. In other wells, cells were treated with 10μM 22ROH in the presence and absence of calpeptin or MDL to determine possible nonspecific effects of calpeptin. Aldosterone levels were assayed by a solid-phase RIA and expressed as fold relative to control (basal) levels. The data represent the mean ± SEM of 3 separate experiments performed in triplicate; *, P < .05; ***, P < .001 vs the control; ƒƒƒ, P < .001; ƒ, P < .05 vs agonist alone (AngII or K+) by ANOVA followed by a Student-Newman-Keuls post hoc test. There was no significant difference in 22ROH-mediated aldosterone production (P > .05) in the presence or absence of calpeptin or MDL.
When bovine ZG cells were treated with MDL similar results were obtained, whereby the increase in the aldosterone secretory response induced by elevated K+ levels and AngII was inhibited by pretreatment with MDL. After a 1-hour treatment with agonist in the presence of MDL, there was an approximately 85% and 45% inhibition of K+- and AngII-elicited aldosterone production, respectively (Figure 1B). Furthermore, the inhibition of agonist-induced steroidogenesis was not a result of cytotoxicity, as indicated by the inability of MDL to inhibit 22ROH-mediated aldosterone production.
The pan-calpain inhibitors, calpeptin and MDL, inhibit agonist-induced aldosterone production in human adrenocortical carcinoma HAC15 cells
To confirm the involvement of calpain in agonist-elicited aldosterone production in another cell model of the ZG (19), we treated a human adrenocortical carcinoma cell line, HAC15 cells, with the pan-calpain inhibitors calpeptin and MDL in the presence and absence of elevated K+ levels and AngII, and monitored aldosterone production after 24 hours. HAC15 cells are a subclone of the H295R cell line (24) that exhibit a greater aldosterone biosynthetic response to AngII (20). Aldosterone production induced by K+ and AngII was inhibited by calpeptin 55% and 40%, respectively, in HAC15 cells (Figure 2A). Similar results were observed with MDL (Figure 2B), although MDL was a more effective inhibitor of agonist-induced aldosterone production (80% for K+; 70% for AngII) than calpeptin in HAC15 cells. Furthermore, the inhibition of aldosterone by the pan-calpain inhibitors was not due to cytotoxicity as there was no effect of the inhibitors on 22ROH-mediated aldosterone production (Supplemental Figure 1) and a trypan blue exclusion assay indicated that there was no significant difference in cell viability among the treatment groups (data not shown). We suggest a potential role for calpain in agonist-induced aldosterone production. Based on the more robust response to AngII, we elected to focus on this agonist in subsequent experiments.
Figure 2.

The pan-calpain inhibitors, calpeptin and MDL, inhibited agonist-induced aldosterone production in human adrenocortical carcinoma (HAC15) cells. HAC15 cells incubated for 20–24 hours in low-serum medium were treated with low-serum medium with or without 10nM AngII or an elevated extracellular K+ concentration (15mM) in the presence and absence of 25μM calpeptin (A) or 30μM MDL (B) (or 0.1% DMSO vehicle) for 24 hours. Aldosterone levels were assayed by a solid-phase RIA and expressed as fold relative to control (basal) levels. The data represent the mean ± SEM of 4 separate experiments performed in duplicate; **, P < .01; ***, P < .001 vs the control; ƒƒƒ, P < .001; ƒ, P < .05 vs agonist alone (either AngII or K+) by ANOVA followed by a Student-Newman-Keuls post hoc test.
AngII increases calpain activity in HAC15 cells
AngII has been reported to increase calpain activity in vitro and in vivo in cardiovascular tissue (15, 16). We employed an in vitro calpain activity assay that uses a fluorescence resonance energy transfer (FRET)-based fluorogenic substrate (DABCYL)-TPLK∼SPPPSPR-(EDANS) (23) to measure calpain activity in HAC15 cells. This substrate is believed to be a more selective substrate for calpains vs other proteases than are other available peptide substrates. After treatment, HAC15 cells were harvested and lysed in a buffer solution as described in Methods, and the relative fluorescence per second due to calpain cleavage of the substrate was monitored as a measure of calpain activity. AngII increased calpain activity to approximately 25 fluorescent units per second when compared with a control of about 19 fluorescent units per second, and this activity was attenuated by calpeptin alone and in the presence of AngII to approximately 5 fluorescent units per second (Figure 3A). Cumulative values obtained from multiple experiments indicated that AngII stimulated calpain activity and this activity was blocked by calpeptin (Figure 3B).
Figure 3.
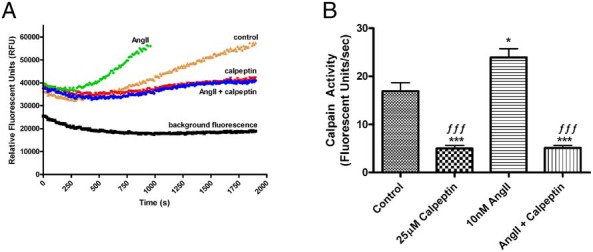
AngII increased calpain activity in human adrenocortical carcinoma (HAC15) cells. HAC15 cells were incubated for 24 hours in the presence and absence of 10nM AngII in low-serum medium. Calpain activity was then determined using 100μM FRET fluorogenic substrate, (DABCYL)-TPLK∼SPPPSPR-(EDANS) in the presence and absence of 25μM calpeptin (or 0.1% DMSO vehicle). The initial velocity of increasing fluorescence was determined using a fluorescent reader at 320-nm excitation and 480-nm emission at 30°C for 30 minutes. A, Representative experiment illustrating relative fluorescence over time; (substrate and buffer in the absence of cell lysate). B, Cumulative data of quantified calpain activity from multiple experiments expressed as fluorescent units per second. The data represent the mean ± SEM of 4 separate experiments performed in duplicate; *, P < .05; ***, P < .001 vs the control; ƒƒƒ, P < .001 vs AngII alone by ANOVA followed by a Student-Newman-Keuls post hoc test.
The pan-calpain inhibitors, calpeptin and MDL, inhibit AngII-induced CYP11B2 expression in HAC15 cells
The 2 key proteins regulating aldosterone production in adrenal glomerulosa cells are StAR, which mediates the initial rate-limiting step of translocation of cholesterol into the mitochondria, and aldosterone synthase (encoded by CYP11B2), which catalyzes the final reactions in aldosterone biosynthesis (4). We determined the effect of pan-calpain inhibitors on the AngII-induced expression of StAR and CYP11B2. Although the expression of the StAR transcript was increased in response to AngII as expected, calpain inhibitors had no effect on this increase (data not shown). In contrast, CYP11B2 mRNA expression was increased upon stimulation with AngII at 24 hours, and calpeptin and MDL inhibited CYP11B2 expression by approximately 60% and 30%, respectively (Figure 4A). The levels of aldosterone synthase in HAC15 cells were also determined using Western blot analysis. AngII increased aldosterone synthase levels, and the calpain inhibitors returned the AngII-stimulated levels to a value not statistically different from the control (Figure 4B), with MDL inhibiting the aldosterone synthase increase by about 30%. Although a 24-hour exposure was selected for these experiments, it is possible that this time point was not optimal, as the inhibitory effect of the calpain inhibitors on CYP11B2 mRNA expression was maximal at 12 hours (Supplemental Figure 2). Collectively, our results suggest that calpain regulates aldosterone production, in part by regulating CYP11B2 expression.
Figure 4.

The pan-calpain inhibitors, calpeptin and MDL, inhibited AngII-induced CYP11B2 expression in human adrenocortical carcinoma (HAC15) cells. HAC15 cells incubated for 20–24 hours in low-serum medium were treated with low-serum medium with or without 10nM AngII in the presence or absence of 30μM MDL or 25μM calpeptin (or 0.1% DMSO vehicle) for 24 hours. RNA was isolated and reverse transcribed for qRT-PCR quantification. qRT-PCR data were normalized to cyclophilin and expressed as fold relative to control (basal) levels (A). Cells were lysed and analyzed by Western blotting for aldosterone synthase levels (B). Western blotting data were normalized to GAPDH protein levels. The data represent the mean ± SEM of at least 3 separate experiments performed in duplicate; *, P < .05; **, P < .01; ***, P < .001 vs the control; ƒƒ, P < .01; ƒ, P < .05 vs AngII alone by ANOVA followed by a Student-Newman-Keuls post hoc test.
The typical calpain inhibitors PD150606 and cell-permeant calpastatin peptide do not inhibit AngII-induced aldosterone production in HAC15 cells
Calpains are classified as either typical or atypical based on the presence or absence of the penta-EF domain in the C terminus. To determine the subclass of the calpain involved in AngII-mediated aldosterone production, we performed additional studies using inhibitors that are more specific to typical calpains. Wang et al (25) identified a class of calpain inhibitors that include PD150606 [3-(4-iodophenyl)-2-mercapto-(Z)-2-propenoic acid], which binds to the penta-EF-hand domain of calpains instead of to the active site of the protease, allowing for selectivity against calpains that possess this penta-EF-domain, ie, typical calpain isozymes. Based on previously reported inhibitor concentrations (26), we treated HAC15 cells with 1μM, 3μM, 10μM, and 30μM PD150606 and found that this agent had no effect on basal aldosterone production except at the 30μM treatment dose, at which concentration it actually increased aldosterone production (Figure 5A). PD150606 had no significant effect on AngII-induced aldosterone production at any dose, whereas the positive control MDL significantly inhibited the response (Figure 5B). In bovine ZG cells, PD150606 had no effect on aldosterone production either in the absence or the presence of AngII (data not shown), providing evidence that the calpain involved in inducing aldosterone production was not a typical calpain.
Figure 5.

The typical calpain inhibitors, PD150606 and calpastatin peptide, did not inhibit AngII-induced aldosterone production in human adrenocortical (HAC15) cells. HAC15 cells incubated for 20–24 hours in low-serum medium were treated with or without 10nM AngII in the presence and absence of PD150606 (A and B) at 1μM, 3μM, 10μM, and 30μM (or 0.1% DMSO vehicle) or 30μM MDL (as a positive control for inhibition) or calpastatin peptide (CS, 2.5μM and 5μM) for 24 hours (C). Aldosterone levels were assayed by a solid-phase RIA and expressed as fold relative to control (basal) levels or relative to stimulation with AngII alone. The data represent the mean ± SEM of at least 3 separate experiments performed in duplicate; ***, P < .001; **, P < .01; *, P < .05 vs the control; ƒƒƒ, P < .001 vs AngII alone by ANOVA followed by a Student-Newman-Keuls post hoc test.
To verify the results with PD150606, we used a synthetic peptide derived from the endogenous calpain inhibitor calpastatin, calpastatin peptide, which is fused to an 11 poly-arginine peptide to allow for improved cell permeability (27). HAC15 cells were treated with calpastatin peptide at 2.5μM and 5μM doses in the presence and absence of AngII. A dose of 2.5μM alone increased aldosterone production 6-fold when compared with control; 5μM calpastatin peptide enhanced basal aldosterone production by approximately 11-fold (Figure 5C). However, in the presence of AngII, there was no effect of calpastatin peptide on aldosterone production at either dose, confirming the importance of an atypical rather than a typical calpain isoform in AngII-induced aldosterone production.
The atypical calpain-5, calpain-7, and calpain-10 are expressed in HAC15 cells
We examined the expression of atypical calpains in HAC15 cells by semiquantitative RT-PCR and found calpain-5, calpain-7, and calpain-10 were expressed (Supplemental Figure 3). Because the mitochondrion is a key organelle in steroidogenesis and calpain-10 exhibits cytosolic, nuclear, and mitochondrial localization (12), we hypothesized that mitochondrial calpain-10 mediates AngII-mediated aldosterone production.
The calpain-10 inhibitor, CYGAK, inhibits AngII-induced aldosterone production in HAC15 cells
The 5 amino acid peptide, cysteine-tyrosine-glycine-alanine-lysine (CYGAK) has been shown to be a potent and efficacious calpain-10 inhibitor in renal proximal tubular cells, inhibiting both cytosolic and mitochondrial calpain-10 (28). Treatment of HAC15 cells with and without 10μM CYGAK in the presence and absence of 10nM AngII for 24 hours significantly inhibited AngII-induced aldosterone production (by approximately 20%) (Figure 6A). Inhibition of calpain-10 by CYGAK also tended to inhibit CYP11B2 mRNA expression in AngII-stimulated cells when compared with AngII alone. A two-way ANOVA analysis of δCt values revealed a small main effect of CYGAK to inhibit CYP11B2 expression, although in individual pairwise comparisons, statistical significance was not achieved due to the sample size (Figure 6B).
Figure 6.

The calpain-10 inhibitor, CYGAK, inhibited AngII-induced aldosterone production in human adrenocortical (HAC15) cells. HAC15 cells incubated for 20–24 hours in low-serum medium were treated with or without 10nM AngII in the presence and absence of 10μM CYGAK (or 0.1% DMSO vehicle) for 24 hours. Aldosterone levels were assayed by a solid-phase RIA (A). RNA and cell lysates were collected for qRT-PCR assay of CYP11B2 expression (B). The data represent the mean ± SEM of at least 3 separate experiments performed in duplicate; **, P < .01; ***, P < .001 vs the control; ƒƒƒ, P < .001 vs AngII alone by ANOVA followed by a Student-Newman-Keuls post hoc test.
Calpain-10 overexpression increases AngII-induced aldosterone production but has no effect on AngII-induced CYP11B2 mRNA expression in HAC15 cells
To verify the calpain-10 inhibitor studies, we infected HAC15 cells with calpain-10-expressing adenovirus vs a GFP-expressing control vector for 6 hours, after which we replaced the media with low-serum medium for an additional 18 hours and then stimulated with or without 10nM AngII for 24 hours. There was an increase in aldosterone production in response to AngII treatment (Figure 7A). Uninfected AngII-treated HAC15 cells and AngII-treated GFP vector-infected cells produced comparable amounts of aldosterone, indicating that there was no effect of adenoviral infection. In the presence of AngII cells overexpressing calpain-10 at different MOIs exhibited a concentration-dependent increase in aldosterone production (Figure 7A).
Figure 7.

Calpain-10 overexpression increased AngII-induced aldosterone production but had no effect on AngII-induced CYP11B2 mRNA expression levels in human adrenocortical (HAC15) cells. Cultured HAC15 cells were incubated for 6 hours with adenovirus expressing GFP or recombinant calpain-10. After 6 hours of infection, low-serum medium was replaced with fresh medium and the cells incubated for an additional 18 hours. Cells were then treated with or without 10nM AngII for 24 hours. Aldosterone levels were assayed by a solid-phase RIA (A) and cells processed for Western blotting using calpain-10 antibody (B). RNA was isolated and reverse transcribed for qRT-PCR assay of the expression of calpain-10 (C) and CYP11B2 (D). All data are expressed as fold over the vector-infected control and represent the mean ± SEM of at least 3 separate experiments performed in duplicate; ***, P < .001; **, P < .01; *, P < .05 vs the vector (GFP-infected); ƒƒƒ, P < .001; ƒƒ, P < .01, vs AngII + vector by ANOVA followed by a Student-Newman-Keuls post hoc test.
Overexpression of calpain-10 was confirmed by monitoring calpain-10 protein and mRNA levels. Calpain-10 was successfully overexpressed at all 3 MOIs. The calpain-10 protein bands at 50 and 54 kDa increased with increasing MOI from 25 to 100 MOI (Figure 7B). Furthermore, infection with calpain-10 adenovirus increased expression of the 75-kDa calpain isoform, which has been reported to be the calpain-10a isoform (29, 30); however, we failed to visualize this 75-kDa band in uninfected or GFP-infected HAC15 cells. At MOIs 25 and 50, there was a 2.5-fold increase in calpain-10 protein expression when compared with GFP vector-infected cells in the absence and presence of AngII. Furthermore, overexpression of calpain-10 at MOI 50 led to a 110-fold increase in calpain-10 mRNA when compared with vector-infected cells (Figure 7C). We also examined the effect of overexpression of calpain-10 on CYP11B2 mRNA levels and found that whereas AngII increased CYP11B2 expression in both vector- and calpain-10-infected cells when compared with untreated cells, there was no significant difference in the AngII-induced increase in CYP11B2 expression with calpain-10 overexpression (Figure 7D). However, calpain-10 overexpression alone resulted in an approximate 5-fold increase in CYP11B2 mRNA levels under basal (untreated) conditions (Figure 7D), with no effect on basal aldosterone production. This disparate outcome likely results from the lack of an effect of calpain-10 overexpression on StAR (data not shown), the early rate-limiting step in steroidogenesis.
Knockdown of calpain-10 inhibits AngII-induced CYP11B2 mRNA expression levels and aldosterone production in transfected HAC15 cells
We also determined the effect of down-regulating calpain-10 using siRNA. HAC15 cells were transfected with pooled-calpain-10 siRNA using an AMAXA Nucleofector, and the cells were allowed to recover for 48 hours before treatment with or without AngII for 24 hours. HAC15 cells transfected with GFP-expressing scrambled siRNA served as control. AngII-mediated aldosterone production was inhibited with calpain-10 knockdown when compared with HAC15 cells transfected with scrambled siRNA followed by AngII treatment (Figure 8A). Expression results indicated successful knockdown of calpain-10 mRNA by more than 50% when compared with HAC15 cells that had been transfected with scrambled siRNA (Figure 8B). Knockdown of calpain-10 siRNA also inhibited AngII-induced CYP11B2 expression when compared with AngII-treated scrambled siRNA (Figure 8C). As the identity between the coding sequence of the other atypical calpains that we found to be expressed in HAC15 cells (calpain-5 and calpain-7) and capain-10 (variant 3) is only approximately 41%–50%, the ability of calpain-10 siRNA to down-regulate calpain-5 or calpain-7 is unlikely, although the possibility of such an effect was not eliminated.
Figure 8.

Knockdown of calpain-10 inhibited AngII-induced CYP11B2 mRNA expression levels and aldosterone production in nucleofected human adrenocortical carcinoma (HAC15) cells. HAC15 cells were transfected by nucleofection with 40nM pooled calpain-10 siRNA or 40nM control scrambled siRNA with GFP and incubated for 48 hours. Before experiments, medium was replaced with low-serum medium overnight followed by treatment in the presence and absence of 10nM AngII for 24 hours. Aldosterone levels were assayed by a solid-phase RIA (A), RNA was isolated and reverse transcribed for qRT-PCR assay of calpain-10 (B) and CYP11B2 (C) expression. The data are expressed as the fold over the scrambled-siRNA control (sc-sirna) and represent the mean ± SEM of at least 3 separate experiments performed in duplicate; ***, P < .001; *, P < .05 vs scrambled siRNA; ƒƒƒ, P < .001; ƒƒ, P < .01; ƒ, P < .05 vs AngII plus scrambled siRNA by ANOVA followed by a Student-Newman-Keuls post hoc test.
Relevance of calpain-10 to adrenal steroidogenesis in vivo
Our results provide evidence of the importance of calpain-10 to regulating aldosterone production in in vitro models of ZG cells. To determine whether these data are potentially relevant to the situation in vivo, we examined calpain-10 protein expression in the human adrenal cortex in situ. Normal adrenal tissue obtained from individuals undergoing adrenalectomy was subjected to immunohistochemical procedures and calpain-10 immunoreactivity determined. Calpain-10 staining was observed throughout the adrenal cortex, with perhaps a slight predominance in the region immediately subjacent to the capsule, which showed no immunoreactivity (Supplemental Figure 4).
Calpain-10 and cortisol production
Immunohistochemistry indicated that calpain-10 protein was distributed throughout the adrenal cortex, suggesting the possibility that it might play a role in the production of other adrenal steroids. HAC15 cells are dedifferentiated and can be induced to produce all of the adrenal steroids; in fact, they produce greater amounts of cortisol than aldosterone (19). To investigate whether calpain-10 regulates adrenal steroiodogenesis in general, cortisol levels were measured in the supernatants from cells treated with and without AngII in the presence or absence of pan-calpain inhibitors. Supplemental Figure 5 illustrates that the pan-calpain inhibitors, calpeptin and MDL, had no effect on cortisol production in response to AngII, suggesting that the involvement of calpain-10 in steroidogenesis may be restricted to the adrenal ZG.
Discussion
We have shown for the first time that the pan-calpain inhibitors calpeptin and MDL reduce aldosterone production in response to elevated extracellular K+ levels and AngII in primary bovine ZG cells and HAC15 cells. Further exploration determined that calpain-10 underlies in part the ability of AngII to stimulate steroidogenesis. Primary cultures of bovine ZG cells respond to agonist treatments with aldosterone secretion within minutes of exposure, whereas the HAC15 cells take hours as basal aldosterone synthase (encoded by CYP11B2) levels are low and expression of this key late rate-limiting enzyme must be induced. Therefore, use of these cells can provide insight into the mechanisms regulating the chronic aldosterone response.
The pan-calpain inhibitors did not completely return AngII-elicited aldosterone production to control levels, suggesting that calpain contributes to the induced aldosterone production but is not the sole molecular signal mediating the response. It is known that AngII stimulates an increase in intracellular Ca2+ levels (reviewed by Ref. 31), which presumably triggers calpain activation to increase aldosterone production. The pan-calpain inhibitors partially inhibited AngII-induced CYP11B2 mRNA expression at all experimental time points (Supplemental Figure 2). Maximal expression of CYP11B2, and maximum inhibition by the calpain inhibitors, was observed at the 12-hour treatment time point but we chose the 24-hour time point for subsequent studies as this period of incubation yielded more robust aldosterone production. In addition, we have also shown that calpain inhibition decreased the CYP11B2 expression induced in response to AngII; calpain inhibitors also reduced the AngII-stimulated protein levels to a value not statistically different from the control. Nevertheless, it is not clear whether an ability to increase CYP11B2 expression is the sole, or even the primary, mechanism by which calpain increases AngII-induced aldosterone production. It seems possible that calpain could activate the aldosterone synthase enzyme encoded by CYP11B2, leading to the observed inhibition of aldosterone production with calpain inhibitors. However, the evidence to date suggests that the activity of aldosterone synthase is regulated predominantly by changes in its expression/levels. It is also possible that there may be actions of calpain at earlier events in steroiodogenesis; this idea is the subject of ongoing studies in the laboratory.
We were also able to show that AngII increased calpain activity in HAC15 cells using a FRET-based calpain-selective substrate, and this activity was inhibited by calpeptin, indicating that AngII activated calpain. However, the calpain substrate is not specific for a particular calpain isoform; therefore, it is difficult to determine which specific calpain isoform(s) is (are) activated. However, in subsequent experiments we showed that the calpain most likely involved in AngII-stimulated aldosterone production was an atypical calpain because the typical calpain inhibitors, PD150606 and calpastatin, had no effect on AngII-elicited aldosterone production in HAC15 cells. Interestingly, PD150606 alone at 30μM induced a slight but significant increase in aldosterone production. Similarly, the synthetic calpastatin peptide, which mimics endogenous calpastatin to inhibit typical calpains, also induced an increase in the aldosterone secretory response in the absence of AngII. We suggest that typical calpains, if involved in glomerulosa cell function, may in fact have an inhibitory role in controlling the basal synthesis of aldosterone rather than AngII-elicited aldosterone production.
Our results indicated that the specific calpain-10 inhibitor, CYGAK, inhibited AngII-induced aldosterone production. The percentage inhibition was approximately 20% of the AngII-elicited maximal response, a slightly lower degree of inhibition than was observed with the pan-calpain inhibitors, although we were somewhat limited in the dosage of CYGAK that we could test as this newly synthesized inhibitor is not commercially available (28, 32). Nevertheless, this result provided evidence for a role of calpain-10 in AngII-elicited aldosterone production, although potential roles for other atypical calpains are not excluded. We also showed that CYGAK tended to inhibit CYP11B2 mRNA expression, suggesting that calpain-10 may be able to regulate AngII-induced CYP11B2 expression.
We were then able to manipulate calpain-10 genetically and showed using adenovirus infection that we could successfully overexpress calpain-10. By Western blot analysis, we observed multiple calpain-10 protein bands. Previously, multiple calpain-10 bands have been suggested to be splice variants (29). However, because the adenovirus expresses a mature calpain-10 cDNA, our result suggests that the multiple bands may instead be proteolytic products of calpain-10. Adenoviral-mediated overexpression enhanced the levels of the 75-kDa protein as well as bands of other molecular weights. The 54-kDa band of calpain-10 has been reported to be the membrane-associated “isoform” that is linked to the cleavage of SNAP25 during insulin secretion from pancreatic β-cells (33). This immunoreactive band was of greatest intensity in the uninfected HAC15 cells; however, the literature does not specify which of the calpain-10 “splice variants” this band represents. Importantly, overexpression of calpain-10 enhanced AngII-induced aldosterone production in a dose-dependent manner. Despite enhancing AngII-induced aldosterone production, calpain-10 overexpression did not increase AngII-induced CYP11B2 expression, which was somewhat unexpected. It is possible that the time point that we selected for monitoring CYP11B2 expression in this experiment was not ideal or that endogenous calpain-10 levels are already optimal for CYP11B2 expression. Nevertheless, CYP11B2 mRNA levels were increased by overexpression of calpain-10 under basal conditions, without a corresponding enhancement in basal aldosterone production. In fact, CYP11B2 expression is the late rate-limiting step in aldosterone production; the early rate-limiting step is the movement of cholesterol from the outer to the inner mitochondrial membrane, requiring the activity of StAR. As we found no effect of calpain-10 inhibition on StAR expression or levels (data not shown), the absence of an enhancement of basal aldosterone production by calpain-10 overexpression likely results from the lack of effect of calpain-10 on this early rate-limiting event in steroidogenesis.
Taking the opposite approach, we showed that siRNA-mediated calpain-10 knockdown inhibited AngII-elicited CYP11B2 mRNA expression and aldosterone production, corroborating our data using the calpain inhibitors. This result indicates that calpain-10 is important for AngII-induced aldosterone production. Therefore, collectively our data suggest that calpain-10 is important in the regulation of chronic aldosterone biosynthesis. Although our results suggest an involvement of calpain-10 in AngII-induced aldosterone production, our data do not directly identify the mechanism by which this enzyme modulates steroidogenesis. Calpains are often referred to as intracellular “modulator proteases” due to their ability to hydrolyze their substrates in a limited fashion to transform or modulate their substrates' structure and/or activity (34). The substrate (or substrates) of calpain-10 that is partially proteolyzed to induce steroidogenesis is unknown, although steroidogenesis activator peptide appears to be derived from glucose-regulated protein 78 by proteolysis (35). It is interesting to speculate that calpain-10 might be the protease that cleaves glucose-regulated protein 78 to generate the approximately 30-kDa steroidogenesis activator peptide, which acts synergistically with GTP to enhance steroidogenesis (36), although the inability of calpain inhibition to affect AngII-stimulated cortisol production argues against this possibility. Calpain's actions on cytoskeletal arrangement or vesicle trafficking could also be involved (14, 37). Thus, calpains have been shown to localize to focal adhesion complexes and cleave substrates, such as myristoylated alanine-rich C kinase substrate, resulting in disruption of its cross-linking ability (38). Future studies will investigate the means by which calpain-10 regulates aldosterone production, although difficulties in delineating these mechanisms will be substantial, due to limitations in the available experimental techniques and reagents that can accurately identify calpain isoforms and/or activities.
In summary, we have shown for the first time that calpains are involved in the regulation of agonist-induced aldosterone production in adrenal glomerulosa cells. In addition, AngII increased calpain activity in these cells. We were able to identify the atypical calpain-10 as an isoform involved in steroidogenesis in adrenal glomerulosa cells. Overexpression of calpain-10 increased AngII-induced aldosterone production and knockdown of calpain-10 by RNA interference inhibited aldosterone production and CYP11B2 expression, validating our results with calpain inhibitors. Therefore, AngII stimulates atypical calpain-10 activity to increase aldosterone production, likely in part by regulating the expression of CYP11B2. Although little is known about the role and mechanism of action of calpain in steroidogenesis, our novel data have shed light on a previously unknown signal regulating aldosterone production. Our results identify calpain-10 and its downstream pathways as potential targets for the development of drugs to inhibit aldosterone production to ultimately lead to new therapeutic treatments for hypertension.
Acknowledgments
We thank Ms Maribeth Johnson for her assistance with the two-way ANOVA statistical analysis and Ms Kimya Jones of GRU's Georgia Esoteric Molecular (GEM) Lab's Histology Services for immunohistochemical assistance. We also thank the GRU Tumor and Tissue Biorepository and the GRU Adrenal Center for providing normal human tissue samples. Finally, we thank Xunsheng (Sara) Chen for her assistance with semiquantitative RT-PCR.
This work was supported in part by the National Institutes of Health Grant HL070046 and Veterans Affairs Merit Review Award BX001344 (to W.B.B.) and by National Institutes of Health Grants GM084147 and ES012239 and Veterans Affairs Merit Review Award BX000851 (to R.G.S.). W.B.B. is also supported by a Veterans Affairs Research Career Scientist award.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AngII
- angiotensin II
- (DABCYL)-TPLK∼SPPPSPR-(EDANS)
- [4-((4-(dimethylamino) phenyl) azo) benzoic acid, succinimidyl ester]-threonine-proline-leucine-lysine∼serine-proline-proline-serine-proline-arginine-[5-((2-aminoethyl) amino) naphthalene-1-sulfonic acid]
- DMSO
- dimethyl sulfoxide
- FRET
- fluorescence resonance energy transfer
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- GFP
- green fluorescent protein
- KRB+
- bicarbonate-buffered Kreb's Ringer containing 2.5 mM sodium acetate
- MDL
- MDL-28170
- MOI
- multiplicity of infection
- 22ROH
- 22R-hydroxycholesterol
- qRT-PCR
- quantitative real-time PCR
- siRNA
- small interfering RNA
- StAR
- steroidogenic acute regulatory protein
- T2D
- type 2 diabetes
- ZG
- zona glomerulosa.
References
- 1. Pitt B. Effect of aldosterone blockade in patients with systolic left ventricular dysfunction: implications of the RALES and EPHESUS studies. Mol Cell Endocrinol. 2004;217:53–58. [DOI] [PubMed] [Google Scholar]
- 2. Ganguly A, Davis JS. Role of calcium and other mediators in aldosterone secretion from the adrenal glomerulosa cells. Pharmacol Rev. 1994;46:417–447. [PubMed] [Google Scholar]
- 3. Miller WL. Steroidogenic acute regulatory protein (StAR), a novel mitochondrial cholesterol transporter. Biochim Biophys Acta. 2007;1771:663–676. [DOI] [PubMed] [Google Scholar]
- 4. Hattangady NG, Olala LO, Bollag WB, Rainey WE. Acute and chronic regulation of aldosterone production. Mol Cell Endocrinol. 2012;350:151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bassett MH, White PC, Rainey WE. The regulation of aldosterone synthase expression. Mol Cell Endocrinol. 2004;217:67–74. [DOI] [PubMed] [Google Scholar]
- 6. Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. [DOI] [PubMed] [Google Scholar]
- 7. Sorimachi H, Hata S, Ono Y. Calpain chronicle–an enzyme family under multidisciplinary characterization. Proc Jpn Acad Ser B Phys Biol Sci. 2011;87:287–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dargelos E, Poussard S, Brulé C, Daury L, Cottin P. Calcium-dependent proteolytic system and muscle dysfunctions: a possible role of calpains in sarcopenia. Biochimie. 2008;90:359–368. [DOI] [PubMed] [Google Scholar]
- 9. Campbell RL, Davies PL. Structure-function relationships in calpains. Biochem J. 2012;447:335–351. [DOI] [PubMed] [Google Scholar]
- 10. Turner MD, Cassell PG, Hitman GA. Calpain-10: from genome search to function. Diabetes Metab Res Rev. 2005;21:505–514. [DOI] [PubMed] [Google Scholar]
- 11. Smith MA, Schnellmann RG. Calpains, mitochondria, and apoptosis. Cardiovasc Res. 2012;96:32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arrington DD, Van Vleet TR, Schnellmann RG. Calpain 10: a mitochondrial calpain and its role in calcium-induced mitochondrial dysfunction. Am J Physiol Cell Physiol. 2006;291:C1159–C1171. [DOI] [PubMed] [Google Scholar]
- 13. Covington MD, Arrington DD, Schnellmann RG. Calpain 10 is required for cell viability and is decreased in the aging kidney. Am J Physiol Renal Physiol. 2009;296:F478–F486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jiang L, Wang M, Zhang J, et al. Increased aortic calpain-1 activity mediates age-associated angiotensin II signaling of vascular smooth muscle cells. PLoS One. 2008;3:e2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Letavernier E, Perez J, Bellocq A, et al. Targeting the calpain/calpastatin system as a new strategy to prevent cardiovascular remodeling in angiotensin II-induced hypertension. Circ Res. 2008;102:720–728. [DOI] [PubMed] [Google Scholar]
- 16. Scalia R, Gong Y, Berzins B, et al. A novel role for calpain in the endothelial dysfunction induced by activation of angiotensin II type 1 receptor signaling. Circ Res. 2011;108:1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barrett PQ, Bollag WB, Isales CM, McCarthy RT, Rasmussen H. Role of calcium in angiotensin II-mediated aldosterone secretion. Endocr Rev. 1989;10:496–518. [DOI] [PubMed] [Google Scholar]
- 18. Betancourt-Calle S, Mann-Blakeney RS, Isales CM, Calle RA, Bollinger Bollag W. Angiotensin II priming of aldosterone secretion with agents that enhance Ca(2+) influx. Mol Cell Endocrinol. 2001;177:61–70. [DOI] [PubMed] [Google Scholar]
- 19. Parmar J, Key RE, Rainey WE. Development of an adrenocorticotropin-responsive human adrenocortical carcinoma cell line. J Clin Endocrinol Metab. 2008;93:4542–4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang T, Rowland JG, Parmar J, Nesterova M, Seki T, Rainey WE. Comparison of aldosterone production among human adrenocortical cell lines. Horm Metab Res. 2012;44:245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bollag WB, Barrett PQ, Isales CM, Rasmussen H. Angiotensin-II-induced changes in diacylglycerol levels and their potential role in modulating the steroidogenic response. Endocrinology. 1991;128:231–241. [DOI] [PubMed] [Google Scholar]
- 22. Betancourt-Calle S, Bollag WB, Jung EM, Calle RA, Rasmussen H. Effects of angiotensin II and adrenocorticotropic hormone on myristoylated alanine-rich C-kinase substrate phosphorylation in glomerulosa cells. Mol Cell Endocrinol. 1999;154:1–9. [DOI] [PubMed] [Google Scholar]
- 23. Tompa P, Buzder-Lantos P, Tantos A, et al. On the sequential determinants of calpain cleavage. J Biol Chem. 2004;279:20775–20785. [DOI] [PubMed] [Google Scholar]
- 24. Wang T, Rainey WE. Human adrenocortical carcinoma cell lines. Mol Cell Endocrinol. 2012;351:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang KK, Nath R, Posner A, et al. An α-mercaptoacrylic acid derivative is a selective nonpeptide cell-permeable calpain inhibitor and is neuroprotective. Proc Natl Acad Sci USA. 1996;93:6687–6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Waters SL, Sarang SS, Wang KK, Schnellmann RG. Calpains mediate calcium and chloride influx during the late phase of cell injury. J Pharmacol Exp Ther. 1997;283:1177–1184. [PubMed] [Google Scholar]
- 27. Wu HY, Tomizawa K, Matsushita M, Lu YF, Li ST, Matsui H. Poly-arginine-fused calpastatin peptide, a living cell membrane-permeable and specific inhibitor for calpain. Neurosci Res. 2003;47:131–135. [DOI] [PubMed] [Google Scholar]
- 28. Smith MA, McInnes C, Whitaker RM, et al. Calpain 10 homology modeling with CYGAK and increased lipophilicity leads to greater potency and efficacy in cells. ACS Chem Biol. 2012;7:1410–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dong B, Liu R. Characterization of endogenous and recombinant human calpain-10. Biochimie. 2008;90:1362–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Norton L, Parr T, Chokkalingam K, et al. Calpain-10 gene and protein expression in human skeletal muscle: effect of acute lipid-induced insulin resistance and type 2 diabetes. J Clin Endocrinol Metab. 2008;93:992–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spät A, Hunyady L. Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev. 2004;84:489–539. [DOI] [PubMed] [Google Scholar]
- 32. Rasbach KA, Arrington DD, Odejinmi S, Giguere C, Beeson CC, Schnellmann RG. Identification and optimization of a novel inhibitor of mitochondrial calpain 10. J Med Chem. 2009;52:181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marshall C, Hitman GA, Partridge CJ, et al. Evidence that an isoform of calpain-10 is a regulator of exocytosis in pancreatic β-cells. Mol Endocrinol. 2005;19:213–224. [DOI] [PubMed] [Google Scholar]
- 34. Ono Y, Ojima K, Torii F, et al. Skeletal muscle-specific calpain is an intracellular Na+-dependent protease. J Biol Chem. 2010;285:22986–22998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li XA, Warren DW, Gregoire J, Pedersen RC, Lee AS. The rat 78,000 dalton glucose-regulated protein (GRP78) as a precursor for the rat steroidogenesis-activator polypeptide (SAP): the SAP coding sequence is homologous with the terminal end of GRP78. Mol Endocrinol. 1989;3:1944–1952. [DOI] [PubMed] [Google Scholar]
- 36. Pedersen RC, Brownie AC. Steroidogenesis-activator polypeptide isolated from a rat Leydig cell tumor. Science. 1987;236:188–190. [DOI] [PubMed] [Google Scholar]
- 37. Potter DA, Tirnauer JS, Janssen R, et al. Calpain regulates actin remodeling during cell spreading. J Cell Biol. 1998;141:647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dulong S, Goudenege S, Vuillier-Devillers K, Manenti S, Poussard S, Cottin P. Myristoylated alanine-rich C kinase substrate (MARCKS) is involved in myoblast fusion through its regulation by protein kinase Cα and calpain proteolytic cleavage. Biochem J. 2004;382:1015–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]