

Abstract
Background:
Transcriptome profiling has helped characterise nodal spread. The interpretation of these data, however, is not without ambiguities.
Methods:
We profiled the transcriptomes of papillary thyroid cancer nodal metastases, associated primary tumours and primary tumours from N0 patients. We also included patient-matched non-cancerous thyroid and lymph node samples as controls to address some limits of previous studies.
Results:
The transcriptomes of patient-matched primary tumours and metastases were more similar than those of unrelated metastases/primary pairs, as previously reported in other organ systems. This similarity partly reflected patient background. Lymphoid tissues in the metastases confounded the comparison of patient-matched primary tumours and metastases. We circumvented this with an original data adjustment, revealing a differential expression of stroma-related gene signatures also regulated in other organs. The comparison of N0 vs N+ primary tumours uncovered a signal irreproducible across independent data sets. This signal was also detectable when comparing the non-cancerous thyroid tissues adjacent to N0 and N+ tumours, suggesting a cohort-specific bias also likely present in previous similarly sized studies. Classification of N0 vs N+ yielded an accuracy of 63%, but additional statistical controls absent in previous studies revealed that this is explainable by chance alone. We used large data sets from The Cancer Genome Atlas: N0 vs N+ classification was not better than random for most cancers. Yet, it was significant, but of limited accuracy (<70%) for thyroid, breast and head and neck cancers.
Conclusions:
The clinical potential of gene expression to predict nodal metastases seems limited for most cancers.
Keywords: transcriptome, papillary thyroid cancer, nodal metastasis, lymph nodes
Thyroid cancers are the most frequent endocrine malignancies. Eighty per cent of them are of the papillary subtype (PTC). In these cancers, regional lymph node metastasis is correlated with poorer survival (Lundgren et al, 2006). Therapeutic dissection of lymph nodes within the central neck area improves disease outcome (Nixon and Shaha, 2013; Venkat and Guerrero, 2013) and is recommended by current international guidelines (Pacini et al, 2006; American Thyroid Association (ATA) Guidelines Taskforce on Thyroid Nodules and Differentiated Thyroid Cancer et al, 2009), whereas the benefit of prophylactic dissection remains a matter of heated debate (Nixon and Shaha, 2013; Venkat and Guerrero, 2013). The biology underlying the nodal dissemination of PTC is poorly understood.
A number of cancer studies in other organ systems have explored with transcriptome profiling the relationship between primary tumours and their associated nodal metastases and the characteristics of those primary tumours producing nodal metastases (Huang et al, 2003; Kikuchi et al, 2003; Nagata et al, 2003; Hao et al, 2004; Takada et al, 2004; Tamoto et al, 2004; Croner et al, 2005; Hoang et al, 2005; O'Donnell et al, 2005; Weigelt et al, 2005; Xi et al, 2005; Bidus et al, 2006; Roepman et al, 2006a; Wang et al, 2006a; Feng et al, 2007; Inamura et al, 2007; Méndez et al, 2007, 2011; Nguyen et al, 2007; Pei et al, 2007; Suzuki and Tarin, 2007; Kashiwazaki et al, 2008; Kroon et al, 2008; Vecchi et al, 2008; Ellsworth et al, 2009; Wong et al, 2009; Xie et al, 2010; Edfeldt et al, 2011; Smeets et al, 2011). A recurrent finding was that molecular phenotypes of nodal metastases are similar to those of the primary tumours they originate from (Perou et al, 2000; Hao et al, 2004; Hoang et al, 2005; O'Donnell et al, 2005; Weigelt et al, 2005; Roepman et al, 2006a; Feng et al, 2007; Inamura et al, 2007; Vecchi et al, 2008; Ellsworth et al, 2009). This has been interpreted as evidence that most cells in the primary tumour have the potential to seed metastases (Hao et al, 2004; Hoang et al, 2005; Inamura et al, 2007; Ellsworth et al, 2009), in contrast with the classical view that a few cells acquire metastatic capacity within the primary tumour (Fidler and Kripke, 1977). Furthermore, several studies defined the biological context of nodal spread by pointing out transcription of genes related to the immune response (Weigelt et al, 2005; Wang et al, 2006a; Vecchi et al, 2008; Ellsworth et al, 2009; Xie et al, 2010) and the stroma (Hao et al, 2004; O'Donnell et al, 2005; Weigelt et al, 2005; Roepman et al, 2006a; Feng et al, 2007; Suzuki and Tarin, 2007; Vecchi et al, 2008; Ellsworth et al, 2009) differing between primary and metastatic samples. Finally, the prediction of nodal spread from primary tumour yielded conflicting results ranging from no classification (Xi et al, 2005; Kroon et al, 2008) to perfect classification of N0 (without apparent nodal spread) and N+ (with nodal spread) diseases (Kikuchi et al, 2003; Tamoto et al, 2004; O'Donnell et al, 2005; Roepman et al, 2005; Inamura et al, 2007; Pei et al, 2007; Kashiwazaki et al, 2008; Méndez et al, 2011). Whether these discrepancies reflect technical issues or a cancer type-specific classification potential remains unclear.
Data presented in these studies are not without ambiguities. Patient's genetic and physiological background could both contribute to the transcriptional similarity between primary tumours and patient-matched nodal metastases. The immune-related signals reported in nodal metastasis could, as noted by several authors, reflect the presence of lymphoid tissues alongside the metastasis. Laser capture microdissection has been proposed to address this ambiguity in some studies (Hoang et al, 2005; Wang et al, 2006a; Inamura et al, 2007; Nguyen et al, 2007; Ellsworth et al, 2009), whereas others have advocated the analysis of bulk tissues because they better reflect the wider context of metastasis (Hoang et al, 2005; O'Donnell et al, 2005; Méndez et al, 2007). The N0 vs N+ classification problem has been approached with a very wide range of statistical methods and validation procedures in addition to differences in lymph node screening protocols.
This study applies the transcriptomic approach to the analysis of nodal metastatic spread in PTCs. Numerous biological and computational controls have been included to address the ambiguities of previous studies.
Materials and methods
Samples and patients
Samples derived from 31 patients diagnosed for PTC were obtained from the Jules Bordet Institute (Brussels, Belgium) and La Pitié-Salpêtrière Hospital (Paris, France). They were selected on the basis of RNA quality rather than clinicopathologic parameters. Protocols were approved by the ethics committees of the institutions.
Extraction and quality assessment
RNA was extracted with RNeasy Mini Kit columns (Qiagen, Hilden, Germany). The histology of each sample was verified using haematoxylin- and eosin-stained sections. Percentages of tumour cells were estimated and, when present, the percentages of adjacent non-tumour cells or lymphocyte infiltration were quantified. Tumours with <70% cancer cells were discarded. RNA concentrations were measured using NanoDrop ND-1000 spectrophotometer (Life Technologies, Grand Island, NY, USA). RNA integrity was assessed using an automated gel electrophoresis system (Experion; Bio-Rad, Hercules, CA, USA), yielding a score from 1 (fully degraded) to 10 (intact RNA): the RNA Quality Index. Only samples with a score of 7 or above were considered.
Microarrays hybridisation
Genome-wide mRNA profiles were obtained through the hybridisation of samples onto Affymetrix GeneChip Human Genome U133 Plus 2.0 Arrays (Affymetrix, Santa Clara, CA, USA). RNA amplification, hybridisation and image scanning were performed according to standard Affymetrix protocols.
Mutational status
We assessed the mutational status of BRAF, RET/PTC1, RET/PTC3, NRAS, HRAS and KRAS in the PTC samples. After a DNase treatment with DNaseI Amplification Grade Kit (Invitrogen, Carlsbad, CA, USA), 1 μg of total RNA was used for reverse transcription using hexamers (3.6 μg μl−1; Roche, Basel, Switzerland) and reverse transcriptase (SuperscriptII RNase H ReverseTranscriptase Kit; Invitrogen). Polymerase chain reactions were performed on 2 μl of cDNA using the recombinant Taq DNA Polymerase Kit (Invitrogen) and appropriate primer pairs (primer sequences and PCR conditions provided in a previous study; Saiselet et al, 2012). Polymerase chain reaction products were purified with the QIAquick PCR Purification Kit (Qiagen) according to the manufacturer's instructions. Sequencing was performed with the BigDye Terminator V3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) with the sequencer ABI PRISM 3130 (Applied Biosystems) and the genetic analysis program 3130-XI.
Microarray analysis
All analyses and images were performed in R v.3.1.0 (R Core Team, 2013).
CEL files were normalized with fRMA (McCall et al, 2010) v.1.6.0 with default parameters and hgu133plus2frmavecs v.1.1.8 annotation. The arrays used contain 54 613 probe sets (probes) representing 20 027 genes, annotated with R packages annotate v.1.32.3 (Gentleman, 2014) and hgu133plus2.db v2.6.3 (Carlson et al, 2014).
Hierarchical clustering was performed using the function hclust with ‘method=ward'. The distance between samples was defined as 1−|ρS|, where ρS is Spearman's correlation coefficient. Principal component analysis was computed with prcomp.
Differential expression was assessed with Rank Product (Breitling et al, 2004), a nonparametric method related to the fold-change method (Hong and Breitling, 2008).
The four normal lymph node (NLN) profiles were used to assess the contaminant fraction in nodal metastasis profiles (LNM) as follows:
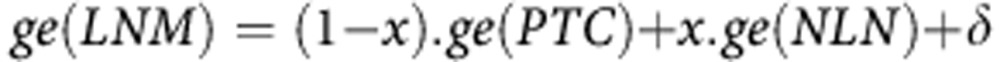 |
where ge(tissue) is the expression profile of a tissue; x is the fraction of NLN contaminant and δ is an unknown signal associated with metastasis and measurement errors. Taking the ratios of expression:
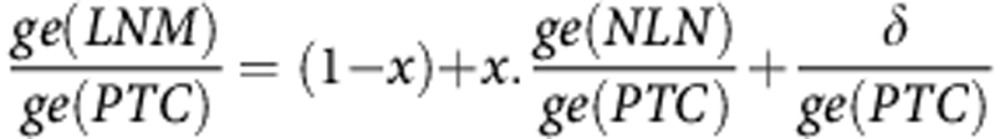 |
Thus, the fraction of contaminant x can be estimated from the slope between LNM/PTC and NLN/PTC ratios. This method was assessed with a data set of tissue mixtures of known contaminant fraction (Supplementary Figure S3). Estimated and real contaminant fractions were similar and highly correlated (Spearman's ρ=0.94; P=1.16 × 10−78). To deconvolute the contaminant signal, for each log2-transformed LNM-PTC ratio, a loess normalization was performed with regard to their NLN-PTC ratio.
Gene set enrichment analysis was run with 1000 label permutations to derive a P-value and its associated false discovery rate for each gene set (Subramanian et al, 2005).
After balancing the number of N0 and N+ samples, nested cross-validation was performed using support vector machine as the classification method (Chang and Lin, 2011) and top scoring pairs (Leek, 2009) for the feature selection. The data sets were divided randomly into five parts of equal size and equal number of samples from each class. One part was set aside as the test set, whereas the training set consisted of the four remaining parts. The training set was used in the inner fivefold cross-validation to select the best number of probes N ∈ {8, 16, 32, 64}, that is, the number of probes maximising the average accuracy of the predictors.
False discovery rates, a confidence measure that accounts for multiple testing, <0.05 were considered significant for differential expression and <0.25 for gene set analysis, as recommended by Subramanian et al (2005).
This microarray data set is available from the Gene Expression Omnibus (Edgar et al, 2002) under accession number GSE60542.
Results
No strong global transcriptional difference between N0 primary tumours, N+ primary tumours and nodal metastases
We profiled the transcriptomes of 11 primary PTCs with no detectable nodal invasion, 17 primary PTCs with nodal invasion and 17 patient-matched nodal metastases. We also profiled a number of control samples. These included 24 patient-matched non-cancerous thyroid tissues (11 from N0, 13 from N+ patients), and 4 normal lymph nodes, and technical and biological replicates including additional nodal metastasis for 3 patients, adjacent blocks for 5 primary tumours, 4 non-cancerous thyroid tissues and 1 nodal metastasis. The non-cancerous thyroid tissues were taken from the thyroid tissues adjacent to the tumor, and in the contralateral thyroid lobe whenever possible.
To compare the global molecular phenotypes of these samples, we projected them on the two first principal components, representing together 46% of the total variance of the data set (Figure 1). Samples were grouped into three clusters: tumour tissues, non-cancerous thyroid tissues and normal lymph nodes. We eliminated from further analysis five presumably mislabelled ‘tumour' samples with global phenotypes akin to non-cancerous tissues (Figure 1, red labels). The tumour cluster included N0, N+ primary tumours and nodal metastases demonstrating the absence of a strong global transcriptional difference between these tissues.
Figure 1.

Principal component analysis of the data set. Samples are projected on the two first components, which explain 28.9% and 16.7% of the total variance of the data set, respectively. Red denotes mislabelled samples. Orange denotes samples with high lymphoid tissue contamination (see Figure 4A).
The transcriptomes of primary tumours and patient-matched lymph node metastases are highly correlated owing to both patient and tumour common backgrounds
Several publications reported that the global transcriptomes of primary tumours are more related to those of their associated metastasis than to the metastasis of other primary tumours from other patients (Perou et al, 2000; Hao et al, 2004; O'Donnell et al, 2005; Weigelt et al, 2005; Roepman et al, 2006a; Feng et al, 2007; Inamura et al, 2007; Vecchi et al, 2008; Ellsworth et al, 2009). To verify if this applied to our data, we ran a hierarchical clustering on all pairs of nodal metastases and matched primary tumours (Figure 2A). Samples clustered by patient for 11 in 13 pairs, confirming earlier studies.
Figure 2.

Correlation between expression profiles of nodal metastases and patient-matched primary tumours. (A) Hierarchical clustering of primary tumours (PT, in grey) and patient-matched lymph node metastases (LNM, in black). Numbers denote patient identity. Patient-matched samples cluster together, except for patients 7 and 8 (orange labels). (B) Panel ‘PT replicates': correlation between blocks from the same primary tumour (brown) and between primary tumours from different patients (grey). Panel ‘PTC-LNM': correlations between primary tumours and patient-matched nodal metastasis (brown) and between primary tumours and unrelated nodal metastasis (grey). Panel ‘N replicates': same as ‘PT replicates, using non-cancerous tissues instead of primary tumours. Panel ‘PTC-N': correlation between primary tumours and patient-matched non-cancerous thyroid tissues (brown) and between primary tumours and unrelated non-cancerous thyroid tissues (grey). Panel ‘LNM-N': same as ‘PTC-N' using nodal metastases instead of primary tumours.
The present and previous analyses provided little quantitative insight and did not investigate the contribution of patient and tumour backgrounds in determining sample similarities. Nodal samples were significantly more correlated with primary tumours from the same patients than with unrelated primary tumours (Figure 2B). The correlations between matched primary/nodal samples and between primary tumour replicates were not significantly different.
Patient-matched non-cancerous and primary tumour samples were significantly more correlated than unmatched primary and non-cancerous samples. Thus, part of the primary tumour phenotype is related to patient background. The former correlation, however, was significantly lower than the correlations between tumour replicates and between non-cancerous tissue replicates. Patient-matched nodal metastasis and non-cancerous thyroid tissues were not more correlated than equivalent unmatched samples. We concluded that the observed pairing of the patient-matched primary and nodal transcriptomes was owing to both tumour- and patient-specific backgrounds.
Normal lymphoid and non-cancerous thyroid tissues likely confounded the expression differences between primary tumours and nodal metastases
The transcriptomes of primary tumours and nodal metastases were undistinguishable on a global scale, yet defined gene expression signature may set them apart and provide insights into the metastatic process. We performed a supervised search for genes differentially expressed between N+ primary tumours and their matched metastases (details in Material and Methods) – an approach widely used in previous studies (Huang et al, 2003; Kikuchi et al, 2003; Nagata et al, 2003; Tamoto et al, 2004; Croner et al, 2005; O'Donnell et al, 2005; Roepman et al, 2005; Xi et al, 2005; Bidus et al, 2006; Inamura et al, 2007; Méndez et al, 2007, 2011; Nguyen et al, 2007; Pei et al, 2007; Kashiwazaki et al, 2008; Kroon et al, 2008; Wong et al, 2009; Smeets et al, 2011).
We detected 1074 probes more expressed in metastases than in primary tumours (Supplementary Table S3). To gain functional insight, we ran a gene set enrichment analysis (GSEA) (Subramanian et al, 2005) using a gene set database composed of the Gene Ontology biological processes based on categories, signatures of primary tumour vs nodal metastasis from previous studies (Nagata et al, 2003; Hao et al, 2004; Hoang et al, 2005; Roepman et al, 2005; Xi et al, 2005; Bidus et al, 2006; Finak et al, 2006; Wang et al, 2006b; Feng et al, 2007; Inamura et al, 2007; Nguyen et al, 2007; Suzuki and Tarin, 2007; Casey et al, 2009; Ellsworth et al, 2009; Edfeldt et al, 2011) and a signature of normal thyroid differentiation (Tomás et al, 2012). Gene sets from previous studies and gene sets associated with the immune system were upregulated in nodal metastasis (Figure 3A). This signal could have resulted from the trivial fact that we compared tissues with two different backgrounds, that is, thyroid and lymphoid. To address this confounding variable problem, we compared the fold changes of probes obtained when comparing nodal metastases vs primary tumours with the fold changes obtained when comparing normal lymph nodes vs non-cancerous thyroid (Figure 3B). The two variables were highly correlated, ρS=0.39 (P<2 × 10−16), demonstrating that the 1074 upregulated probes and the gene sets were not informative about the metastatic process, but were due to the confounding effect of normal lymphoid cells.
Figure 3.

Gene expression differences between nodal metastases and patient-matched primary tumours. (A) A gene set enrichment analysis (Subramanian et al, 2005) for the nodal metastases vs patient-matched primary tumours comparison with gene sets from the Gene Ontology Biological Processes database (black) and curated gene sets from previous studies investigating nodal metastasis or stromal gene expression (Hoang et al, 2005; Wang et al, 2005; Feng et al, 2007; Suzuki and Tarin, 2007; Casey et al, 2009; Ellsworth et al, 2009; blue, only significant gene sets are shown (see Supplementary Table S2 for the full list of investigated gene sets) and a signature of thyroid differentiation (Tomás et al, 2012; green). Red bars denote increased expression in metastases compared with primary tumours and green bars denote decreased expression. (B) Each point denotes a gene with expression average fold change between normal lymph node (NLN) and non-cancerous thyroid (Thyroid) on the x axis and average fold change between nodal metastases (LNM) and primary tumours (PTC) on the y axis. Spearman's correlation coefficient between these two variables is 0.39 (P<2 × 10−16).
A lower number of probes, 27, were downregulated in metastases compared with primary tumours (Supplementary Table S3). Gene set enrichment analysis detected the thyroid differentiation signature as the unique downregulated gene set (Figure 3A). This raised the possibility that expression negatively associated with nodal metastases was confounded by the absence of normal, differentiated, thyroid cells in lymph nodes.
Correcting for contaminant reveals an increased stromal signal in nodal metastases compared with matched primary
The normal lymph node transcriptomes enabled estimating lymphoid cell fraction in the nodal metastasis (Material and Methods and Supplementary Figure S1). It was below the 30% limit as determined by the pathologic examination in all but two samples (Figure 4A). These two samples were precisely those not clustering with their matched primary tumour in Figure 2A, strongly suggesting that these two exceptions to the general clustering pattern were caused by low tumour content.
Figure 4.

Correcting for lymphoid content reveals an upregulation of stroma-related genes in nodal metastasis compared with primary tumours. (A) Estimate of the fraction of normal lymphoid tissues in nodal metastasis samples (calculation explained in Material and Methods and illustrated in Supplementary Figure S1). (B) Same as in Figure 3A, except that expression profiles of nodal metastases were adjusted for normal lymphoid tissue content.
Expression data were adjusted for lymphoid cell fraction (Materials and Methods and Supplementary Figure S1) and gene set enrichment was investigated as described in the previous section (Figure 4B; see Supplementary Table S3 for the list of differentially expressed genes). Two signatures of nodal metastasis from breast cancer studies (Suzuki and Tarin, 2007; Ellsworth et al, 2009), and one from gastric cancer using laser capture microdissection (Wang et al, 2006b), were upregulated in PTC nodal metastases. Two signatures obtained by comparing stroma with other cell types (Finak et al, 2006; Casey et al, 2009) were also upregulated in the PTC metastases.
Surprisingly, the three cancer-derived signatures were downregulated in nodal metastases in the original publications (Wang et al, 2006b; Suzuki and Tarin, 2007; Ellsworth et al, 2009). To further investigate why the same signatures had an opposite regulation in PTC metastases, we estimated their expression in primary tumour RNA-seq data from The Cancer Genome Atlas (TCGA). All three signatures had a significantly higher median expression in breast than thyroid tumours (Supplementary Figure S2). Thus, the referential defining of ‘up-' and ‘nodal up-' and ‘downregulation' is different in these two types of cancer. An equal expression of stroma-related genes in nodal metastases could lead to opposite senses of differential expression when comparing with related primary tumours. We noted, however, that this tentative explanation could not apply to gastric cancer because it had the lowest stroma-related expression among all TCGA cancers.
Differential expression between N0 and N+ tumours is reproducible in patient-matched non-cancerous tissues, but not in independent PTC data sets
The unsupervised analysis (Figure 1) revealed no global difference between the transcriptomes of N0 and N+ primary tumours. Again, we searched for specific signatures of nodal status with a supervised analysis.
A preliminary analysis determined that nodal status was not confounded with any other recorded clinical variables in our samples (variables listed in Supplementary Table S1). Mutational status was not associated with nodal status: 16 out of 26, that is ∼60% of PTC were BRAF mutated (8 N0 and 8 N+). We detected 129 probes more expressed in N+ primary tumours compared with N0 and 11 less expressed (Supplementary Table S3). Gene set enrichment analysis uncovered a single significant gene set, a signature upregulated in nodal metastases in a previous breast cancer study (Suzuki and Tarin, 2007), but downregulated in N+ samples in our data set. As we will see below, such discrepancy is typical.
The same analysis was performed with the non-cancerous tissues adjacent to the N+ and N0 tumours. Eight hundred and forty-seven probes were detected as upregulated and 8 as downregulated in non-cancerous tissues adjacent to N+ tumours (Supplementary Table S3). Three gene sets reached significance with GSEA: calcium-independent cell–cell adhesion was upregulated; two signatures of upregulated genes in nodal metastases elsewhere (Feng et al, 2007; Suzuki and Tarin, 2007) were downregulated. Hence, the categories associated with nodal infiltration seemed similar in the tumour and matched non-cancerous tissues. To confirm this impression, we estimated with GSEA the expression of the tumour-derived N+ signature in the non-cancerous tissues (Figure 5A and B), and conversely, of the non-cancerous tissue-derived N+ signature in the tumours (Figure 5C and D). Both had a consistent expression in the alternate patient-matched tissue. In other words, there was high similarity in transcriptional signals associated with nodal status in the non-cancerous and tumour tissues. This is a straightforward consequence of the established fact (Figure 2) that transcriptomes of a tumour and its matched non-cancerous tissues were correlated on a global scale.
Figure 5.

Coherence of N0 vs N+ differential expression across tumour and non-cancerous tissues and across data sets. (A–D) The same N0 vs N+ differential expression is present in tumour and patient-matched non-cancerous tissues. We use the procedure and standard graphical representation of GSEA (Subramanian et al, 2005). Genes are ranked from the most upregulated in N+ to the most downregulated. Genes belonging to the investigated signature are denoted with black bars at the bottom of each panel. The green curve denotes the enrichment score. The more biased on the left, resp. right, the more the signature is upregulated in N+, resp. N0. (A) A signature comprising the 200 most upregulated genes in N+ primary tumours compared with N0 tumours, which was evaluated in non-cancerous thyroid tissues: it is upregulated as well. (B) Same as in (A) with the 200 most downregulated genes. (C and D) Same as in (A and B), except that the signatures were extracted from non-cancerous tissue data and then evaluated in primary tumours. (E) The N0 vs N+ differential expression is irreproducible across independent data sets (Delys et al, 2007; Detours et al, 2007; Dom et al, 2012). Rows depict data set and column N0 vs N+ signatures extracted from specific data sets. Signatures up- and downregulated in N+ are shown in different panels. Colours represent statistical significance, with black denoting P>0.05, red denoting significant downregulation and green significant upregulation. For example, the third column, Delys et al (2007), Down corresponds to the genes downregulated in N+ in the data set of Delys et al (2007). Hence, the colour of diagonal elements was set to grey. Delys et al (2007) signature, however, is not associated with nodal status in the present study nor in the study of Dom et al (2012), yet is significantly upregulated in N+ tumours in the TCGA thyroid data set. Thus, this signature is not corroborated in two data sets and is expressed in the opposite direction in a third data set. Similarly, discordant results were obtained in a negative control obtained by running the exact same calculations on the same data, but in which N0 and N+ class labels were permuted randomly across the samples of each study.
To validate these results, we considered the 129 and 11 genes sets as signatures and evaluated their expression in three published PTC data sets with nodal status information. These signatures were not consistently regulated. To assess comprehensively the discrepancies between data sets, we selected the 200 top genes ordered by their fold change of expression and associated with nodal status in each one of the three data sets and in our data set. We then tested all these signatures in all data sets (Figure 5C). No signature showed consistent expression across data sets.
Taken together, these results suggest that the signals associated with nodal status are cohort-specific – all the PTC cohorts are relatively small, except TCGA – and are associated with patient rather than tumour background.
Nodal invasion can be predicted from primary tumour transcriptomes for some, but not all cancer types
A number of studies present predictors of nodal status from primary tumour gene expression (Kikuchi et al, 2003; Tamoto et al, 2004; O'Donnell et al, 2005; Roepman et al, 2005; Inamura et al, 2007; Méndez et al, 2007; Pei et al, 2007; Kashiwazaki et al, 2008). Although the previous section ruins any hopes to find a relevant signal in our data, we conducted a comparable analysis to illustrate a widely overlooked statistical point. Using state-of-the-art machine learning methods and a nested cross-validation protocol immune to feature and model selection biases (Material and Methods), nodal status could be predicted with accuracy of 63%.
However, error estimation is variable depending on the specific split of the data into training and validation samples (Ambroise and McLachlan, 2002) – an issue rarely controlled for. We reran the above analysis 100 times on our data, but each time the N0/N+ labels were shuffled randomly among the samples. Twelve per cent of the runs produced accuracy estimates >63% (Figure 6A) in our data set, that is, a 63% accuracy has a high probability to occur by chance. We extended this analysis to other published PTC data sets and obtained similar results (Figure 6A), except for the TCGA thyroid data set.
Figure 6.

Classification accuracy of N0 vs N+. (A) Colour bars denote the fraction of correctly classified samples in each data sets. The black density lines denote the null distribution of this fraction in data sets in which the N+ and N0 class labels have been permuted randomly across samples. Accuracy reached significance only for the TCGA thyroid data set. (B) The same information as in (A) is represented in a compact format for 14 TCGA data sets. Each row stands for a specific type of cancer. Black shapes represent the null distribution of the N+ vs N0 classification accuracy, red bar the 95% confidence limits and blue squares the accuracy obtained on the original, non-permuted, data. Significant classification accuracies were obtained for breast, thyroid and head and neck cancers. PAAD, pancreatic cancer; STAD, stomach cancer; PRAD, prostate cancer; KIRC, kidney clear cell cancer; READ, rectal cancer; BLCA, bladder cancer; COAD, colon cancer; LUSC, lung small cell cancers; HNSC, head and neck cancer; LUAD, lung adenocarcinomas; THCA, thyroid cancer (same as in A); BRCA, breast cancer. Each data set was balanced as to have an equal number of N+ and N0 samples.
This problem is more likely with small data sets. With 340 PTC samples, the TCGA made it possible to overcome the small size of our data set. We ran the classification analysis, including the random shuffling control, on the TCGA PTC data set and all other TCGA data sets with nodal status information (Figure 6B). The N0 vs N+ classification accuracy was significant in thyroid, breast and head and neck, but not high (<70%). No evidence for N0 vs N+ classification accuracy better than random was found in 11 other cancers. Note that significant classification accuracies were obtained for large data sets. Thus, data set size and cancer type influenced the classification potential of expression data.
Discussion
Nodal metastases tended to be more similar to their patient-matched primary tumours than to unrelated primary tumours (Figure 2). This trend, also present in previous studies (Perou et al, 2000; Hao et al, 2004; O'Donnell et al, 2005; Weigelt et al, 2005; Roepman et al, 2006a; Feng et al, 2007; Inamura et al, 2007; Vecchi et al, 2008; Ellsworth et al, 2009), has been interpreted in the context of the stepwise and the ab initio models of metastatic progression. In contrast to the stepwise model, the ab initio model posits that metastatic potential is acquired early during tumour expansion and is detectable in bulk samples. It is also compatible with a small or no difference between primary and nodal tumours (Inamura et al, 2007; Ellsworth et al, 2009). However, the evidence provided by the expression profile comparison is limited, as it may depend on the cell types present in the samples (Roepman et al, 2006b). Here, we found that all the mismatched nodal samples were highly contaminated by lymphoid tissues. Another potential confounder is the presence of multiple primary foci (Weigelt et al, 2005). The interpretation of similarity results is further complicated by our findings that primary tumours are more similar to their contralateral non-cancerous thyroid tissues than to unrelated non-cancerous thyroid tissues. Thus, the stronger similarity between primary and nodal tumours is owing to both acquired tumour-specific features and patient-specific genetics and biology.
The global transcriptional profiles of primary PTCs and nodal metastases were very similar (Figure 1). Yet, a supervised analysis revealed hundreds of differentially expressed genes, most of them suggesting an important immune component in the metastases (Figure 3). Again, this result echoes studies conducted in other cancer types (Weigelt et al, 2005; Wang et al, 2006b; Vecchi et al, 2008; Ellsworth et al, 2009; Xie et al, 2010). A number of these studies have addressed or discussed the contamination of nodal samples by lymphoid tissues (Hoang et al, 2005; O'Donnell et al, 2005; Weigelt et al, 2005; Suzuki and Tarin, 2007; Vecchi et al, 2008; Ellsworth et al, 2009; Méndez et al, 2011). We proposed an original approach to the problem. We demonstrated that in our data fold changes were correlated to those obtained from the comparison of normal lymph nodes and non-cancerous thyroids (Figure 3), suggesting an overwhelming confounding effect of lymphoid tissues. Adjusting for lymphoid tissue content revealed the upregulation of five published stroma-related signatures in other tissues (Finak et al, 2006; Wang et al, 2006b; Suzuki and Tarin, 2007; Casey et al, 2009; Ellsworth et al, 2009). Four of five of these signatures were derived from the transcriptional profiles of laser capture microdissected primary and nodal tumours (Wang et al, 2006b; Ellsworth et al, 2009), or epithelium and stroma (Finak et al, 2006; Casey et al, 2009), suggesting that our statistical adjustment removed lymphoid contamination, but not the biologically relevant signal. Three of 9 and 8 of 13 genes were shared between two studies of microdissected lung and breast primary, respectively, with associated nodal tumours (Hoang et al, 2005; Ellsworth et al, 2009) and our unadjusted gene list, which mostly reflects immune infiltration. These overlaps could reflect imperfect microdissection or a small-amplitude immune signal unrelated to contamination. Finally, non-cancerous thyroid cells present in the primary tissue blocks could have produced the spurious downregulation of thyroid differentiation genes (Figure 3). Because the adjustment for the lymphoid signal also removed the thyroid signal (Figure 4B), the latter was probably an indirect consequence of lymphoid contamination: the transcriptomes of bulk tissues contaminated by lymphoid tissues are obviously less thyroid-like.
We compared the expression profiles of primary PTCs, which are associated or not with nodal metastasis. We found a signal associated with nodal invasion, in agreement with studies in other cancer types (Kikuchi et al, 2003; Tamoto et al, 2004; O'Donnell et al, 2005; Roepman et al, 2005; Inamura et al, 2007; Méndez et al, 2007; Pei et al, 2007; Kashiwazaki et al, 2008). To further gauge the significance of these results, we compared the signatures associated with N0/N+ obtained from four thyroid cancer data sets. The signature obtained from one data set was typically not associated with N0/N+ in the three others (Figure 5). Thus, our N0/N+ signature and, presumably, those published in other studies of comparable size, are not general markers of nodal invasion. Anecdotal comparisons of signatures already hinted at this conclusion (Méndez et al, 2007). Intriguingly, the same N0/N+ transcriptional signal was also present in the patient-matched non-cancerous tissues in our data (Figure 5). To the best of our knowledge, no other study presented this control. It suggests that the signature captures patient, rather than tumour-specific characteristics. This could arise from a predisposition to develop nodal metastases. However, the inconsistency of the signals suggests a cohort-specific bias. Only 2 (Takada et al, 2004; Kroon et al, 2008) among more than 20 studies we surveyed tested whether the N0/N+ status was associated with other available clinical variables. We searched for such association in our data, but did not find any. Yet, association with unobserved patient characteristics cannot be ruled out and is not unlikely given the limited sample size that characterizes the studies at hand.
Accuracy of N0 apart from N+ primary tumours classification was 63% in our thyroid cancer data set. However, a random permutation control demonstrated that an equal or higher accuracy might result from chance alone with a probability of 0.12. None of the studies we surveyed presented this control. It is particularly needed for small studies because accuracy estimates have a high variance (Figure 6). Published N0/N+ classification accuracies range from near perfect to near random. Several factors may account for this variation, including the type of cancer investigated, the comprehensiveness of lymph node investigation and technical details of the statistical procedure. Feature selection bias is an endemic flaw in the ‘omic' literature that results in near-perfect – but grossly inflated – classification accuracies (Ambroise and McLachlan, 2002). It occurs when the marker genes are determined using the entire data set and then when the model is adjusted and ‘validated' with classical cross-validation. Such approach breaks the statistical principle of separation of training and validation data. Five studies reporting accuracies of 89–100% presented this flaw (Takada et al, 2004; Tamoto et al, 2004; Inamura et al, 2007; Méndez et al, 2007, 2011). Studies without feature selection bias reported less successful classification. Focusing on the classification of N0 vs N+ with more than 10 nodes invaded in breast primary tumours, Huang et al (2003) obtained an accuracy of 90%. However, a less contrasted class definition yielded a modest 62% accuracy (Smeets et al, 2011), in line with our analysis of the TCGA data (57%, P=0/100, N=606; Figure 6) and in line with the fact that nodal status is a strong survival predictor that is independent of gene expression-based prognostic signatures in this disease (Van de Vijver et al, 2002; Van't Veer et al, 2002; Ramaswamy et al, 2003; Wang et al, 2005; Ivshina et al, 2006). In penile cancer accuracy was 54%, that is, near random (Kroon et al, 2008). It was 62–67% in colon cancer (Croner et al, 2005), but significance could not be reached in TCGA (56%, P=6/100, N=136). Excluding studies with feature selection bias (Takada et al, 2004; Inamura et al, 2007) and a study with only four validation samples (Kikuchi et al, 2003), N0/N+ lung cancers could not be classified (Xi et al, 2005). We did not reach significance with TCGA lung adenocarcinomas (54%, P=6/100, N=230) and small-cell carcinoma (56%, P=11/100, N=184). In oral cancer, Roepman et al (2005) reported a 77% correct classification and Nguyen et al (2007) reported a 92% correct classification using microdissected samples. The Cancer Genome Atlas head and neck cancer samples could be classified but with lower accuracy (62%, P=0/100, N=198). Overall, our TCGA analyses show that N0 and N+ tumours cannot be classified for most cancers: accuracies are typically in the range 50–60% and not significantly better than permutation controls. Papillary thyroid carcinomas are among the most amenable to N0/N+ classification, but accuracy is still limited (67%, P=0/100, N=340).
We have presented new biological and statistical controls that expose some of the limits of published studies of the relationship between primary tumours and nodal metastases. The potential clinical relevance of gene expression to predict nodal metastases seems limited for most cancers.
Acknowledgments
We thank Françoise Lallemand, Sandy Haussy, Samira Majjaj for their contribution, Chantal Degraef for her technical assistance, Andrea Karambelas for final proofreading and Jacques E Dumont for his advice. VD was funded by FNRS Grants 14657170 and 19556102. MT and MS were supported by FRIA/FNRS grant. Other supports include Welbio, Fondation Contre le Cancer, Fonds de la Recherche Scientifique Médicale (FRSM), Fonds National de la Recherche Scientifique (FNRS), Plan Cancer (action 29), Télévie, Les Amis de l'Institut Bordet and Fondation Van Buuren.
Author contributions
CM conceived the experiments; CM, MS, CT, CH, DL and GA collected the data and carried out experiments; MT analysed the data and generated the figures; MT, CM and VD interpreted the results; MT and VD wrote the manuscript.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on British Journal of Cancer website (http://www.nature.com/bjc)
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Material
References
- Ambroise C, McLachlan GJ. Selection bias in gene extraction on the basis of microarray gene-expression data. Proc Natl Acad Sci USA. 2002;99:6562–6566. doi: 10.1073/pnas.102102699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Thyroid Association (ATA) Guidelines Taskforce on Thyroid Nodules and Differentiated Thyroid Cancer. Cooper DS, Doherty GM, Haugen BR, Hauger BR, Kloos RT, Lee SL, Mandel SJ, Mazzaferri EL, McIver B, Pacini F, Schlumberger M, Sherman SI, Steward DL, Tuttle RM. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid. 2009;19:1167–1214. doi: 10.1089/thy.2009.0110. [DOI] [PubMed] [Google Scholar]
- Bidus MA, Risinger JI, Chandramouli GVR, Dainty LA, Litzi TJ, Berchuck A, Barrett JC, Maxwell GL. Prediction of lymph node metastasis in patients with endometrioid endometrial cancer using expression microarray. Clin Cancer Res. 2006;12:83–88. doi: 10.1158/1078-0432.CCR-05-0835. [DOI] [PubMed] [Google Scholar]
- Breitling R, Armengaud P, Amtmann A, Herzyk P. Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 2004;573:83–92. doi: 10.1016/j.febslet.2004.07.055. [DOI] [PubMed] [Google Scholar]
- Carlson M, Falcon S, Pages H, Li N.2014. hgu133plus2.db: Affymetrix Human Genome U133 Plus 2.0 Array annotation data (chip hgu133plus2), v2.14.0, , http://www.bioconductor.org/packages/release/data/annotation/html/hgu133plus2.db.html .
- Casey T, Bond J, Tighe S, Hunter T, Lintault L, Patel O, Eneman J, Crocker A, White J, Tessitore J, Stanley M, Harlow S, Weaver D, Muss H, Plaut K. Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast Cancer Res Treat. 2009;114:47–62. doi: 10.1007/s10549-008-9982-8. [DOI] [PubMed] [Google Scholar]
- Chang C-C, Lin C-J.2011LIBSVM: a library for support vector machines ACM Trans Intell Syst Technol 2271–27:27. [Google Scholar]
- Croner RS, Peters A, Brueckl WM, Matzel KE, Klein-Hitpass L, Brabletz T, Papadopoulos T, Hohenberger W, Reingruber B, Lausen B. Microarray versus conventional prediction of lymph node metastasis in colorectal carcinoma. Cancer. 2005;104:395–404. doi: 10.1002/cncr.21170. [DOI] [PubMed] [Google Scholar]
- Delys L, Detours V, Franc B, Thomas G, Bogdanova T, Tronko M, Libert F, Dumont JE, Maenhaut C. Gene expression and the biological phenotype of papillary thyroid carcinomas. Oncogene. 2007;26:7894–7903. doi: 10.1038/sj.onc.1210588. [DOI] [PubMed] [Google Scholar]
- Detours V, Delys L, Libert F, Weiss Solís D, Bogdanova T, Dumont JE, Franc B, Thomas G, Maenhaut C. Genome-wide gene expression profiling suggests distinct radiation susceptibilities in sporadic and post-Chernobyl papillary thyroid cancers. Br J Cancer. 2007;97:818–825. doi: 10.1038/sj.bjc.6603938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dom G, Tarabichi M, Unger K, Thomas G, Oczko-Wojciechowska M, Bogdanova T, Jarzab B, Dumont JE, Detours V, Maenhaut C. A gene expression signature distinguishes normal tissues of sporadic and radiation-induced papillary thyroid carcinomas. Br J Cancer. 2012;107:994–1000. doi: 10.1038/bjc.2012.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edfeldt K, Björklund P, Åkerström G, Westin G, Hellman P, Stålberg P. Different gene expression profiles in metastasizing midgut carcinoid tumors. Endocr Relat Cancer. 2011;18:479–489. doi: 10.1530/ERC-10-0256. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucl Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellsworth RE, Seebach J, Field LA, Heckman C, Kane J, Hooke JA, Love B, Shriver CD. A gene expression signature that defines breast cancer metastases. Clin Exp Metast. 2009;26:205–213. doi: 10.1007/s10585-008-9232-9. [DOI] [PubMed] [Google Scholar]
- Feng Y, Sun B, Li X, Zhang L, Niu Y, Xiao C, Ning L, Fang Z, Wang Y, Zhang L, Cheng J, Zhang W, Hao X. Differentially expressed genes between primary cancer and paired lymph node metastases predict clinical outcome of node-positive breast cancer patients. Breast Cancer Res Treat. 2007;103:319–329. doi: 10.1007/s10549-006-9385-7. [DOI] [PubMed] [Google Scholar]
- Fidler IJ, Kripke ML. Metastasis results from preexisting variant cells within a malignant tumor. Science. 1977;197:893–895. doi: 10.1126/science.887927. [DOI] [PubMed] [Google Scholar]
- Finak G, Sadekova S, Pepin F, Hallett M, Meterissian S, Halwani F, Khetani K, Souleimanova M, Zabolotny B, Omeroglu A, Park M. Gene expression signatures of morphologically normal breast tissue identify basal-like tumors. Breast Cancer Res. 2006;8:R58. doi: 10.1186/bcr1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman R.2014. annotate: Annotation for microarrays, v1.42.1, , http://www.bioconductor.org/packages/release/bioc/html/annotate.html .
- Hao X, Sun B, Hu L, Lähdesmäki H, Dunmire V, Feng Y, Zhang S-W, Wang H, Wu C, Wang H, Fuller GN, Symmans WF, Shmulevich I, Zhang W. Differential gene and protein expression in primary breast malignancies and their lymph node metastases as revealed by combined cDNA microarray and tissue microarray analysis. Cancer. 2004;100:1110–1122. doi: 10.1002/cncr.20095. [DOI] [PubMed] [Google Scholar]
- Hoang CD, Guillaume TJ, Engel SC, Tawfic SH, Kratzke RA, Maddaus MA. Analysis of paired primary lung and lymph node tumor cells: a model of metastatic potential by multiple genetic programs. Cancer Detect Prev. 2005;29:509–517. doi: 10.1016/j.cdp.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Hong F, Breitling R. A comparison of meta-analysis methods for detecting differentially expressed genes in microarray experiments. Bioinformatics. 2008;24:374–382. doi: 10.1093/bioinformatics/btm620. [DOI] [PubMed] [Google Scholar]
- Huang E, Cheng SH, Dressman H, Pittman J, Tsou MH, Horng CF, Bild A, Iversen ES, Liao M, Chen CM, West M, Nevins JR, Huang AT. Gene expression predictors of breast cancer outcomes. Lancet. 2003;361:1590–1596. doi: 10.1016/S0140-6736(03)13308-9. [DOI] [PubMed] [Google Scholar]
- Inamura K, Shimoji T, Ninomiya H, Hiramatsu M, Okui M, Satoh Y, Okumura S, Nakagawa K, Noda T, Fukayama M, Ishikawa Y. A metastatic signature in entire lung adenocarcinomas irrespective of morphological heterogeneity. Hum Pathol. 2007;38:702–709. doi: 10.1016/j.humpath.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Ivshina AV, George J, Senko O, Mow B, Putti TC, Smeds J, Lindahl T, Pawitan Y, Hall P, Nordgren H, Wong JEL, Liu ET, Bergh J, Kuznetsov VA, Miller LD. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006;66:10292–10301. doi: 10.1158/0008-5472.CAN-05-4414. [DOI] [PubMed] [Google Scholar]
- Kashiwazaki H, Hassan NMM, Hamada J-I, Moriuchi T, Yamazaki Y, Tei K, Totsuka Y, Inoue N. Gene expression profile changes correlated with lymph node metastasis in oral squamous cell carcinoma. Odontology. 2008;96:38–43. doi: 10.1007/s10266-008-0084-1. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, Daigo Y, Katagiri T, Tsunoda T, Okada K, Kakiuchi S, Zembutsu H, Furukawa Y, Kawamura M, Kobayashi K, Imai K, Nakamura Y. Expression profiles of non-small cell lung cancers on cDNA microarrays: identification of genes for prediction of lymph-node metastasis and sensitivity to anti-cancer drugs. Oncogene. 2003;22:2192–2205. doi: 10.1038/sj.onc.1206288. [DOI] [PubMed] [Google Scholar]
- Kroon BK, Leijte JAP, van Boven H, Wessels LFA, Velds A, Horenblas S, van't Veer LJ. Microarray gene-expression profiling to predict lymph node metastasis in penile carcinoma. BJU Int. 2008;102:510–515. doi: 10.1111/j.1464-410X.2008.07697.x. [DOI] [PubMed] [Google Scholar]
- Leek JT. The tspair package for finding top scoring pair classifiers in R. Bioinformatics. 2009;25:1203–1204. doi: 10.1093/bioinformatics/btp126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren CI, Hall P, Dickman PW, Zedenius J. Clinically significant prognostic factors for differentiated thyroid carcinoma: a population-based, nested case-control study. Cancer. 2006;106:524–531. doi: 10.1002/cncr.21653. [DOI] [PubMed] [Google Scholar]
- McCall MN, Bolstad BM, Irizarry RA. Frozen robust multiarray analysis (fRMA) Biostatistics. 2010;11:242–253. doi: 10.1093/biostatistics/kxp059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez E, Fan W, Choi P, Agoff SN, Whipple M, Farwell DG, Futran ND, Weymuller EA, Jr, Zhao L-P, Chen C. Tumor-specific genetic expression profile of metastatic oral squamous cell carcinoma. Head Neck. 2007;29:803–814. doi: 10.1002/hed.20598. [DOI] [PubMed] [Google Scholar]
- Méndez E, Lohavanichbutr P, Fan W, Houck JR, Rue TC, Doody DR, Futran ND, Upton MP, Yueh B, Zhao LP, Schwartz SM, Chen C. Can a metastatic gene expression profile outperform tumor size as a predictor of occult lymph node metastasis in oral cancer patients. Clin Cancer Res. 2011;17:2466–2473. doi: 10.1158/1078-0432.CCR-10-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata M, Fujita H, Ida H, Hoshina H, Inoue T, Seki Y, Ohnishi M, Ohyama T, Shingaki S, Kaji M, Saku T, Takagi R. Identification of potential biomarkers of lymph node metastasis in oral squamous cell carcinoma by cDNA microarray analysis. Int J Cancer. 2003;106:683–689. doi: 10.1002/ijc.11283. [DOI] [PubMed] [Google Scholar]
- Nguyen ST, Hasegawa S, Tsuda H, Tomioka H, Ushijima M, Noda M, Omura K, Miki Y. Identification of a predictive gene expression signature of cervical lymph node metastasis in oral squamous cell carcinoma. Cancer Sci. 2007;98:740–746. doi: 10.1111/j.1349-7006.2007.00454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon IJ, Shaha AR. Management of regional nodes in thyroid cancer. Oral Oncol. 2013;49:671–675. doi: 10.1016/j.oraloncology.2013.03.441. [DOI] [PubMed] [Google Scholar]
- O'Donnell RK, Kupferman M, Wei SJ, Singhal S, Weber R, O'Malley B, Cheng Y, Putt M, Feldman M, Ziober B, Muschel RJ. Gene expression signature predicts lymphatic metastasis in squamous cell carcinoma of the oral cavity. Oncogene. 2005;24:1244–1251. doi: 10.1038/sj.onc.1208285. [DOI] [PubMed] [Google Scholar]
- Pacini F, Schlumberger M, Dralle H, Elisei R, Smit JWA, Wiersinga W, European Thyroid Cancer Taskforce European consensus for the management of patients with differentiated thyroid carcinoma of the follicular epithelium. Eur J Endocrinol. 2006;154:787–803. doi: 10.1530/eje.1.02158. [DOI] [PubMed] [Google Scholar]
- Pei H, Zhu H, Zeng S, Li Y, Yang H, Shen L, Chen J, Zeng L, Fan J, Li X, Gong Y, Shen H. Proteome Analysis and Tissue Microarray for Profiling Protein Markers Associated with Lymph Node Metastasis in Colorectal Cancer. J Proteome Res. 2007;6:2495–2501. doi: 10.1021/pr060644r. [DOI] [PubMed] [Google Scholar]
- Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lønning PE, Børresen-Dale AL, Brown PO, Botstein D. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- R Core Team 2013. R: A Language and Environment for Statistical Computing.
- Roepman P, de Jager A, Koerkamp MJAG, Kummer JA, Slootweg PJ, Holstege FCP. Maintenance of Head and Neck Tumor Gene Expression Profiles upon Lymph Node Metastasis. Cancer Res. 2006;66:11110–11114. doi: 10.1158/0008-5472.CAN-06-3161. [DOI] [PubMed] [Google Scholar]
- Roepman P, de Koning E, van Leenen D, de Weger RA, Kummer JA, Slootweg PJ, Holstege FCP. Dissection of a metastatic gene expression signature into distinct components. Genome Biol. 2006;7:R117. doi: 10.1186/gb-2006-7-12-r117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepman P, Wessels LFA, Kettelarij N, Kemmeren P, Miles AJ, Lijnzaad P, Tilanus MGJ, Koole R, Hordijk G-J, Vliet PC, van der, Reinders MJT, Slootweg PJ, Holstege FCP. An expression profile for diagnosis of lymph node metastases from primary head and neck squamous cell carcinomas. Nat Genet. 2005;37:182–186. doi: 10.1038/ng1502. [DOI] [PubMed] [Google Scholar]
- Saiselet M, Floor S, Tarabichi M, Dom G, Hébrant A, van Staveren WCG, Maenhaut C. Thyroid cancer cell lines: an overview. Front Endocrinol (Lausanne) 2012;3:133. doi: 10.3389/fendo.2012.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeets A, Daemen A, Vanden Bempt I, Gevaert O, Claes B, Wildiers H, Drijkoningen R, Van Hummelen P, Lambrechts D, De Moor B, Neven P, Sotiriou C, Vandorpe T, Paridaens R, Christiaens MR. Prediction of lymph node involvement in breast cancer from primary tumor tissue using gene expression profiling and miRNAs. Breast Cancer Res Treat. 2011;129:767–776. doi: 10.1007/s10549-010-1265-5. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Tarin D. Gene expression profiling of human lymph node metastases and matched primary breast carcinomas: clinical implications. Mol Oncol. 2007;1:172–180. doi: 10.1016/j.molonc.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada M, Tada M, Tamoto E, Kawakami A, Murakawa K, Shindoh G, Teramoto K, Matsunaga A, Komuro K, Kanai M, Fujiwara Y, Shirata K, Nishimura N, Miyamoto M, Okushiba S, Kondo S, Hamada J, Katoh H, Yoshiki T, Moriuchi T. Prediction of lymph node metastasis by analysis of gene expression profiles in non-small cell lung cancer1. J Surg Res. 2004;122:61–69. doi: 10.1016/j.jss.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Tamoto E, Tada M, Murakawa K, Takada M, Shindo G, Teramoto K, Matsunaga A, Komuro K, Kanai M, Kawakami A, Fujiwara Y, Kobayashi N, Shirata K, Nishimura N, Okushiba S, Kondo S, Hamada J, Yoshiki T, Moriuchi T, Katoh H. Gene-expression profile changes correlated with tumor progression and lymph node metastasis in esophageal cancer. Clin Cancer Res. 2004;10:3629–3638. doi: 10.1158/1078-0432.CCR-04-0048. [DOI] [PubMed] [Google Scholar]
- Tomás G, Tarabichi M, Gacquer D, Hébrant A, Dom G, Dumont JE, Keutgen X, Fahey TJ, 3rd, Maenhaut C, Detours V. A general method to derive robust organ-specific gene expression-based differentiation indices: application to thyroid cancer diagnostic. Oncogene. 2012;31:4490–4498. doi: 10.1038/onc.2011.626. [DOI] [PubMed] [Google Scholar]
- Vecchi M, Confalonieri S, Nuciforo P, Viganò MA, Capra M, Bianchi M, Nicosia D, Bianchi F, Galimberti V, Viale G, Palermo G, Riccardi A, Campanini R, Daidone MG, Pierotti MA, Pece S, Di Fiore PP. Breast cancer metastases are molecularly distinct from their primary tumors. Oncogene. 2008;27:2148–2158. doi: 10.1038/sj.onc.1210858. [DOI] [PubMed] [Google Scholar]
- Van't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AAM, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Venkat R, Guerrero MA. Recent advances in the surgical treatment of differentiated thyroid cancer: a comprehensive review. Scientific World J. 2013;2013:425136. doi: 10.1155/2013/425136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Vijver MJ, He YD, van 't Veer LJ, Dai H, Hart AAM, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH, Bernards R. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhu J-S, Song M-Q, Chen G-Q, Chen J-L. Comparison of gene expression profiles between primary tumor and metastatic lesions in gastric cancer patients using laser microdissection and cDNA microarray. World J Gastroenterol. 2006;12:6949–6954. doi: 10.3748/wjg.v12.i43.6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhu J-S, Song M-Q, Chen G-Q, Chen J-L. Comparison of gene expression profiles between primary tumor and metastatic lesions in gastric cancer patients using laser microdissection and cDNA microarray. World J Gastroenterol. 2006;12:6949–6954. doi: 10.3748/wjg.v12.i43.6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Klijn JGM, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, Jatkoe T, Berns EMJJ, Atkins D, Foekens JA. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- Weigelt B, Wessels LFA, Bosma AJ, Glas AM, Nuyten DSA, He YD, Dai H, Peterse JL, van't Veer LJ. No common denominator for breast cancer lymph node metastasis. Br J Cancer. 2005;93:924–932. doi: 10.1038/sj.bjc.6602794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong F-H, Huang C-YF, Su L-J, Wu Y-C, Lin Y-S, Hsia J-Y, Tsai H-T, Lee S-A, Lin C-H, Tzeng C-H, Chen P-M, Chen Y-J, Liang S-C, Lai J-M, Yen C-C. Combination of microarray profiling and protein–protein interaction databases delineates the minimal discriminators as a metastasis network for esophageal squamous cell carcinoma. Int J Oncol. 2009;34:117–128. [PubMed] [Google Scholar]
- Xie HL, Li ZY, Gan RL, Li XJ, Zhang QL, Hui M, Zhou XT. Differential gene and protein expression in primary gastric carcinomas and their lymph node metastases as revealed by combined cDNA microarray and tissue microarray analysis. J Dig Dis. 2010;11:167–175. doi: 10.1111/j.1751-2980.2010.00432.x. [DOI] [PubMed] [Google Scholar]
- Xi L, Lyons-Weiler J, Coello MC, Huang X, Gooding WE, Luketich JD, Godfrey TE. Prediction of lymph node metastasis by analysis of gene expression profiles in primary lung adenocarcinomas. Clin Cancer Res. 2005;11:4128–4135. doi: 10.1158/1078-0432.CCR-04-2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.