

Abstract
Background:
This study aimed to investigate copy number variations (CNVs) of CYP17A1 and androgen receptor (AR) genes in serum cell-free DNA collected before starting abiraterone in 53 consecutive patients with castration-resistant prostate cancer (CRPC).
Methods:
Serum DNA was isolated and CNVs were analysed for AR and CYP17A1 genes using Taqman copy number assays. The association between CNVs and progression-free/overall survival (PFS/OS) was evaluated by the Kaplan–Meier method and log-rank test.
Results:
Median PFS of patients with AR gene gain was 2.8 vs 9.5 months of non-gained cases (P<0.0001). Patients with CYP17A1 gene gain had a median PFS of 2.8 months vs 9.2 months in the non-gained patients (P=0.0014). A lower OS was reported in both cases (AR: P<0.0001; CYP17A1: P=0.0085). Multivariate analysis revealed that PSA decline ⩾50%, AR and CYP17A1 CNVs were associated with shorter PFS (P<0.0001, P=0.0004 and P=0.0450, respectively), while performance status, PSA decline ⩾50%, AR CNV and DNA concentration were associated with OS (P=0.0021, P=0.0014, P=0.0026 and P=0.0129, respectively).
Conclusions:
CNVs of AR and CYP17A1 genes would appear to be associated with outcome of CRPC patients treated with abiraterone.
Keywords: abiraterone, androgen receptor, castration-resistant prostate cancer, circulating cell-free DNA, CYP17A1
The majority of patients with recurrent prostate cancer have previously received androgen deprivation therapy and, despite an initial response, become castration-resistant after 2–3 years of treatment. The mechanism by which the tumour acquires castration resistance is still not completely understood, but recent evidence that even low levels of testosterone can activate androgen receptor (AR) and its signalling cascade has led to the search for novel therapeutic strategies that specifically target members of AR pathway (Bishr and Saad, 2013; Karantanos et al, 2013).
Abiraterone is a potent and selective inhibitor of cytochrome P450 17 α-hydrolase (CYP17A1) enzyme that blocks androgen synthesis in the adrenal glands, testicles and tumour microenvironment (O'Donnell et al, 2004; Attard et al, 2008; Ferraldeschi et al, 2013). A randomised phase III trial of abiraterone vs placebo in patients with metastatic castration-resistant prostate cancer (CRPC) previously treated with docetaxel showed a significant increase in progression-free survival (PFS) and overall survival (OS) (de Bono et al, 2011; Fizazi et al, 2012). Moreover, a randomised phase III trial of abiraterone vs placebo in CRPC patients in the pre-docetaxel space reported an advantage in PFS and OS from abiraterone treatment (Ryan et al, 2013, 2015). These findings led to a large-scale use of abiraterone in clinical practice, but it has now become essential to identify the mechanisms of drug resistance so that patients who are most likely to benefit can be selected for treatment.
CYP17A1 and AR would seem to be the two most important actors in the AR signalling axis in the acquisition of resistance to abiraterone treatment (Friedlander et al, 2012; Lin et al, 2013). The CYP17A1enzyme is the specific target of abiraterone and catalyses key reactions in sex-steroid biosynthesis by mediating 17α-hydrolase and 17,20-lyase activities. Its gene is located on chromosome 10q24.3 (Waterman and Keeney, 1992). Intratumoral CYP17A1 overexpression has been detected in prostate cancer tissue biopsies from patients treated with abiraterone, suggesting that upregulation of the enzyme could have a key role in resistance to treatment (Cai et al, 2011; Lin et al, 2013). The AR gene is located on chromosome Xq12. It is known to be frequently amplified in prostate cancer tissue, especially in CRPC, and correlates with high protein expression, resulting in a putative escape mechanism from therapies targeting AR signalling (Edwards et al, 2003; Friedlander et al, 2012). The role of these genes as potential circulating biomarkers has yet to be explored.
The prognostic potential of circulating tumour DNA (liquid biopsy) has also been evaluated in prostate cancer in an attempt to identify biomarkers that are capable of predicting outcome and of monitoring the effect of specific therapies (Schwarzenbach et al, 2009; Ellinger et al, 2011). We hypothesised that genomic changes in CYP17A1 and AR genes may influence the sensitivity of CRPC cells to abiraterone.
In the present study, we analysed copy number variations (CNVs) of CYP17A1 and AR genes in serum cell-free DNA of CRPC patients treated with abiraterone and then correlated them with clinical outcome.
Materials and methods
Study design
Patients with metastatic CRPC without neuroendocrine differentiation in progression after docetaxel and treated with abiraterone were included in this retrospective study. Inclusion criteria were as follows: baseline serum testosterone <50 ng dl−1; Eastern Cooperative Oncology Group (ECOG) performance status ⩽2; adequate cardiac, hepatic, renal and bone marrow function; serum potassium level ⩾3.5 mmol l−1; and ongoing androgen deprivation therapy. Prior treatment with ketoconazole was not permitted. Patient serum was collected before the start of abiraterone treatment. Blood samples were drawn into 10-ml tubes without anticoagulant, which were maintained at room temperature, processed within 30 min, and stored at −80 °C. The study protocol was approved by IRST Ethical Committee and all patients signed informed consent.
Antitumour therapy consisted of 28-day cycles of daily abiraterone acetate 1000 mg with twice-daily prednisone 5 mg. Abiraterone was continued until there was evidence of progressive disease (PD) or unacceptable toxicity. Before starting therapy, patients underwent a baseline blood PSA assessment and a chest and abdominal CT scan. PSA response and toxicity were evaluated on a monthly basis. A CT scan was then performed every 3 months during treatment with abiraterone. Progressive disease was defined on the basis of Prostate Cancer Working Group 2 (PCWG2) criteria (Scher et al, 2008). Adverse events were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), version 3.
Detecting copy number gain of AR and CYP17A1 in blood from CRPC patients
CNV analyses of the two target genes were performed for all samples by duplex TaqMan quantitative real-time PCR (qPCR) assay (Applied Biosystems, Foster City, CA, USA). Androgen receptor and CYP17A1 genes were each evaluated in two loci using different assays. RNaseP, located on 14q11, was selected as the internal standard reference gene. This region is not known to be aberrant in prostate cancer. Serum DNA was extracted using QIAamp DNA Mini Kit (Qiagen, Milan, Italy), according to the manufacturer's instructions. DNA quality and concentration were determined using spectrophotometry quantification (NanoDrop ND-1000, Celbio, Milan, Italy). For each sample, 20 ng of DNA was analysed in triplicate using TaqMan Genotyping Master Mix and the primers for target and reference sequences. Three serum DNAs from healthy males over 40 years were singly tested, and then pooled and used as a calibrator, sample with no CNVs of target gene (Supplementary Figure S1). In the same run, each sample was evaluated for the two loci of the target gene (TaqMan Copy Number Assays ID: Hs04107225 (AR 1) and Hs04511283 (AR 2); Hs01583974 (CYP17A1 1) and Hs05126409 (CYP17A1 2)). Standard deviation results are reported in Supplementary Table S1.
Copy number variation analysis was performed using relative quantitation method by CopyCaller Software (Applied Biosystems). Final results were calculated as the average between the copy number values of the two gene loci. For CYP17A1, values >2.5 were considered as amplification and <1.5 as deletion. For AR, the cutoffs were >1.5 for amplification and <0.5 for deletion. Cutoffs were selected considering the copy number calculated for the healthy sample replicate group (Supplementary Figure S1) and of LNCap cell line (Supplementary Figure S2).
We calculated the copy number corrected, using the formula:
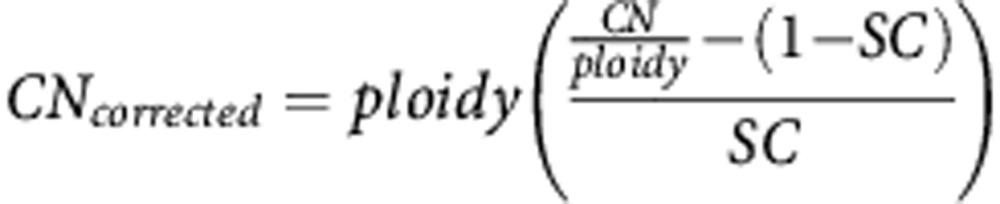 |
where CN=copy number variation observed; ploidy=1 for AR and 2 for CYP17A1; SC=sample content: percentage of DNA from patient in respect to total serum DNA.
Validation data with digital PCR
We validated samples for AR and CYP17A1 CNV with QuantStudio3D digital PCR system (Life Technologies, Carlsbad, CA, USA). The total PCR volume was 15 μl, containing serum DNA (15 ng), 0.9 μl of TaqMan assay for the genes of interest, 0.9 μl of the probe for the reference gene (RNaseP), and 9 μl of QuantStudio 3D Master mix (Life Technologies). The PCR was analysed by QuantStudio 3D software (Life Technologies). Poisson distribution was used to estimate the average number of copies per reaction microlitres. Ratio between target copies and reference copies was calculated for each sample. The results were compared with the healthy DNA pool.
Statistical analysis
Progression-free survival was defined as the time between the first day of treatment with abiraterone and the date of PD or death (whichever came first). Patients who had not progressed at database closure were censored at the final follow-up or discontinuation of treatment due to toxicity. Overall survival was defined as the time between the first day of abiraterone treatment and the date of death from any cause or the date of the last follow-up visit.
The association between CNVs and clinical outcome was evaluated by the Kaplan–Meier method and log-rank test. The percentage of early PD during treatment was calculated after 4 months by AR and CYP17A1 CNVs and compared using the Chi-Square test. A Cox regression model was used to estimate hazard ratios (HRs) and 95% confidence intervals (CIs) for PFS and OS. The multivariable Cox models included all factors that were significantly associated in the univariate models. The area under the receiver operating characteristic (ROC) curves and sensitivity and specificity indices were calculated to assess the diagnostic performance of CNVs in detecting progression and death. An area under the ROC curve close to one indicated high diagnostic accuracy. All P-values were two-sided and a P<0.05 was considered as statistically significant. Statistical analyses were performed with SAS 9.3 software (SAS Institute, Cary, NC, USA).
Results
AR and CYP17A1 copy number gain in CRPC
Fifty-three consecutive patients with metastatic CRPC who previously received docetaxel-based chemotherapy were treated with abiraterone between March 2011 and August 2012. Serum samples were collected at baseline and analysed for CNVs of AR and CYP17A1 genes. The median DNA concentration extracted was 6.8 ng μl−1 (range: 2.5–14.3 ng μl−1). Androgen receptor was gained in 16 cases and CYP17A1 in 15. Ten patients showed gain of both genes. No gene losses were detected. Data were validated by digital PCR (dPCR) experiments (Supplementary Figure S3). Approach sensitivity was tested using different ratio between DNA from patients, with AR and CYP17A1 gain, and DNA from healthy volunteers mixed together: the approach proved capable of detecting CNVs even when very low amounts of patients DNA were used. In particular, when the gene copy number was high, only 0.375% of DNA from patients was needed to detect the gain, for example, sample 2 (Figure 1A). We also calculated the correct CNV in relation with the percentage of patient DNA (Figure 1B).
Figure 1.

Approach sensitivity. (A) CNV analysis of AR and CYP17A1 genes from a healthy male and CRPC patient serum DNA pool. Blue line: cutoff for AR (1.5); red line: cutoff for CYP17A1 (2.5). We tested series of different ratios of DNA from patient (gain of AR and CYP17A1 gene) and healthy males mixed together for each sample. Patient DNA percentages were 0.375, 0.75, 1.5, 3, 6, 12, 25, 50 and 100, in respect to total serum DNA. The same healthy DNA was used as a calibrator. (B) Copy number corrected for AR (sample 1) and CYP17A1 (sample 3). Note: sample 2 and sample 4 were analysed only for the points described due to insufficient DNA.
The clinical characteristics of patients in relation to differences in CNVs of AR and CYP17A1 are reported in Table 1. Patients with an ECOG performance status of 0-1 vs 2 showed a statistically significant association with CNVs of AR and CYP17A1 genes and the combination of them (P=0.0209; P=0.0150; P=0.0023, respectively). Median baseline PSA, ALP and LDH levels were significantly associated with CNVs in both genes (Table 1).
Table 1. Patient characteristics.
|
AR |
CYP17A1 |
AR+CYP17A1 combined |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Characteristics | No. of cases (%) | N | A | P-value | N | A | P-value | NN | AA | AN | P-value |
| No. (%) | No. (%) | No. (%) | No. (%) | No. (%) | No. (%) | No. (%) | |||||
| Total | 53 (100) | 37 (100) | 16 (100) | – | 38 (100) | 15 (100) | – | 32 (100) | 10 (100) | 11 (100) | – |
| Median age, years (range) | 74 (57–87) | 74 (57–87) | 74 (57–85) | 0.6787 | 73 (57–87) | 76 (57–86) | 0.3139 | 74 (57–87) | 77 (57–85) | 72 (61–86) | 0.3465 |
|
Gleason score | |||||||||||
| 6–7 | 23 (43) | 18 (49) | 5 (31) | 0.3137 | 16 (42) | 7 (47) | 0.6112 | 14 (44) | 3 (30) | 6 (55) | 0.6232 |
| 8–9 | 29 (55) | 19 (51) | 10 (63) | 22 (58) | 7 (47) | 18 (56) | 6 (60) | 5 (45) | |||
| Unknown | 1 (2) | – | 1 (6) | – | 1 (7) | – | 1 (10) | – | |||
|
ECOG PS | |||||||||||
| 0–1 | 46 (87) | 35 (95) | 11 (69) | 0.0209 | 36 (95) | 10 (67) | 0.0150 | 30 (94) | 5 (50) | 11 (100) | 0.0023 |
| 2 | 7 (13) | 2 (5) | 5 (31) | 2 (5) | 5 (33) | 2 (6) | 5 (50) | – | |||
|
Bone metastases | |||||||||||
| No | 9 (17) | 8 (22) | 1 (6) | 0.2484 | 7 (18) | 2 (13) | 1.0000 | 6 (19) | – | 3 (27) | 0.2656 |
| Yes | 44 (83) | 29 (78) | 15 (94) | 31 (82) | 13 (87) | 26 (81) | 10 (100) | 8 (73) | |||
|
Visceral metastases | |||||||||||
| No | 41 (77) | 29 (78) | 12 (75) | 1.000 | 30 (79) | 11 (73) | 0.7220 | 25 (78) | 7 (70) | 9 (82) | 0.8087 |
| Yes | 12 (23) | 8 (22) | 4 (25) | 8 (21) | 4 (27) | 7 (22) | 3 (30) | 2 (18) | |||
|
No. of previous chemotherapeutic lines | |||||||||||
| 1 | 25 (47) | 19 (51) | 6 (37.5) | 0.3537 | 20 (53) | 5 (33) | 0.2049 | 18 (56) | 4 (40) | 3 (27) | 0.2217 |
| 2 or more | 28 (53) | 18 (49) | 10 (62.5) | 18 (47) | 10 (67) | 14 (44) | 6 (60) | 8 (73) | |||
| Median baseline PSA level, ng ml−1 (range) | 33.1 (1–1501) | 19.7 (1–1501) | 152.9 (13.2–1435) | 0.0064 | 21.9 (1–1501) | 292.4 (13.8–1435) | 0.0006 | 15.9 (1–1501) | 278.3 (13.8–1435) | 129.7 (13.2–1083) | 0.0007 |
| Median baseline ALP level, mU ml−1 (range) | 117 (35–1144) | 83 (35–1144) | 454 (68–1071) | 0.0017 | 93.5 (36–1144) | 383.5 (35–1071) | 0.0219 | 86 (36–1144) | 590 (84–1071) | 181 (35–1018) | 0.0032 |
| Median baseline LDH level, mU ml−1 (range) | 201 (48–999) | 192 (48–542) | 268.5 (121–999) | 0.0290 | 192 (48–961) | 250 (166–999) | 0.0173 | 189 (48–542) | 287 (203–999) | 224.5 (121–961) | 0.0154 |
Abbreviations; ALP=alkaline phosphatase; A=amplified; AR=androgen receptor; CYP17A1=cytochrome P450 17 α-hydrolase; ECOG=Eastern Cooperative Oncology Group; LDH=lactate dehydrogenase; N=normal; PS=performance status; PSA=prostate-specific antigen.
Correlation with PSA decline and early progressive disease
A PSA decline of ⩾50% was reported in 26 (49%) of the 53 patients (5 out of 16 (31%) with AR gene gain and 21 out of 37 (57%) individuals with no gain) (P=0.0881). Furthermore, PSA decline was significantly correlated with CNV of CYP17A1 present in 4 (27%) of the 15 patients with CYP17A1 gain and in 22 (58%) of the 38 individuals with no gain (P=0.0405).
Early PD, defined according to the PCWGC2 as treatment interruption occurring within 4 months of the start of abiraterone, was observed in 18 (34%) of the 53 patients. It was significantly correlated with CNVs of both AR and CYP17A1, occurring in 10 (62.5%) of the 16 patients with AR gene gain and in 8 (22%) of the 37 individuals with no gain (P=0.0039), and in 11 (73%) of the 15 patients with CYP17A1 gain and in 7 (18%) of the 38 individuals with no gain (P=0.0001).
Association with outcome
At the time of analysis, 44 of the 53 patients had PD and 30 patients had died. The median PFS and OS for the overall population were 6.8 months (95% CI: 4.6–9.7) and 17.6 months (95% CI: 10.8–21.9), respectively (Supplementary Figure S4). Overall, abiraterone was well tolerated, the majority of side effects reported as grade 1 or 2. Treatment-related complications requiring the interruption of abiraterone were heart failure (1 patient) and grade 3 hypertransaminase (1). The drug was also suspended for non treatment-related infective diarrhea (1 patient), pulmonary thromboembolism (1) and renal failure (1).
The median PFS of patients with AR gene gain was 2.8 months (95% CI: 2.2–5.6) vs 9.5 months (95% CI: 6.7–12.7) of those with no CNVs (P<0.0001). Patients with CYP17A1 gene gain had a median PFS of 2.8 months (95% CI: 0.9–4.3) vs 9.2 months (95% CI: 6.6–12.7) of those with two copies of the gene (P=0.0014). Considering the two gene CNVs together, we observed an increase in the difference between the median PFS values of patients with both genes gained compared to those with normal gene copy numbers: 2.5 months (95% CI: 0.3–2.9) vs 10.1 months (95% CI: 6.7–12.7), respectively. Individuals with only AR or CYP17A1 gain showed a median PFS of 5.3 months (95% CI: 2.4–14.8) (P<0.0001) (Figure 2).
Figure 2.

PFS—Kaplan Meier curves for AR (A), CYP17A1 (B) and combined AR+CYP17A1 (C). A=gene amplification; N=no gene aberration; AA=two-gene amplification; AN=only one gene amplification; NN=no aberrations for either gene.
The median OS of patients with AR gene gain was 5.0 months (95% CI: 2.7–12.3) compared with 21.9 months (95% CI: 14.4–NR) for individuals with one copy of the gene (P<0.0001). Patients with CYP17A1 gain showed a median OS of 4.3 months (95% CI: 1.8–18.6) vs 19.0 months of individuals with no gain (95% CI: 12.2–NR) (P=0.0085). The OS curve for the two genes combined showed 2.9 months (95% CI: 0.8–12.3) for the patients with two-gene gain and 21.9 months (95% CI: 14.4–NR) for individuals with normal copy number. A median OS of 10.8 months (95% CI: 3.2–NR) was observed for only one gene gain (P=0.0001) (Figure 3).
Figure 3.

OS—Kaplan Meier curves for AR (A), CYP17A1 (B) and combined AR+CYP17A1 (C). A=gene amplification; N=no aberration gene; AA=both genes amplification; AN=only one gene amplification; NN=no aberrations for either gene.
Both raw and adjusted hazard ratios for PFS and OS are summarised in Table 2. Multivariate analysis for PFS showed a significant association with AR and CYP17A1 CNVs (P=0.0004 and 0.0450, respectively), and PSA decline ⩾50% (P<0.0001). The same analysis for OS revealed a significant association with ECOG performance status (P=0.0021), PSA decline ⩾50% (P=0.0014), AR CNV (P=0.0026) and DNA concentration (P=0.0129). We also analysed the sensitivity and specificity of CNVs in PFS and OS (Supplementary Table S2).
Table 2. Univariate and multivariate analyses for progression-free survival and for overall survival.
|
Progression-free survival |
Overall survival |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Raw HR (95% CI) | P | Adjusted HR (95% CI) | P | Raw HR (95% CI) | P | Adjusted HR (95% CI) | P | ||
| Age, years | <75 vs ⩾75 | 1.11 (0.61–2.02) | 0.7259 | 0.94 (0.46–1.94) | 0.8755 | ||||
| Visceral metastases | Yes vs no | 0.05 (0.47–1.97) | 0.9038 | 1.04 (0.44–2.48) | 0.9293 | ||||
| No. of previous lines of treatment | ⩾2 vs 1 | 1.50 (0.82–2.75) | 0.1858 | 2.01 (0.92–4.41) | 0.0804 | ||||
| ECOG PS | 2 vs 0–1 | 1.91 (0.84–4.35) | 0.1236 | 3.29 (1.32–8.22) | 0.0107 | 5.37 (1.84–15.69) | 0.0021 | ||
| PSA decline⩾50% | No vs yes | 4.90 (2.49–9.62) | <0.0001 | 6.24 (2.91–13.37) | <0.0001 | 4.34 (1.95–9.67) | 0.0003 | 4.23 (1.74–10.24) | 0.0014 |
| AR CNV | A vs N | 3.73 (1.95–7.13) | <0.0001 | 4.06 (1.86–8.86) | 0.0004 | 4.68 (2.17–10.10) | <0.0001 | 3.59 (1.38–9.31) | 0.0026 |
| CYP17A1 CNV | A vs N | 2.79 (1.45–5.35) | 0.0021 | 2.20 (1.02–4.77) | 0.0450 | 2.61 (1.24–5.48) | 0.0111 | 0.92 (0.37–2.29) | 0.8576 |
| DNA concentration | ng μl−1 | 1.14 (1.02–1.28) | 0.0263 | 1.06 (0.93–1.21) | 0.3911 | 1.24 (1.09–1.42) | 0.0015 | 1.22 (1.04–1.43) | 0.0129 |
Abbreviations: A=amplified; AR=androgen receptor; CI=confidence interval; CNV=copy number variation; CYP17A1=cytochrome P450 A1; ECOG=Eastern Cooperative Oncology Group; HR=hazard ratio; PS=performance status; PSA=prostate-specific antigen; N=normal.
Discussion
In the present study, we showed a significant correlation between serum CNVs of AR and CYP17A1 genes and the clinical outcome of metastatic CRPC patients treated with abiraterone. Androgen receptor and CYP17A1 CNVs were associated with poor ECOG performance status, higher levels of PSA, ALP and LDH at baseline (Table 1). The majority of patients with AR or CYP17A1 gain showed early PD that occurred within 4 months of the start of abiraterone treatment, indicating that CNVs of AR and CYP17A1 genes are predictive of early resistance to therapy. Moreover, patients with CNVs of AR and/or CYP17A1 genes had poor PFS and OS (Figures 2 and 3). Multivariate analysis for PFS revealed a significant association with CNVs of AR and CYP17A1 and PSA decline ⩾50%, while the same analysis for OS showed a significant association with ECOG performance status, AR CNV, PSA decline ⩾50% and DNA concentration (Table 2). Thus, DNA concentration would seem to have a prognostic role in these patients. The lack of a significant association between CYP17A1 CNV and OS may be attributable to the effect of treatments administered after abiraterone, in particular enzalutamide, which is an inhibitor of AR but not of CYP17A1, so a potential impact on OS should be considered for these patients. If confirmed, then our results on CNVs of the CYP17A1 gene could influence the decision to personalise treatment in these patients. However, the most effective therapy sequence has yet to be defined for these patients (Loriot et al, 2013; Noonan et al, 2013; Schrader et al, 2014).
The main limitations of this study were the small sample size and the absence of an independent validation set. Our results should thus be considered preliminary and hypothesis-generating. It can be hypothesised that abiraterone-resistant patients with CYP17A1 gain but a normal AR copy number may benefit from treatment with enzalutamide and from other therapies that do not target CYP17A1 (with a favourable impact on the OS). It would be interesting to evaluate the role of AR and CYP17A1 CNVs in well-designed prospective studies on larger series of patients.
Of note, we evaluated AR and CYP17A1 genes in serum circulating-free DNA. This ‘liquid biopsy' provides valuable information on the characteristics of disease at the time of blood sample collection, thus eliminating the need for biopsies at difficult metastatic sites (Crowley et al, 2013; Murtaza et al, 2013). We analysed CNVs of specific regions: intron 1 and exon 3 of CYP17A1 and the AR ligand-binding domain. Literature data report different regulation mechanisms and genomic alterations for CYP17A1 (Taylor et al, 2010; Friedlander et al, 2012; Grasso et al, 2012; Mitsiades et al, 2012; Murtaza et al, 2013). Friedlander et al (2012) showed that, although around 50% of metastatic CRPC exhibited CYP17A1 heterozygous deletions, tumours were capable of upregulating CYP17A1 through methylation of CpG sites in the gene body. Conversely, we observed that CYP17A1 was gained in about 28% of treated patients and hypothesised that this gain was correlated with higher expression. There may, in fact, be different mechanisms that regulate the expression of this enzyme involved in androgen synthesis (Friedlander et al, 2012). When CYP17A1 is overexpressed, only a part of CYP17A1 molecules are inhibited by abiraterone and the tumour microenvironment may nonetheless continue to stimulate testosterone synthesis (Mitsiades et al, 2012). At the same time, AR amplification, leading to increased protein expression, could lead to AR axis activation in the absence of testosterone stimulation. These two resistance mechanisms could help to explain why patients with AR or CYP17A1 gene amplification appear to be resistant to abiraterone. The AR gain is not a novel concept in the pathogenesis of prostate cancer and is known to be present in about 30% of CRPC patients, correlating with higher protein expression (Tsao et al, 2012). A recent study similarly showed that AR copy number gain and AR ligand binding domain point mutations associate with resistance to treatment (Carreira et al, 2014). Recently, an association has been found between a splice-variant of AR (AR-V7) and resistance to both abiraterone and enzalutamide, detecting in circulating tumour cells of CRPC patients (Antonarakis et al, 2014). Androgen receptor gene CNV and alternative splicing could be associated as resistance mechanisms.
In this era of personalised medicine, the importance of identifying new predictive markers for targeted therapies is now widely accepted in CRPC (Logothetis et al, 2013; De Giorgi et al, 2014). Copy number variations will have an important role and new techniques such as next-generation sequencing (NGS) could accurately allow their estimation together with sequencing of somatic point mutations (Carreira et al, 2014). Our results highlighted AR and CYP17A1 CNVs as non-invasive markers capable of identifying patients who are likely to benefit from treatment with abiraterone. Larger prospective studies are warranted to validate these circulating biomarkers as predictors of outcome to treatment with abiraterone and possibly other hormone therapies in patients with CRPC.
Acknowledgments
We thank Ursula Elbling for editing the manuscript. Vincenza Conteduca acknowledges support from an ESMO Translational Research Fellowships 2014–2015.
UG and VC received speaker honorarium from Janssen-Cilag. The ICR developed abiraterone and therefore has a commercial interest in this agent. GA is on the ICR list of rewards to inventors for abiraterone. GA has received honoraria, consulting fees or travel support from Astellas, Medivation, Janssen, and grant support from Janssen.
Footnotes
Supplementary Information accompanies this paper on British Journal of Cancer website (http://www.nature.com/bjc)
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Results of this study have been presented in part at the European Society of Medical Oncology (ESMO) Cancer Congress 2014 in Madrid, Spain, 26 September–30 September, 2014 (poster discussion session).
Supplementary Material
References
- Antonarakis ES, Lu C, Wang HN, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, Isaacs JT, Isaacs WB, Nadal R, Paller CJ, Denmeade SR, Carducci MA, Eisenberger MA, Luo J. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371 (11:1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, Settatree S, Barrett M, Parker C, Martins V, Folkerd E, Clark J, Cooper CS, Kaye SB, Dearnaley D, Lee G, de Bono JS. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26 (28:4563–4571. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- Bishr M, Saad F. Overview of the latest treatments for castration-resistant prostate cancer. Nat Rev Urol. 2013;10 (9:522–528. doi: 10.1038/nrurol.2013.137. [DOI] [PubMed] [Google Scholar]
- Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, Marck B, Matsumoto AM, Simon NI, Wang H, Chen S, Balk SP. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011;71 (20:6503–6513. doi: 10.1158/0008-5472.CAN-11-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira S, Romanel A, Goodall J, Grist E, Ferraldeschi R, Miranda S, Prandi D, Lorente D, Frenel JS, Pezaro C, Omlin A, Rodrigues DN, Flohr P, Tunariu N, de Bono JS, Demichelis F, Attard G. Tumor clone dynamics in lethal prostate cancer. Sci Transl Med. 2014;6 (254:254ra125. doi: 10.1126/scitranslmed.3009448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;10 (8:472–484. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Jr, Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Fléchon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364 (21:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Giorgi U, Caroli P, Burgio SL, Menna C, Conteduca V, Bianchi E, Fabbri F, Carretta E, Amadori D, Paganelli G, Matteucci F. Early outcome prediction on 18F-fluorocholine PET/CT in metastatic castration-resistant prostate cancer patients treated with abiraterone. Oncotarget. 2014;5 (23:12448–12458. doi: 10.18632/oncotarget.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards J, Krishna NS, Grigor KM, Bartlett JM. Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br J Cancer. 2003;89 (3:552–556. doi: 10.1038/sj.bjc.6601127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinger J, Müller SC, Stadler TC, Jung A, von Ruecker A, Bastian PJ. The role of cell-free circulating DNA in the diagnosis and prognosis of prostate cancer. Urol Oncol. 2011;29 (2:124–129. doi: 10.1016/j.urolonc.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Ferraldeschi R, Sharifi N, Auchus RJ, Attard G. Molecular Pathways: Inhibiting steroid biosynthesis in prostate cancer. Clin Cancer Res. 2013;19 (13:3353–3359. doi: 10.1158/1078-0432.CCR-12-0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fizazi K, Scher HI, Molina A, Logothetis CJ, Chi KN, Jones RJ, Staffurth JN, North S, Vogelzang NJ, Saad F, Mainwaring P, Harland S, Goodman OB, Jr, Sternberg CN, Li JH, Kheoh T, Haqq CM, de Bono JS. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012;13 (10:983–992. doi: 10.1016/S1470-2045(12)70379-0. [DOI] [PubMed] [Google Scholar]
- Friedlander TW, Roy R, Tomlins SA, Ngo VT, Kobayashi Y, Azameera A, Rubin MA, Pienta KJ, Chinnaiyan A, Ittmann MM, Ryan CJ, Paris PL. Common structural and epigenetic changes in the genome of castration-resistant prostate cancer. Cancer Res. 2012;72 (3:616–625. doi: 10.1158/0008-5472.CAN-11-2079. [DOI] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, Asangani IA, Ateeq B, Chun SY, Siddiqui J, Sam L, Anstett M, Mehra R, Prensner JR, Palanisamy N, Ryslik GA, Vandin F, Raphael BJ, Kunju LP, Rhodes DR, Pienta KJ, Chinnaiyan AM, Tomlins SA. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487 (7406:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013;32 (49:5501–5511. doi: 10.1038/onc.2013.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Gout PW, Wang Y. Lessons from in-vivo models of castration resistant prostate cancer. Curr Opin Urol. 2013;23 (3:214–219. doi: 10.1097/MOU.0b013e32835e9f07. [DOI] [PubMed] [Google Scholar]
- Logothetis CJ, Gallick GE, Maity SN, Kim J, Aparicio A, Efstathiou E, Lin SH. Molecular classification of prostate cancer progression: foundation for marker-driven treatment of prostate cancer. Cancer Discov. 2013;3 (8:849–861. doi: 10.1158/2159-8290.CD-12-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loriot Y, Bianchini D, Ileana E, Sandhu S, Patrikidou A, Pezaro C, Albiges L, Attard G, Fizazi K, De Bono JS, Massard C. Antitumour activity of abiraterone acetate against metastatic castration-resistant prostate cancer progressing after docetaxel and enzalutamide (MDV3100) Ann Oncol. 2013;24 (7:1807–1812. doi: 10.1093/annonc/mdt136. [DOI] [PubMed] [Google Scholar]
- Mitsiades N, Sung CC, Schultz N, Danila DC, He B, Eedunuri VK, Fleisher M, Sander C, Sawyers CL, Scher HI. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res. 2012;72 (23:6142–6152. doi: 10.1158/0008-5472.CAN-12-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, Marass F, Humphray S, Hadfield J, Bentley D, Chin TM, Brenton JD, Caldas C, Rosenfeld N. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497 (7447:108–112. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- Noonan KL, North S, Bitting RL, Armstrong AJ, Ellard SL, Chi KN. Clinical activity of abiraterone acetate in patients with metastatic castration-resistant prostate cancer progressing after enzalutamide. Ann Oncol. 2013;24 (7:1802–1807. doi: 10.1093/annonc/mdt138. [DOI] [PubMed] [Google Scholar]
- O'Donnell A, Judson I, Dowsett M, Raynaud F, Dearnaley D, Mason M, Harland S, Robbins A, Halbert G, Nutley B, Jarman M. Hormonal impact of the 17alpha-hydroxylase/C (17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br J Cancer. 2004;90 (12:2317–2325. doi: 10.1038/sj.bjc.6601879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, Fizazi K, Mainwaring P, Piulats JM, Ng S, Carles J, Mulders PF, Basch E, Small EJ, Saad F, Schrijvers D, Van Poppel H, Mukherjee SD, Suttmann H, Gerritsen WR, Flaig TW, George DJ, Yu EY, Efstathiou E, Pantuck A, Winquist E, Higano CS, Taplin ME, Park Y, Kheoh T, Griffin T, Scher HI, Rathkopf DE. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368 (2:138–148. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan CJ, Smith MR, Fizazi K, Saad F, Mulders PF, Sternberg CN, Miller K, Logothetis CJ, Shore ND, Small EJ, Carles J, Flaig TW, Taplin ME, Higano CS, de Souza P, de Bono JS, Griffin TW, De Porre P, Yu MK, Park YC, Li J, Kheoh T, Naini V, Molina A, Rathkopf DE. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015;16 (2:152–160. doi: 10.1016/S1470-2045(14)71205-7. [DOI] [PubMed] [Google Scholar]
- Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, Carducci MA, Eisenberger MA, Higano C, Bubley GJ, Dreicer R, Petrylak D, Kantoff P, Basch E, Kelly WK, Figg WD, Small EJ, Beer TM, Wilding G, Martin A. Prostate Cancer Clinical Trials Working Group. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol. 2008;26 (7:1148–1159. doi: 10.1200/JCO.2007.12.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader AJ, Boegemann M, Ohlmann Ch, Schnoeller TJ, Krabbe LM, Hajili T, Jentzmik F, Stoeckle M, Schrader M, Herrmann E, Cronauer MV. Enzalutamide in castration-resistant prostate cancer patients progressing after docetaxel and abiraterone. Eur Urol. 2014;65 (1:30–36. doi: 10.1016/j.eururo.2013.06.042. [DOI] [PubMed] [Google Scholar]
- Schwarzenbach H, Alix-Panabières C, Müller I, Letang N, Vendrell JP, Rebillard X, Pantel K. Cell-free tumor DNA in blood plasma as a marker for circulating tumor cells in prostate cancer. Clin Cancer Res. 2009;15 (3:1032–1038. doi: 10.1158/1078-0432.CCR-08-1910. [DOI] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18 (1:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao CK, Galsky MD, Small AC, Yee T, Oh WK. Targeting the androgen receptor signalling axis in castration-resistant prostate cancer (CRPC) BJU Int. 2012;110 (11:1580–1588. doi: 10.1111/j.1464-410X.2012.11445.x. [DOI] [PubMed] [Google Scholar]
- Waterman MR, Keeney DS. Genes involved in androgen biosynthesis and the male phenotype. Horm Res. 1992;38 (5-6:217–221. doi: 10.1159/000182546. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.