

Abstract
NO-aspirin (NO-ASA), consisting of aspirin and a nitric oxide-releasing group, is safer than aspirin and effective in colon cancer prevention. Here, we examined the mechanism of action of NO-ASA by focusing primarily on its effects on the cell cycle. NO-ASA reduced the growth of several cell lines from colon, pancreas, skin, cervix and breast cancer much more potently than aspirin, with 24-h IC50 values of 133-268 μM, while those of ASA were >1,000 μM. NO-ASA elevated the intracellular levels of reactive oxygen species, generating a state of oxidative stress. In all cell lines examined, NO-ASA induced cell cycle arrest in the G2/M phase transition accompanied by altered expression of G2/M transition-related proteins. In SW480 colon cancer cells NO-ASA modulated proteins controlling this transition. Thus, it markedly increased the levels of cyclin B1, decreased the expression of cyclin D1 and Cdc25C, and increased the Thr14/Tyr15-phosphorylation of Cdk1 while leaving unchanged its protein levels. These changes, including the G2/M arrest, were prevented by pretreating the cells with the anti-oxidant N-acetyl-cysteine, indicating that redox signaling is likely responsible for the cell cycle changes, a conclusion consi stent with the known redox regulation of these proteins. Collectively, these results confirm the profound cytokinetic effect of NO-ASA and provide strong evidence that it regulates cell cycle transitions through its ability to induce oxidative stress, which activates redox signaling in the target cell.
Keywords: nitric oxide, aspirin, cell cycle
Introduction
Cancer is a group of diseases characterized by uncontrolled growth and spread of abnormal cells. The American Cancer Society estimates that in 2011 about 101,340 new cancer cases are expected to be diagnosed in USA and about 49,380 Americans are expected to die of cancer (1). A large body of evidence has shown that aspirin and other non-steroidal anti-inflammatory drugs (NSAIDs) possess the ability to prevent colon and other cancers (2). The limited efficacy of NSAIDs coupled with significant adverse effects has provided the impetus to develop agents that are safer and more efficacious (3,4). Nitric oxide-donating NSAIDs (NO-NSAIDs) represent such an approach. NO-NSAIDs consist of a traditional NSAID that bears a nitric oxide-releasing moiety which reduces gastric toxicity (5).
Nitric oxide-donating aspirin (NO-ASA), the most widely studied NO-NSAID (Fig. 1), has been shown to be very effective in preventing colon and pancreatic cancers in in vitro and in animal tumor models (6,7). In terms of safety, animal data have provided evidence of the superior safety of NO-ASA (7-9). In addition, the gastroduodenal toxicity of NO-ASA in healthy volunteers was reported to be equivalent to that of placebo (10). It is now clear that NO-ASA has a significant potential for the prevention and perhaps the treatment of colon cancer. The mechanism of action of NO-ASA on cancer is, however, not yet fully understood. The anti-proliferative effect of NO-ASA has been observed in colon cancer cell lines (10,11) and a cisplatin-sensitive human ovarian cancer cell line (12). NO-ASA induced apoptosis in cisplatin-sensitive human ovarian cancer cells by activating Bax and releasing cytochrome c (13).
Figure 1.
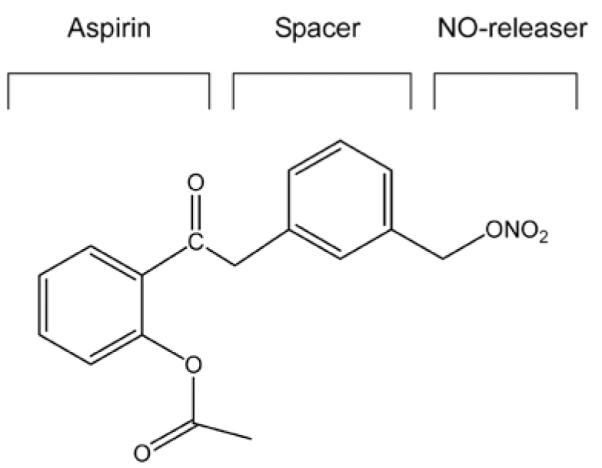
The chemical structure of NO-ASA. NO-ASA consists of aspirin, a spacer moiety and a nitric oxide-releasing moiety (−ONO2). This is the meta-isomer of NO-ASA in terms of the position of the −ONO2 moiety with respect to its closest carboxylic ester.
Given the potential of NO-ASA as an anti-cancer agent, we assessed its effect on cell cycle. The present study demonstrates that NO-ASA treatment causes a G2/M phase cell cycle arrest in cultured human cancer cell lines associated with marked changes in the expression of proteins regulating this phase transition.
Materials and methods
Reagents
NO-ASA [2-(acetyloxy)benzoic acid 3-(nitrooxymethyl)phenyl ester] was provided by Nicox, SA, Sophia Antipolis, France. Dihydroethidium (DHE) and 2′,7′-dichlorofluorescine diacetate (H2DCFDA) were obtained from Calbiochem. DMSO was from Fisher Scientific, Fair Lawn, NJ. N-acetyl-L-cysteine (NAC) and aspirin were from Sigma Chemical, St. Louis, MO. Primary antibodies were from the following sources (the final concentrations used are indicated in parentheses): rabbit polyclonal antibodies against caspase 3 (1:500), PARP (1:500), Cdc25C (1:500), p-Cdc2 (Thr14/Tyr15), CDC2 and mouse monoclonal antibody against cyclin B1 (1:500) were all from Santa Cruz Biotechnology, Santa Cruz, CA. The rabbit polyclonal antibody against cyclin D1 (1:1,000) was from Upstate Cell Signaling Solution, Lake Placid, NY. The mouse monoclonal antibody against α-tubulin (1:1,500) was from Oncogene Research Products, San Diego, CA. Stock solutions of NO-ASA (100 mM), aspirin (100 mM), and NAC (250 mM) were prepared in DMSO and stored at −20°C.
Cell culture
SW480, HT-29, HCT-15, and LoVo human colon adenocarcinoma cells, BxPC-3 human pancreatic adenocarcinoma cell, A431 human skin carcinoma cell, HeLa human cervix adenocarcinoma cell, and MCF-7 human breast adenocarcinoma cell were obtained from American Type Culture Collection. The SW480, HCT-15, BxPC-3 cells were grown in RPMI-1640; A431, HeLa and MCF-7 cells were grown in Dulbecco’s modified Eagle’s medium; LoVo cells were grown in F-12; and the colon HT-29 cells in McCoy 5A medium. All media were supplemented with 10% fetal calf serum (Mediatech, Herndon, VA), penicillin (50 U/ml), and streptomycin (50 μg/ml) (Invitrogen, Carlsbad, CA).
Cell growth assay
For each cell line, 8,300 cells/well (3,000 cells/well for A431) were seeded in 96-well plate and allowed to attach overnight, the medium was replaced with fresh complete medium containing the various concentrations of NO-ASA. After 48 h of treatment, cell viability was measured by the MTT assay from Boehringer Mannheim (Roche Diagnostics Corp., Indianapolis, IN) according to the instructions of the manufacturer.
Cell cycle analysis
For each cell line, 2.5×105 cells/well were seeded into 6-well plate and allowed to attach overnight. The medium was replaced with fresh complete medium containing NO-ASA or aspirin at the indicated concentrations. At the indicated time points, cells were harvested and fixed in 70% ethanol. Cells were then pelleted and resuspended in 0.5 ml of 50 μg/ml propidium iodide in PBS containing 20 μg/ml RNase for 30 min. The stained cells were analyzed using a flow cytometer. In some experiments, the cells were treated with NO-ASA in the presence of NAC.
Immunoblotting
After treatment with the NO-ASA, cells were harvested and lysed for 15 min on ice in 50 mM Tris-Cl buffer (pH 7.5) containing 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.2% SDS, 1 mM phenylmethylsulfonyl fluoride, 1 mM sodium fluoride, 1 mM sodium orthovanadate, aprotinin 10 μg/ml and leupeptin 10 μg/ml. The lysates were centrifuged at 10,000 × g for 15 min at 4°C. The extracts were subjected to SDS-10% or 15% polyacrylamide gel electrophoresis and transferred to nitrocellulose by electroblotting. Proteins were probed with primary antibody and visualized using an ECL kit (Amersham) according to the manufacturer’s instructions.
Determination of reactive oxygen species (ROS)
Cells were pretreated with either 5 μM 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) or 5 μM dihydroethidium (DHE) in RPMI medium without FCS or red phenol for 1 h, as described (11). Then cells were incubated at indicated concentrations of NO-ASA for 4 h. H2DCFDA (non-fluorescent) can be oxidized to its fluorescent product, dichlorofluorescein. H2DCFDA is a probe for H2O2 and other peroxides and thus their generation is measured by monitoring the increasing fluorescence. DHE is the intracellular probe that preferentially measures superoxide anion (O2·−); DHE is oxidized by O2·− to the fluorogenic ethidium. Fluorescence of cells was measured by flow cytometry.
Results
NO-ASA inhibits cell growth and induces G2/M arrest
The effects of NO-ASA were evaluated on colon, pancreas, skin, breast and cervical cancer cell lines. Following a 48-h treatment of these cell lines, NO-ASA inhibited their growth with similar potency (IC50 from 133 to 268 μM; Table I). In contrast, even at the maximum tested concentration (1 mM), we were not able to obtain an IC50 value for aspirin.
Table I.
Effect of NO-ASA on cell growth and cell cycle in human cancer cell lines.
| Cell cycle distribution | |||||
|---|---|---|---|---|---|
| Cell lines | IC50 (μM) | G0/G1 | S | G2M | |
| HT-29 colon | 174 | Control | 63.2 | 16.3 | 20.5 |
| Treated | 39.4 | 14.8 | 45.8 | ||
| SW480 colon | 256 | Control | 63.7 | 17.9 | 18.5 |
| Treated | 8.4 | 25.7 | 65.9 | ||
| HCT-15 colon | 257 | Control | 45.4 | 32.9 | 21.7 |
| Treated | 15.4 | 12.3 | 72.3 | ||
| LoVo colon | 175 | Control | 43.6 | 23.1 | 33.3 |
| Treated | 31.9 | 18.7 | 49.4 | ||
| BxPC-3 pancreas | 175 | Control | 50.9 | 25.0 | 24.1 |
| Treated | 18.4 | 17.3 | 64.3 | ||
| A431 skin | 133 | Control | 59.8 | 18.1 | 22.1 |
| Treated | 18.9 | 25.9 | 55.3 | ||
| HeLa cervix | 240 | Control | 45.9 | 24.6 | 29.5 |
| Treated | 27.4 | 20.1 | 52.2 | ||
| MCF-7 breast | 268 | Control | 63.2 | 6.6 | 30.2 |
| Treated | 47.7 | 17.2 | 35.1 | ||
To elucidate the mechanism of cell growth inhibition by NO-ASA, fixed and PI stained cells were subjected to flow cytometric analysis. In all cases, cell lines treated for 24-h with NO-ASA resulted in a significant G2/M arrest, which was accompanied by a significant decrease of the proportion of cells in the G0/G1 phase (Table I). These results suggest a generalized growth inhibitory effect of NO-ASA on cell lines derived from cancers from various tissue types which is at least in part caused by the G2/M cell cycle arrest.
NO-ASA modulates the expression of G2/M phase-related regulators
Eukaryotic cell cycle progression is regulated by sequential activation of Cdks, whose activity is dependent upon their association with regulatory cyclins (15-17). In most organisms the complex between Cdk1 (also known as CDC2) and cyclin B1 is important for entry into mitosis. The activity of Cdk1/cyclin B1 is negatively regulated by reversible phosphorylation at the Thr14 and Tyr15 residues of Cdk1. Dephosphorylation of the Thr14 and Tyr15 residues of Cdk1, and hence activation of Cdk1/cyclin B1 kinase complex, is catalyzed by the Cdc25 dual specificity phosphatases (Cdc25B and Cdc25C). These reactions are believed to be the rate-limiting step for entry into mitosis.
To examine the mechanism for G2/M arrest by NO-ASA, we determined the effect of NO-ASA on the levels of the G2/M phase regulating proteins cyclin B1, Cdk1 (CDC2) and Cdc25C in SW480 cells treated with 200 μM NO-ASA for up to 72 h. Cyclin B1, which is generally detected in the G2 and early mitotic phases of the cell cycle, was markedly increased after 1 h of treatment (maximum level at 12 h), maintaining a strong expression after 24 h (Fig. 2A). This effect coincided with the accumulation of cells in the G2/M fraction by flow cytometry. After 48 h of treatment, cyclin B1 decreased, reaching almost zero at 72 h (Fig. 2A). This finding implies a G2/M phase arrest. The expression of Cdk1 did not change throughout the treatment period. Cyclin D1 expression, generally detected in the G1 phase, decreased in a time-dependent manner; its expression was reduced to approximately one tenth of its value at baseline (0 h; Fig. 2A). This also coincided with reduction of the cells in G1 phase. Similarly, 24-h treatment of NO-ASA increased the expression of cyclin B1 in a concentration-dependent manner, an effect accompanied by decreased expression of cyclin D1. Treatment with NO-ASA for 24 h did not affect the level of Cdk1 (Fig. 2B). On the other hand, exposure of SW480 cells to NO-ASA resulted in a significant decrease in the level of Cdc25C in a concentration-dependent fashion (Fig. 2B). These results suggest that the NO-ASA-induced G2/M cell cycle arrest in SW480 was at least in part due to the marked decline of the levels of CDC25C.
Figure 2.

Effect of NO-ASA on proteins regulating the G2/M transition in SW480 cells. SW480 cells were treated with 200 μM NO-ASA at the indicated times (A) and with various concentrations of NO-ASA for 24 h (B). Cellular proteins were extracted and the levels of those indicated were assayed by immunoblotting. Loading control: actin and α-tubulin.
Because Cdc25C, whose levels were reduced markedly in NO-ASA-treated cells, plays critical roles in the dephosphorylation of Cdk1, the NO-ASA-induced decrease in the level of Cdc25C may cause accumulation of phosphorylated Cdk1 (inactive) at Tyr15 and Thr14 residues (18). Thus, we examined the phosphorylated state of Cdk1 using an antibody specific for phosphor-Cdk1 (Thr14/Tyr15). As shown in Fig. 2B, NO-ASA increased the Thr14/Tyr15-phosphorylated Cdk1 in a concentration-dependent manner.
We then examined whether the correlation between NO-ASA-mediated decrease of Cdc25C and G2/M cell cycle arrest was restricted to this cell line. Thus, we determined the effect of NO-ASA on Cdc25C protein levels in HT-29 colon adenocarcinoma cells. Similar to SW480, NO-ASA markedly decreases Cdc25C levels in HT-29 cells at 24 h (Fig. 3). As expected, NO-ASA treatment caused an increase in the protein level of cyclin B1 in HT-29 cells as well.
Figure 3.
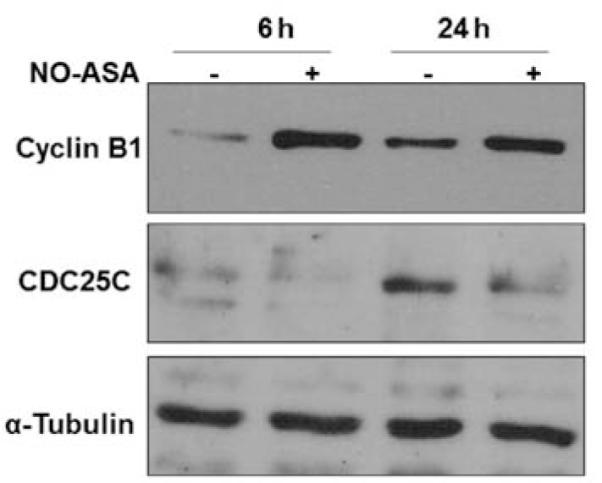
Effect of NO-ASA on proteins regulating G2/M transition in HT-29 cells. HT-29 cells were treated with 200 μM NO-ASA for the indicated periods of time. Cellular proteins were extracted and assayed by immunoblotting. Loading control: α-tubulin.
NO-ASA-induced G2/M arrest is linked to the generation of oxidative stress
DNA damage caused by oxidative stress is known to lead to G2/M arrest (12). NO-ASA caused a 50% reduction in the levels of the cellular anti-oxidant glutathione in cisplatin-sensitive human ovarian cancer cells (12) and also in HT-29 colon cancer cells (13). These observations raised the question whether NO-ASA is able to generate oxidative stress. Therefore, we determined the level of reactive oxygen species (ROS) in SW480 cells in response to NO-ASA. SW480 cells loaded with one of two molecular probes for the detection of ROS were treated with 200 μM NO-ASA for 4 h. The probes used were DHE, which preferentially measures superoxide anion, and H2DCFDA, which probes for over 10 individual species (14). Both DHE- and H2DCFDA-derived fluorescence was increased after treatment with NO-ASA, indicating the generation of oxidative stress (Fig. 4A). To evaluate whether NO-ASA-induced oxidative stress plays a role in cell cycle arrest, SW480 cells were pretreated with NAC, a potent anti-oxidant. NAC almost completely blocked the G2/M arrest induced by NO-ASA (Fig. 4B).
Figure 4.

Effect of NO-ASA-induced ROS on G2/M arrest in S480 cells. (A), SW480 cells were pre-loaded with H2DCFDA or DHE for 1 h followed by 200 μM NO-ASA for 4 h. Fluorescence was measured by flow cytometry in single-cell suspensions of the attached cells. (B), SW480 cells were treated with or without 2.5 mM NAC for 1 h before the start of treatment with 200 μM NO-ASA for 24 h. Cells were stained with PI and analyzed by flow cytometry. The table presents the corresponding cell cycle results from triplicate experiments. Values: mean ± SEM.
NO-ASA induces apoptosis in SW480 cells
Irreversible cell cycle arrest leads to apoptosis (15). To determine whether G2/M arrest induced by NO-ASA leads to apoptosis, SW480 cells were exposed to DMSO (control) or 200 μM of NO-ASA for up to 72 h. At the indicated times, cells were analyzed for cell cycle distribution. Consistent with the results shown in Table I, treatment with NO-ASA for 24 h induced significant G2/M arrest. Apoptotic cells were detected as a sub-G0/G1 fraction in the DNA histogram. NO-ASA-induced cell death was not significant until 24-h treatment, but 48-h treatment increased the sub-G0/G1 fraction from 4.5% to 11.5%, reaching 20.8% after 72 h. This indicated that at least some cells arrested in G2/M lost their ability to reenter the cell cycle and therefore underwent cell death.
In order to confirm that the observed cell death is due to apoptosis, expression levels of two proteins of apoptotic hallmark, caspase-3 and PARP, were determined. As shown in Fig. 5, treatment with NO-ASA (48 h) induced the cleavage of procaspase-3 into activated caspase-3 and of PARP (116 kDa) into an 85 kDa fragment, confirming the induction of apoptotic cell death by NO-ASA in SW480 cells. G2/M arrest by NO-ASA has been observed in three human colon cancer cell lines, but the mechanism of G2/M arrest was not investigated in that study (11).
Figure 5.

NO-ASA-induced apoptosis in SW480 cells. (A), After SW480 cells were treated with either 200 μM NO-ASA or vehicle for the indicated periods of time, they were stained with PI and analyzed by flow cytometry. (B), SW480 cells were treated with 200 μM NO-ASA for the indicated periods of time, when proteins were extracted and analyzed by immunoblotting. Loading control: α-tubulin.
Discussion
The present study indicates that NO-ASA displays a profound growth inhibitory effect on cancer cell lines from different tissues, which is far greater than that by conventional ASA. In all cancer cell lines, this growth inhibitory effect was accompanied by an arrest of these cells in the G2/M cell cycle phase. This effect is brought about by an elaborate array of changes in the expression of proteins controlling the cell cycle machinery.
The cyclin B1/Cdk1 kinase complex is considered a master regulator of G2/M progression and inhibition of its activity results in G2/M arrest. The activity of cyclin B1/Cdk1 kinase is dependent upon the formation of this bimolecular complex. Phosphorylation of Cdk1 at Thr14 and Tyr15 is required for complex formation; Cdc25C plays a critical role in the dephosphorylation of these two amino acid residues of Cdk1, being thus a critical regulator. We examined the status of this complex in response to NO-ASA. As summarized in Fig. 6, NO-ASA caused the G2/M arrest through a coordinated effect on the cyclin B1/Cdk1 complex by: a) increasing the levels of cyclin B, b) increasing the phosphorylation of cdk1, and c) suppressing the levels of cdc25, which could dephosphorylate cdk1. This effect is apparently significantly enhanced by the suppression of the expression of cyclin D1 by NO-ASA. Such an effect decelerates the progression of the cell from G2/M towards the next phase, G1. NO-ASA ultimately leads the target cell to apoptosis as evidenced by our flow cytometry data, the cleavage of PARP and the activation of the execution caspase-3 (16-19).
Figure 6.
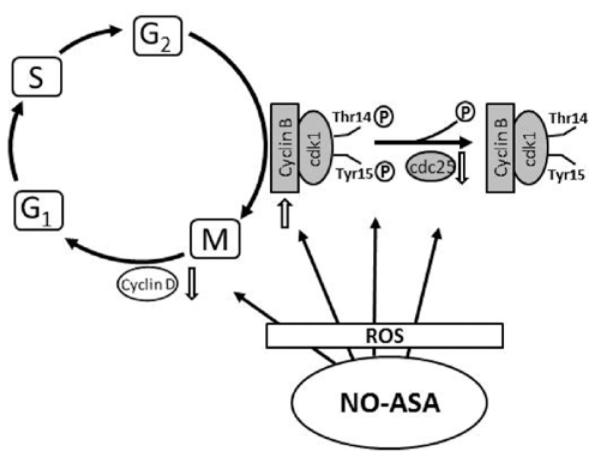
The mechanism of G2/M arrest by NO-ASA. NO-ASA increases intracellular ROS levels, inducing a state of oxidative stress. Redox signaling is then activated, altering the expression of proteins that control the G2/M transition, as indicated here. The end result is cell cycle arrest that contributes to the anticancer effect of NO-ASA.
The common denominator of these changes is likely the induction of oxidative stress by NO-ASA. Indeed, NO-ASA markedly enhanced the levels of ROS in the cell as indicated by the general ROS probe DCFDA, which responds to more than 10 individual reactive species and also by the increased levels of superoxide anion. That oxidative stress was relevant to the induction of cell cycle arrest by NO-ASA was documented by the finding that it was abolished by pretreatment with the anti-oxidant NAC. NO-ASA induces oxidative stress through multiple molecular effects, including reduction of the cellular levels of glutathione (GSH), a major anti-oxidant system in mammalian cells (20). In the absence of a compensatory mechanism, a consequence of the reduction of GSH level in mammalian cells is the induction of oxidative stress. This is further supported by the notion that NO-ASA reduces the levels of GSH in cisplatin-sensitive human ovarian cancer cells (12). NO-ASA reacts with cellular GSH forming a conjugate between its spacer moiety and GSH in HT-29 colon cancer cells (21). Interestingly, oxidative stress has been recently proposed as a general mechanism of action of modified NSAIDs such as NO-ASA (13,22).
Oxidative stress is known to inactivate key cell cycle regulators and to impair cell cycle progression through mitosis. Indeed, ROS through cyclic oxidation/reduction of cysteine residues in kinases, phosphatases, and other regulatory factors regulate the strength and duration of cell cycle signaling (23). Such evidence includes the following: Cdc25 phosphatases contain an active-site cysteine that is sensitive to oxidation by H2O2 (24); H2O2 inactivates Cdc25C by inducing a disulfide bond in Cdc25C between the active-site Cys377 and the invariant Cys330 (24); and, in turn, this disulfide bond can be reduced by Vitamin C (a modulator of the cellular redox status) by inhibiting activation of the cyclin B/CDK1 complex by Cdc25C (25). In addition, ROS stimulate mitogenic pathways controlling the activity of CDKs (26). For example, redox modification of cyclin D1 at Cys285 may result in conformational changes that affect the phosphorylation of Thr286 and/or −288, which in turn affecting its degradation (27). The regulation of cyclin D1 by redox is supported by the ability of NAC to suppress cyclin D1 levels (28).
Collectively, these results confirm the profound cytokinetic effect of NO-ASA and provide strong evidence that it regulates cell cycle transitions through its ability to induce oxidative stress which activates redox signaling in the target cell.
Acknowledgements
This work was supported by the grant K01CA106604 and R01CA140487.
References
- 1.American Cancer Society . Cancer Facts and Figures 2011. American Cancer Society; Atlanta, GA: 2011. [Google Scholar]
- 2.Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94:252–266. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- 3.Rayyan Y, Williams J, Rigas B. The role of NSAIDs in the prevention of colon cancer. Cancer Invest. 2002;20:1002–1011. doi: 10.1081/cnv-120005917. [DOI] [PubMed] [Google Scholar]
- 4.Baron JA. Epidemiology of non-steroidal anti-inflammatory drugs and cancer. Prog Exp Tumor Res. 2003;37:1–24. doi: 10.1159/000071364. [DOI] [PubMed] [Google Scholar]
- 5.Rigas B, Kashfi K. Nitric-oxide-donating NSAIDs as agents for cancer prevention. Trends Mol Med. 2004;10:324–330. doi: 10.1016/j.molmed.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 6.Kashfi K, Borgo S, Williams JL, et al. Positional isomerism markedly affects the growth inhibition of colon cancer cells by nitric oxide-donating aspirin in vitro and in vivo. J Pharmacol Exp Ther. 2005;312:978–988. doi: 10.1124/jpet.104.075994. [DOI] [PubMed] [Google Scholar]
- 7.Ouyang N, Williams JL, Tsioulias GJ, et al. Nitric oxide-donating aspirin prevents pancreatic cancer in a hamster tumor model. Cancer Res. 2006;66:4503–4511. doi: 10.1158/0008-5472.CAN-05-3118. [DOI] [PubMed] [Google Scholar]
- 8.Bak AW, McKnight W, Li P, et al. Cyclooxygenase-independent chemoprevention with an aspirin derivative in a rat model of colonic adenocarcinoma. Life Sci. 1998;62:PL367–PL373. doi: 10.1016/s0024-3205(98)00191-x. [DOI] [PubMed] [Google Scholar]
- 9.Williams JL, Kashfi K, Ouyang N, Del Soldato P, Kopelovich L, Rigas B. NO-donating aspirin inhibits intestinal carcinogenesis in Min (APC(Min/+)) mice. Biochem Biophys Res Commun. 2004;313:784–788. doi: 10.1016/j.bbrc.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 10.Fiorucci S, Santucci L, Gresele P, Faccino RM, Del Soldato P, Morelli A. Gastrointestinal safety of NO-aspirin (NCX-4016) in healthy human volunteers: a proof of concept endoscopic study. Gastroenterology. 2003;124:600–607. doi: 10.1053/gast.2003.50096. [DOI] [PubMed] [Google Scholar]
- 11.Troyano A, Fernandez C, Sancho P, De Blas E, Aller P. Effect of glutathione depletion on antitumor drug toxicity (apoptosis and necrosis) in U-937 human promonocytic cells. The role of intracellular oxidation. J Biol Chem. 2001;276:47107–47115. doi: 10.1074/jbc.M104516200. [DOI] [PubMed] [Google Scholar]
- 12.Chang DK, Goel A, Ricciardiello L, et al. Effect of H(2)O(2) on cell cycle and survival in DNA mismatch repair-deficient and -proficient cell lines. Cancer Lett. 2003;195:243–251. doi: 10.1016/s0304-3835(03)00145-9. [DOI] [PubMed] [Google Scholar]
- 13.Gao J, Liu X, Rigas B. Nitric oxide-donating aspirin induces apoptosis in human colon cancer cells through induction of oxidative stress. Proc Natl Acad Sci USA. 2005;102:17207–17212. doi: 10.1073/pnas.0506893102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009;16:1040–1052. doi: 10.1038/cdd.2009.49. [DOI] [PubMed] [Google Scholar]
- 15.Aylon Y, Oren M. p53: guardian of ploidy. Mol Oncol. 2011;5:315–323. doi: 10.1016/j.molonc.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams JM. Ways of dying: multiple pathways to apoptosis. Genes Dev. 2003;17:2481–2495. doi: 10.1101/gad.1126903. [DOI] [PubMed] [Google Scholar]
- 17.Hay BA, Huh JR, Guo M. The genetics of cell death: approaches, insights and opportunities in Drosophila. Nat Rev Genet. 2004;5:911–922. doi: 10.1038/nrg1491. [DOI] [PubMed] [Google Scholar]
- 18.Boyce M, Degterev A, Yuan J. Caspases: an ancient cellular sword of Damocles. Cell Death Differ. 2004;11:29–37. doi: 10.1038/sj.cdd.4401339. [DOI] [PubMed] [Google Scholar]
- 19.Degterev A, Boyce M, Yuan J. A decade of caspases. Oncogene. 2003;22:8543–8567. doi: 10.1038/sj.onc.1207107. [DOI] [PubMed] [Google Scholar]
- 20.Kalgutkar AS, Crews BC, Rowlinson SW, Garner C, Seibert K, Marnett LJ. Aspirin-like molecules that covalently inactivate cyclooxygenase-2. Science. 1998;280:1268–1270. doi: 10.1126/science.280.5367.1268. [DOI] [PubMed] [Google Scholar]
- 21.Gao J, Niwa K, Sun W, et al. Non-steroidal anti-inflammatory drugs inhibit cellular proliferation and upregulate cyclooxygenase-2 protein expression in endometrial cancer cells. Cancer Sci. 2004;95:901–907. doi: 10.1111/j.1349-7006.2004.tb02200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Y, Huang L, Mackenzie GG, Rigas B. Oxidative stress mediates through apoptosis the anticancer effect of phosphononsteroidal anti-inflammatory drugs: implications for the role of oxidative stress in the action of anticancer agents. J Pharmacol Exp Ther. 2011;338:775–783. doi: 10.1124/jpet.111.183533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vogt A, Tamura K, Watson S, Lazo JS. Antitumor imidazolyl disulfide IV-2 causes irreversible G(2)/M cell cycle arrest without hyperphosphorylation of cyclin-dependent kinase Cdk1. J Pharmacol Exp Ther. 2000;294:1070–1075. [PubMed] [Google Scholar]
- 24.Sohn J, Rudolph J. Catalytic and chemical competence of regulation of cdc25 phosphatase by oxidation/reduction. Biochemistry. 2003;42:10060–10070. doi: 10.1021/bi0345081. [DOI] [PubMed] [Google Scholar]
- 25.Thomas CG, Vezyraki PE, Kalfakakou VP, Evangelou AM. Vitamin C transiently arrests cancer cell cycle progression in S phase and G2/M boundary by modulating the kinetics of activation and the subcellular localization of Cdc25C phosphatase. J Cell Physiol. 2005;205:310–318. doi: 10.1002/jcp.20405. [DOI] [PubMed] [Google Scholar]
- 26.Burhans WC, Heintz NH. The cell cycle is a redox cycle: linking phase-specific targets to cell fate. Free Radic Biol Med. 2009;47:1282–1293. doi: 10.1016/j.freeradbiomed.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 27.Menon SG, Goswami PC. A redox cycle within the cell cycle: ring in the old with the new. Oncogene. 2007;26:1101–1109. doi: 10.1038/sj.onc.1209895. [DOI] [PubMed] [Google Scholar]
- 28.Menon SG, Sarsour EH, Spitz DR, et al. Redox regulation of the G1 to S phase transition in the mouse embryo fibroblast cell cycle. Cancer Res. 2003;63:2109–2117. [PubMed] [Google Scholar]