

Abstract
To fulfill systems biology’s promise of providing fundamental new insights will require the development of quantitative and predictive models of whole cells. In this issue, Karr et al. present the first integrated and dynamic computational model of a bacterium that accounts for all of its components and their interactions.
Scientific disciplines must, from time to time, challenge the standards by which they measure understanding. The absolute standard in classical mechanics, for example, is the ability to predict the dynamic behavior of a physical system. Will biology ever achieve this level of understanding, even for a single cell? Perhaps it is now time to test our current state of understanding by attempting to build a whole-cell simulator that captures everything that we know. The devil in such an undertaking lies in the thousands of details that must be properly accounted for in order to represent an entire cell with any degree of accuracy. In this issue, Karr et al. (2012) present a whole-cell computational model for the bacterium Mycoplasma genitalium that accounts for the actions of all known genes and gene products and allows simulation of the entire cell cycle.
The model presented by the authors is the first truly integrated effort to simulate the workings of a free-living microbe, and it should be commended for its audacity alone. This is a tremendous task, involving the interpretation and integration of a massive amount of data, largely incorporated by proxy from other microbes such as Mycoplasma pneumonia and Escherichia coli. It is arguable whether the current scale and depth of our knowledge of the workings of individual components is adequate to attempt a whole-cell reconstruction at this point. Undoubtedly, some will claim that the sparsity of this knowledge, even for an organism with close to a minimal genome, dooms any such attempt for failure. A completely accurate and detailed fine-grained simulation certainly remains a distant dream. Yet, in light of the model presented by Karr et al. (2012), the more pertinent question is: do we currently know enough to attempt a mesoscale simulation that will produce nontrivial results? The only way to address this question is to produce whole-cell models that generate testable predictions. It is in this spirit that the authors have produced a first-draft framework for asking and answering systems-level questions by using quantitative cell-scale models.
The model presented by Karr et al. (2012) is not the first attempt to provide a quantitative, predictive cell-scale computational framework. Prior efforts have used nonlinear differential equation-based models of coarse-grained biological networks (for example, Castellanos et al., 2004) or constraint-based modeling, typified by flux balance analysis (FBA) and related models (Orth et al., 2010). Of all previous approaches to modeling cell-scale metabolic and regulatory networks, FBA deserves particular attention due to the breadth of successful applications of this family of techniques; constraint-based models have proven useful and qualitatively correct in applications ranging from the prediction and rationalization of evolutionary trajectories to metabolic engineering (recently reviewed by Feist and Palsson, 2008). Despite their numerous successes, however, such approaches have generally been limited to treating only a subset of phenomena in the cell due to the absence of any single computational framework appropriate for modeling all of the diverse reactions and interactions occurring in a living cell. In addition, constraint-based models in particular generally yield steady-state, rather than dynamic, information, although some recent efforts have been made to integrate FBA into dynamic simulations (such as Lee et al., 2008).
In order to model the heterogeneous collection of mechanisms and timescales present in living cells, Karr et al. (2012) use a clever modular design in which many active processes function and interact with each other. The modules, each of which represents a single class of processes (e.g., transcription or metabolism), are all separately formulated, parameterized, and tested, representing their components at various levels of coarse-graining. The modules interact and exchange variables (which together specify the internal state of the cell) at 1 s intervals; propagation of this model through time allows simulation of an entire M. genitalium cell cycle. The model certainly proves accurate on a number of basic points; the authors show that their simulation produces metabolite abundances that are on the order of magnitude observed in real cells and are able to predict the essentiality of M. genitalium genes with ~80% accuracy. Far more impressively, the model provides an entirely original hypothesis for the regulation of Mycoplasma cell-cycle duration: that genomic replication is eventually rate limited by deoxyribonucleotide tri-phosphate (dNTP) synthesis and that cells in which early stages of the cell cycle are prolonged are able to catch up with those that initiate replication earlier due to the accumulation of a larger dNTP pool at the onset of replication, thus reducing the variance of overall cell-cycle duration within a population.
Presented with this technological advance, it is crucial to consider both what one wishes to learn from whole-cell models and how they will interact with the rest of biology. We consider both points in Figure 1; as we see it, modern biology acts at the intersection of broad, qualitative underlying principles, global systems-level quantitative measurements from high-throughput experiments, and system-specific quantitative measurements and models from more focused investigations. Quantitative cell-scale modeling offers the promise of a principled framework for combining these disparate sources of information. In the short term, the development and optimization of such models are critical challenges by themselves, and modelers may simply make use of any discrepancies between their predictions and known experimental data to refine the structure and contents of their models. In the longer term, however, these models must offer fundamentally new, experimentally-testable predictions. We envision two primary types of predictions that will be particularly useful: (1) the discovery of new organizing principles that help frame our intellectual understanding of biological systems (the “physicist’s perspective”); and (2) the development of sufficiently accurate computational models to supplant experiment during at least early stages of compound screening or bioengineering applications (the “engineer’s perspective”). The unique potential for detailed cell-scale simulations to provide insight unobtainable through experiment arises because they can provide arbitrarily detailed, single-cell trajectories of the internal state of cells and can be easily perturbed as needed to investigate a phenomenon of interest.
Figure 1. The Role of Whole-Cell Simulations in Modern Biology.
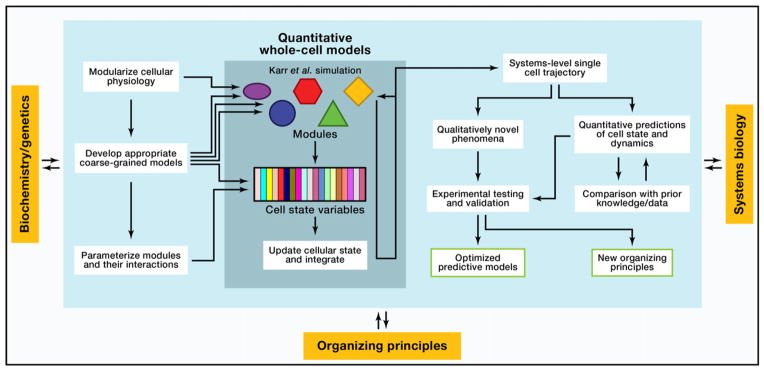
As they mature, whole-cell models will integrate conceptual knowledge, low-throughput, and systems-level experimental information as inputs (the details of the modeling method used by Karr et al. (2012) is depicted as an example), and they will provide as output both quantitative predictions of unspecified parameters and qualitative information on previously unobserved behaviors. Initially, the primary focus of modelers must be to refine their models through a feedback loop of comparing predictions to both old and new focused experiments; as time goes on, however, model predictions will allow the proposal of new, testable hypotheses for previously unobserved organizing principles. In addition, the quantitative predictions of more refined whole-cell models should become increasingly useful in bioengineering applications.
The highly sophisticated multiscale model presented by Karr et al. (2012) is a crucial step in the development of useful and reliable cell-scale simulations. It is impressive that this extremely complex and ambitious preliminary model can provide both rough quantitative agreement with a variety of experimentally measured parameters and new insight into the regulation of a biological process. Nevertheless, we should emphasize that this is far from a platonically ideal simulation of M. genitalium. For every module, there will likely be some expert who will present a fair criticism of the module’s mathematical representation or parameter estimation, even though at present they appear to represent the best available attempt at balancing realism, computational complexity, and number of free parameters. As the authors themselves acknowledge, the present model must be seen as a first draft, more important as a starting point for future refinement than as a productive model in its own right. In the future, it will be crucial for modelers to investigate the points of failure in the current model and to determine what alterations to parameters or structure are needed; as the authors note, expansion to a more experimentally tractable model organism such as E. coli is also highly desirable. In addition, to provide a complete cell-scale reconstruction, modelers will need to either treat or justify the neglect of several lurking complexities that do not appear to be addressed at present, such as the possible presence of fairly pervasive genome-wide antisense transcription (Dornenburg et al., 2010), effects of spatial heterogeneity (Roberts et al., 2011), and enzyme multifunctionality (Khersonsky and Tawfik, 2010). It is also unclear how vulnerable these massive cell-scale models will be to the documented “sloppiness” present in the parameter sensitivities of systems biology models (Gutenkunst et al., 2007), both in terms of how well overall behaviors will be predicted from a collection of separately obtained parameters and to what extent the individual internal state variables of simulated cells may be relied upon even if the overall behavior of the cell appears correct.
References
- Castellanos M, Wilson DB, Shuler ML. Proc Natl Acad Sci USA. 2004;101:6681–6686. doi: 10.1073/pnas.0400962101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dornenburg JE, Devita AM, Palumbo MJ, Wade JT. MBio. 2010;1:e00024–e10. doi: 10.1128/mBio.00119-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feist AM, Palsson BØ. Nat Biotechnol. 2008;26:659–667. doi: 10.1038/nbt1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst RN, Waterfall JJ, Casey FP, Brown KS, Myers CR, Sethna JP. PLoS Comput Biol. 2007;3:1871–1878. doi: 10.1371/journal.pcbi.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karr JR, Sanghvi JC, Macklin DN, Gutschow MV, Jacobs JM, Bolival B, Assad-Garcia N, Glass JI, Covert MW. Cell. 2012;150:389–401. doi: 10.1016/j.cell.2012.05.044. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky O, Tawfik DS. Annu Rev Biochem. 2010;79:471–505. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- Lee JM, Gianchandani EP, Eddy JA, Papin JA. PLoS Comput Biol. 2008;4:e1000086. doi: 10.1371/journal.pcbi.1000086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth JD, Thiele I, Palsson BØ. Nat Biotechnol. 2010;28:245–248. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts E, Magis A, Ortiz JO, Baumeister W, Luthey-Schulten Z. PLoS Comput Biol. 2011;7:e1002010. doi: 10.1371/journal.pcbi.1002010. [DOI] [PMC free article] [PubMed] [Google Scholar]