

Abstract
Background
The mitochondrial genomes (mitogenomes) of flatfishes (Pleuronectiformes) exhibit highly diversified types of large-scale gene rearrangements. We have reported that the mitogenomes of Crossorhombus azureus (Bothidae), Samariscus latus (Samaridae) and Cynoglossus fishes (Cynoglossidae) show different types of gene rearrangements.
Results
In the present study, the complete mitogenomes of two Symphurus species (Cynoglossidae), Symphurus plagiusa and Symphurus orientalis, were determined. The gene order in the S. plagiusa mitogenome is the same as that of a typical vertebrate (without any gene rearrangements). Surprisingly, large-scale gene rearrangements have occurred in S. orientalis. In the rearranged fragment from the control region (CR) to the WANCY tRNA cluster (tRNA cluster of tRNA-W, tRNA-A, tRNA-N, tRNA-C and tRNA-Y) in the S. orientalis mitogenome, tRNA-V and tRNA-M have been translocated to the 3’ end of the 16S rRNA gene, with six large intergenic spacers over 20 bp in length. In addition, an origin for light-strand replication (OL) structure that is typically located in the WANCY region was absent in both the S. plagiusa and S. orientalis mitogenomes. It is generally recognized that a sequence in the WANCY region that encodes tRNAs forms a hairpin structure (OL-like structure) and can act as the OL when the typical locus is lost. Moreover, an additional OL-like structure was identified near the control region in the S. plagiusa mitogenome.
Conclusions
The positions of the intergenic spacers and the rearranged genes of the S. orientalis mitogenome strongly indicate that the mechanism underlying the rearrangement of this mitogenome was Tandem Duplication and Random Loss. Additionally, two OL-like regions substituting for the typical locus were found in the S. plagiusa mitogenome. We speculate that the ancestral mitogenomes of S. plagiusa and S. orientalis also had this characteristic, such that if both OL-like structures functioned during mitochondrial replication, they could initiate duplicate replications of the light strand (L-strand), leading to duplication of the region between the two structures. We consider that this mechanism may account for the gene duplication that occurred during the gene rearrangement process in the evolution of the ancestral mitogenome to the S. orientalis mitogenome.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1581-6) contains supplementary material, which is available to authorized users.
Keywords: Flatfish, Mitogenome, Gene rearrangement, OL-like structure, Mitochondrial replication
Background
Vertebrate mitochondrial genomes (mitogenomes) typically contain the same 37 genes [1]. The order of these genes is generally considered conservative in most vertebrate genomes; however, gene rearrangements also have been found in many taxa, such as birds [2-4], reptiles [5,6], amphibians [7,8], and fishes [9-11]. Teleosts, with the largest number of published complete mitogenome sequences, show only a few gene rearrangement events [9-13]. In most cases, a teleostean group has only one type or a set of similar gene rearrangements [9-14]. However, the flatfish (Pleuronectiformes) mitogenomes exhibit the most diversified types of large-scale gene rearrangements. In the mitogenomes of Cynoglossus fishes (tongue soles, Cynoglossidae), the control region is translocated, and a tRNA (transfer ribonucleic acid) gene is inverted [10]. In contrast, no gene rearrangements have been found in soles (Soleidae), the closest family to Cynoglossidae fishes [15-17]. The mitogenome of Crossorhombus azureus (Bothidae) contains genomic-scale gene rearrangements characterized by the protein-coding gene ND6 and seven tRNA genes encoded on the light strand (L-strand; H- versus L-strands are defined by studies on AT- and GC-skewing [18,19]) that are clustered together [20]. A third type of gene rearrangement was detected in the Samariscus latus (Samaridae) mitogenome [21]. Distinct from the above-mentioned flatfishes, the gene rearrangement in this species is characterized by the duplication and translocation of the control region (CR); simultaneously, the genes located between the two CRs are divided into two clusters in which their relative gene orders have been maintained [21].
Several models have been proposed to explain gene rearrangements in animal mitogenomes. The Recombination model involves the breakage and rejoining of participating DNA strands [22]. The Tandem Duplication and Random Loss (TDRL) model posits that rearrangements of mitochondrial gene order occurred via tandem duplications of certain genes followed by random deletion of some of the duplications [23,24]. Two additional hypotheses are described in the Tandem Duplication and Non-random Loss (TDNL) [25] and tRNA mis-priming models [26,27]. For the gene rearrangements in flatfishes, none of the models mentioned above can provide a perfect explanation. Thus, Kong et al. [10] developed a model of inverse duplication and deletion of redundant genes to explain the gene rearrangements in tongue soles. Subsequently, Shi et al. [20] proposed the Dimer-Mitogenome and Non-Random Loss model (DMNR), which inferred the course of gene rearrangements in C. azureus. Recently, for the rearrangement events in the S. latus mitogenome, Shi et al. [21] proposed the Double Replications and Random Loss model.
Of these mechanisms proposed to explain mitochondrial gene rearrangements, the TDRL model is generally considered the most popular and important in vertebrates [9,23,24,28,29]. Generally, when decrypting gene rearrangements with the TDRL model, it is always necessary to propose multiple duplication and loss steps. It is therefore difficult to trace which steps preserved the functional genes and which DNA segments degenerated to pseudogenes or intergenic spacers. In other words, large-scale gene rearrangements cannot readily yield integrated evidence for the TDRL model [9,23,24,28,29]. San Mauro et al. [29] also indicated that the evidence for this model in the form of duplicated genes that either remain functional or have become pseudogenes in the process of being eliminated is rather limited.
In the present study, the complete mitogenomes of two flatfishes, Symphurus plagiusa and Symphurus orientalis, were sequenced. Surprisingly, the gene order of the S. plagiusa mitogenome resembles that of a typical vertebrate (un-rearranged gene order), whereas that of S. orientalis shows large-scale gene rearrangements. This is the first report of mitogenomes with a typical gene order and large-scale gene rearrangements within the same teleost genus. The characteristics of the gene order and intergenic spacers in S. orientalis provide clear evidence for the TDRL model, accounting for the gene rearrangements in the S. orientalis mitogenome.
Methods
Ethics statement
Ethical approval was not required for the present study because the examined specimens were commonly captured marine economic fishes, and all of the fish specimens were already dead when we obtained them and were sourced from commercial fisheries. Additionally, these species were not included in the IUCN list of endangered species (http://www.iucnredlist.org).
Sampling, DNA extraction, PCR and sequencing
Specimens of S. plagiusa and S. orientalis were collected from Tampa Bay, Florida (USA) and Taiwan (China), respectively. A portion of the epaxial musculature was excised from fresh specimens and immediately stored at −70°C. Total genomic DNA was extracted using the SQ Tissue DNA Kit (OMEGA) following the manufacturer’s protocol. Based on alignments and comparisons of complete mitochondrial sequences from flatfishes, dozens of primer pairs were designed for amplification of the mtDNA genomes (Additional file 1: Table S1 and Additional file 2: Table S2). More than 30 bp of overlapping fragments between tandem regions were used to ensure correct assembly and integrity of the complete sequence.
PCR (polymerase chain reaction) was performed in a 25 μl reaction volume containing 2.0 mM MgCl2, 0.4 mM of each dNTP, 0.5 μM of each primer, 1.0 U Taq polymerase (Takara, China), 2.5 μl of 10x Taq buffer, and approximately 50 ng of DNA template. PCR cycling conditions included an initial denaturation at 95°C for 3 min, followed by 30–35 cycles of denaturation at 94°C for 45 s, annealing at 45–55°C for 45 s, and elongation at 68–72°C for 1.5–5 min. The PCR reactions were completed by a final extension at 72°C for 5 min. The PCR products were purified with the Takara Agarose Gel DNA Purification Kit (Takara, China) and used directly as templates for cycle sequencing reactions. Sequence-specific primers were further designed and used as walking primers for both strands of each fragment on an ABI 3730 DNA sequencer (Applied Biosystems, USA). The mtDNA sequences of S. plagiusa and S. orientalis have been submitted to GenBank under the accession numbers JQ639061 and KP992899, respectively.
Sequence analysis
Sequenced fragments were assembled to create complete mitochondrial genomes using CodonCode Aligner v3 and BioEdit v7 [30]. During the processing of large fragments and walking sequences, regular manual examinations were performed to ensure reliable assembly of the genome sequence. Annotation and boundary determination of protein-coding and rRNA (Ribosomal ribonucleic acid) genes were performed using NCBI-BLAST (http://blast.ncbi.nlm.nih.gov). TRNA genes and their secondary structures were identified using tRNAscan-SE 1.21 [31], setting the cut-off values to 1 when necessary.
Results
Features of the genomes
The complete mitogenomes of S. plagiusa and S. orientalis were 17040 bp and 17498 bp in length, respectively, and contained 13 protein-coding genes, 22 tRNA genes, and 2 rRNA genes as well as one CR. Most of these genes were encoded by the heavy strand (H-strand), except ND6 and eight tRNA genes, which were encoded on the L-strand (Table 1). The 22 tRNA genes were interspersed among rRNAs and protein-coding genes, and all tRNAs can be folded into typical cloverleaf structures. The location of the CR was between the tRNA-P and tRNA-F genes, as is typical for teleosts. Compared with the CR sequences of the other flatfishes, the symbolic structures of the two Symphurus CRs were present as in other bony fishes [16,20,21,32,33]. The typical origin for L-strand replication (OL), which is usually located inside the WANCY cluster (tRNA cluster of tRNA-W, tRNA-A, tRNA-N, tRNA-C and tRNA-Y), was not found in either of the two Symphurus mitogenomes at this location.
Table 1.
Features of the mitogenomes of Symphurus plagiusa (Left) and Symphurus orientalis (Right)
| Gene | Position | Length (bp) | Intergenic region* | Strand | Gene | Position | Length (bp) | Intergenic region | Strand | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| From | To | From | To | ||||||||
| tRNA-Phe (F) | 1 | 69 | 69 | 0 | H | tRNA-Phe | 1 | 71 | 71 | 0 | H |
| 12S | 70 | 1016 | 947 | 0 | H | 12S | 72 | 1020 | 949 | 65 | H |
| tRNA-Val (V) | 1017 | 1086 | 70 | 0 | H | 16S | 1086 | 2796 | 1711 | 98 | H |
| 16S | 1087 | 2793 | 1707 | 0 | H | tRNA-Met | 2895 | 2964 | 70 | 100 | H |
| tRNA-Leu UUA (L1) | 2794 | 2866 | 73 | 0 | H | tRNA-Val | 3065 | 3136 | 72 | 31 | H |
| ND1 | 2867 | 3841 | 975 | 2 | H | tRNA-Leu UUA | 3168 | 3240 | 73 | 0 | H |
| tRNA-Ile (I) | 3844 | 3913 | 70 | -2 | H | ND1 | 3241 | 4212 | 972 | 7 | H |
| tRNA-Gln (Q) | 3912 | 3982 | 71 | -1 | L | tRNA-Ile | 4220 | 4289 | 70 | -2 | H |
| tRNA-Met (M) | 3982 | 4050 | 69 | 124 | H | tRNA-Gln | 4288 | 4358 | 71 | 117 | L |
| ND2 | 4175 | 5227 | 1053 | 42 | H | ND2 | 4476 | 5522 | 1047 | 37 | H |
| tRNA-Trp (W) | 5270 | 5339 | 70 | 0 | H | tRNA-Trp | 5560 | 5628 | 69 | 0 | H |
| tRNA-Ala (A) | 5340 | 5408 | 69 | 0 | L | tRNA-Ala | 5629 | 5697 | 69 | 0 | L |
| tRNA-Asn (N) | 5409 | 5479 | 71 | 2 | L | tRNA-Asn | 5698 | 5770 | 73 | 3 | L |
| tRNA-Cys (C) | 5482 | 5546 | 65 | 3 | L | tRNA-Cys | 5774 | 5839 | 66 | 4 | L |
| tRNA-Tyr (Y) | 5550 | 5616 | 67 | 1 | L | tRNA-Tyr | 5844 | 5910 | 67 | 10 | L |
| COI | 5618 | 7168 | 1551 | 0 | H | COI | 5921 | 7471 | 1551 | 0 | H |
| tRNA-Ser UCA (S1) | 7169 | 7239 | 71 | 19 | L | tRNA-Ser UCA | 7472 | 7542 | 71 | 19 | L |
| tRNA-Asp (D) | 7259 | 7327 | 69 | 4 | H | tRNA-Asp | 7562 | 7630 | 69 | 5 | H |
| COII | 7332 | 7925 | 594 | 96 | H | COII | 7636 | 8326 | 691 | 0 | H |
| tRNA-Lys (K) | 8022 | 8097 | 76 | 1 | H | tRNA-Lys | 8327 | 8400 | 74 | 2 | H |
| ATP8 | 8099 | 8266 | 168 | -10 | H | ATP8 | 8403 | 8570 | 168 | -10 | H |
| ATP6 | 8257 | 8940 | 684 | 2 | H | ATP6 | 8561 | 9244 | 684 | -1 | H |
| COIII | 8943 | 9731 | 789 | 65 | H | COIII | 9244 | 10029 | 786 | -1 | H |
| tRNA-Gly (G) | 9797 | 9864 | 68 | 0 | H | tRNA-Gly | 10029 | 10096 | 68 | 0 | H |
| ND3 | 9865 | 10215 | 351 | -2 | H | ND3 | 10097 | 10447 | 351 | -2 | H |
| tRNA-Arg (R) | 10214 | 10282 | 69 | 0 | H | tRNA-Arg | 10446 | 10514 | 69 | 0 | H |
| ND4L | 10283 | 10579 | 297 | -7 | H | ND4L | 10515 | 10811 | 297 | -7 | H |
| ND4 | 10573 | 11953 | 1381 | 0 | H | ND4 | 10805 | 12184 | 1380 | 1 | H |
| tRNA-His (H) | 11954 | 12022 | 69 | 0 | H | tRNA-His | 12186 | 12253 | 68 | 0 | H |
| tRNA-Ser UGC (S2) | 12023 | 12090 | 68 | 2 | H | tRNA-Ser UGC | 12254 | 12321 | 68 | 2 | H |
| tRNA-Leu GUA (L2) | 12093 | 12164 | 72 | 0 | H | tRNA-Leu GUA | 12324 | 12397 | 74 | 0 | H |
| ND5 | 12165 | 13985 | 1821 | -4 | H | ND5 | 12398 | 14233 | 1836 | 4 | H |
| ND6 | 13982 | 14500 | 519 | 0 | L | ND6 | 14238 | 14756 | 519 | 0 | L |
| tRNA-Glu (E) | 14501 | 14569 | 69 | 2 | L | tRNA-Glu | 14757 | 14825 | 69 | 3 | L |
| Cytb | 14572 | 15711 | 1140 | 1 | H | Cytb | 14829 | 15969 | 1141 | 0 | H |
| tRNA-Thr (T) | 15713 | 15781 | 69 | -1 | H | tRNA-Thr | 15970 | 16042 | 73 | -1 | H |
| tRNA-Pro (P) | 15781 | 15849 | 69 | 0 | L | tRNA-Pro | 16042 | 16110 | 69 | 0 | L |
| OL-like Seq. | 16983 | 17034 | 52 | / | L | D-loop | 16111 | 17498 | 1388 | 0 | H |
| D-loop | 15850 | 17040 | 1191 | 0 | H | ||||||
*Intergenic region: non-coding bases between the feature on the same line and the line below, with a negative number indicating an overlap.
Two different mitochondrial gene orders in the genus Symphurus
The gene order of the S. plagiusa mitogenome is the same as that of a typical vertebrate, while that of S. orientalis contains large-scale gene rearrangements. This phenomenon is rare in vertebrates and the first report in teleosts. The difference between the two mitogenomes lies between the CR and the WANCY region. The gene order of this region in the S. plagiusa mitogenome is CR-F-12S-V-16S-L1-ND1-I-Q-M-ND2-WANCY, which is the same as that of a typical vertebrate, while this region has been rearranged to CR-F-12S-16S-M-V-L1-ND1-I-Q-ND2-WANCY in the S. orientalis mitogenome.
In the rearranged fragment spanning the region from the CR to the WANCY region in the S. orientalis mitogenome, six large intergenic spacers greater than 20 bp in length remain: Gap A, between 12S and 16S (65 bp); Gap B, between 16S and tRNA-M (98 bp); Gap C, between tRNA-M and tRNA-V (100 bp); Gap D, between tRNA-V and tRNA-L1 (31 bp); Gap E, between tRNA-Q and ND2 (117 bp); and Gap F, between ND2 and tRNA-W (37 bp).
Discussion
Which mechanisms account for the gene rearrangements in S. orientalis mitogenome?
To date, approximately 1,500 complete mtDNA sequences have been determined in teleosts, and several types of gene rearrangements have been reported [17,33-36]. In the taxa showing gene rearrangements, species across the entire taxon typically show the same or similar rearrangement events [4,6,14]. That is, at the intra-family or intra-genus level, few groups include species with the typical (unchanged) gene order together with species with large-scale gene rearrangements. Nevertheless, this unique phenomenon occurs in the flatfish genus Symphurus, as described above.
Of the models that have been proposed to explain gene rearrangements in animal mitogenomes, which model most likely applies to the S. orientalis mitogenome? The recombination model is only suitable for block interchanges of small fragments, and this model is quite rare in the mitochondrial genome. As for the tRNA mis-priming [26,27] and TDNL models [25], there are no obvious corresponding model rules in the S. orientalis mitogenome.
Several species within three flatfish groups (Bothidae, Samaridae and Cynoglossidae) have been reported to possess different types of gene rearrangements. Among them, Cynoglossus fishes belong to Cynoglossidae, the same family as Symphurus. However, the rearrangements in the Cynoglossus mitogenomes are characterized by inverted tRNA genes, which were not present in the S. orientalis mitogenome. The rearrangement characteristics of the other flatfish groups also differ from those of S. orientalis.
We numbered the gene order of the S. plagiusa mitogenome (typical gene order) from CR to WANCY in the following series: CR–F (1)–12S (2)–V (3)–16S (4)–L1 (5)–ND1 (6)–I (7)–Q (8)–M (9)–ND2 (10)–W (11) –A (12) –N (13) CY. Following this scheme, the corresponding sequence of S. orientalis would be CR–F (1)–12S (2)–16S (4)–M (9)–V (3)–L1 (5)–ND1 (6)–I(7)–Q (8)–ND2 (10)–W (11) –A (12) –N (13) CY. Based on this numeric order, the genes from F to N (1–13) in S. orientalis can be divided into two gene clusters: 1–2–(4–9) and (3–5–6–7–8)–10–11–12–13, each of which retains the conserved relative gene order, from low to high. It is reasonable to assume that the two clusters were derived from a tandem-duplicated DNA fragment that spanned genes from at least V (3) to M (9) in the typical gene order. Thereafter, one of each pair of duplicated genes was randomly lost: 1– 2 –(3 –4 –5 –6 –7 –8 –9) (3 –4 –5 –6 –7 –8 –9)–10 –11 –12 –13. Within the scope of our current knowledge, this rearrangement process represents the most parsimonious and reasonable hypothesis.
The process of gene duplication
How did the duplication occur? Moreover, was the fragment from V (3) to M (9) or a longer fragment duplicated? Many studies have reported similar duplications. For example, Fujita et al. [37] found that several lineages of parthenogenetic lizards harbor large, tandem duplications, which are hypothesized to represent intermediate stages in gene rearrangement. These authors suggested that the slipped-strand mispairing mechanism could have been responsible for generating the duplications in the mitogenomes of these lizards.
A special phenomenon observed in the Symphurus mitogenomes indicates that the underlying mechanism is different from slipped-strand mispairing and can help us to explain this duplication process. Generally, there are only two stable non-coding regions in vertebrate mitogenomes: the CR and an approximately 40 nucleotide-long segment containing the origin for L-strand replication (OL), which is usually located inside the WANCY cluster at approximately two thirds of the genomic distance away from the CR. At OL, the parental H-strand is displaced as a single strand by the nascent H-strand and adopts a stable hairpin structure (Figure 1A) that serves as the initiation site for L-strand DNA synthesis. We searched all of the complete mitogenome sequences of flatfishes in the GenBank database and found that most contained conserved OL sequences, including Cynoglossus fishes (Figure 1A), which are in the same family as the Symphurus species and also show gene rearrangements. However, neither of the two Symphurus fish mitogenomes contains a typical OL region in the WANCY cluster.
Figure 1.
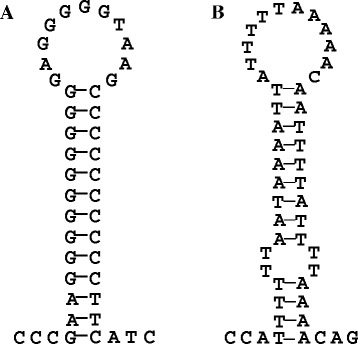
Typical teleostean OL structure and OL-like structure in the Symphurus plagiusa mitogenome. (A) Stem-loop structure of the OL in the Cynoglossus semilaevis mitogenome; (B) the OL-like structure in the Symphurus plagiusa mitogenome.
OL sequence loss has been reported in some other vertebrate mitogenomes, and it was suggested that a sequence in the WANCY region that encodes a tRNA forms a hairpin structure (OL-like structure) and acts as the OL [38-40]. Seligmann and Labra [41] tested whether a natural absence of an OL is associated with a greater capacity for the formation of OL-like structures by WANCY tRNA genes in lepidosaurian taxa. These authors concluded that WANCY tRNA genes form more OL-like structures in the absence of a regular OL than in its presence.
More interestingly, in the S. plagiusa mitogenome, within the CR and very close to tRNA-F, we found a hairpin structure that is very similar to the typical OL of vertebrate mitogenomes (Table 1 and Figure 1B). This finding means that while no typical OL region is present in the S. plagiusa mitogenome, there are two regions with the potential ability to form OL-like structures and initiate the replication of the L-strand. The existence of multiple OLs in vertebrate mitochondria has been demonstrated in many studies [42-44]. These features of multiple replication origins most likely also appeared in the mitogenome of the common ancestor of S. plagiusa and S. orientalis and were inherited by S. plagiusa due to its stable mitogenome structure. In contrast, these features would have been lost in the S. orientalis mitogenome because of the large-scale gene rearrangements. Therefore, in the ancestral mitogenome with the aforementioned dual OL-like structures, if only one is functioning, mitochondrial replication would occur normally, but if both structures functioned at the same time, the region between the two structures could be duplicated.
Accordingly, we speculate that the two ends of the duplicated fragment fall in the WANCY region and at the 3’ end of the CR, which both contain OL-like sequences. The duplication process would occur when both OL-like structures are functional. During a mitochondrial replication, after the replication fork arising from the initiation of H-strand synthesis (Figure 2A) passed the WANCY region, where the typical OL is located in most vertebrates, the parental H-strand in this region was exposed as a single strand, and the WANCY tRNA sequence formed an OL-like structure (OL-like structure 1 in Figure 2B). This structure then initiated an L-strand DNA synthesis event at this site (Nascent L-strand 1 in Figure 2B). Coincidentally, when the replication fork continued to expand and passed the CR, the other OL-like sequence was exposed and also formed a OL-like structure (OL-like structure 2 in Figure 2C), which then initiated a second round of L-strand DNA synthesis (Nascent L-strand 2 in Figure 2C). If both L-strand DNA synthesis events terminated at the WANCY region, the normal termination site, the fragment between the two OL-like sequences would be synthesized twice (Figure 2D). By a circular closure event or mitochondrial repair, the 5’ end of Nascent L-strand 1 would connect to the 3’ end of Nascent L-strand 2, while the 3’ end of Nascent L-strand 1 would connect to the 5’ end of Nascent L-strand 2 (Figure 2E). In the next round of mitochondrial replication, the duplication would be made permanent (Figure 2F).
Figure 2.
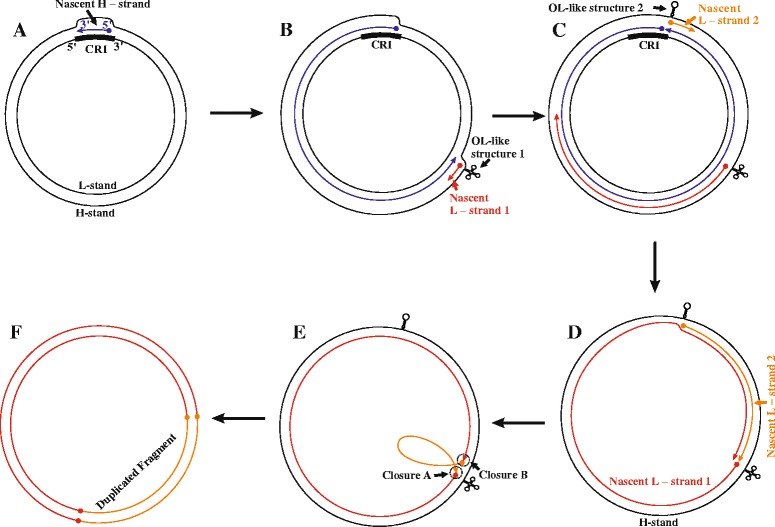
A duplication between the CR and the N gene caused by two OL-like structures. (A) H-strand synthesis is initiated at an origin for light-strand replication (OH). (B) L-strand synthesis is initiated at OL-like structure 1 when the replication fork reaches approximately two thirds of the genomic distance from the CR. (C) H-strand synthesis is terminated, and OL-like structure 2 initiates a second round of L-strand synthesis. (D) Both L-strand synthesis events are terminated at the WANCY region. (E) Connection of the 5’ end of Nascent L-strand 1 to the 3’ end of Nascent L-strand 2 and of the 3’ end of Nascent L-strand 1 to the 5’ end of Nascent L-strand 2. (F) The genes between the CR and N are, thus, duplicated.
Speculated TDRL process
The process of tandem duplication followed by random loss discussed above is typical of the TDRL model.
Therefore, we applied the TDRL model to describe the rearrangement events that altered the typical gene order (as found in S. plagiusa) to that observed in the S. orientalis mitogenome. The hypothesized intermediate steps are as follows. First, the above-mentioned double OL-like structures initiated DNA synthesis twice during mitochondrial replication, causing tandem duplication of the genes located between the CR and the WANCY region (1–13) (Figure 3 A, B) in the ancestral mitogenome (Figure 3A: typical gene order). In this case, the mitogenome would then have contained two sets of the same gene cluster (Figure 3B: 1–13 and 1’–13’). Because the mitogenome only maintains one set of functional genes, during subsequent evolutionary events, one of each of the 13 duplicated gene pairs randomly lost its function and became a pseudogene (Figure 3B; gray boxes). These pseudogenes then accumulated additional mutations to become shorter non-coding sequences or even be lost from the genome. Eventually, the existing gene order of the S. orientalis mitogenome was established (Figure 3C).
Figure 3.
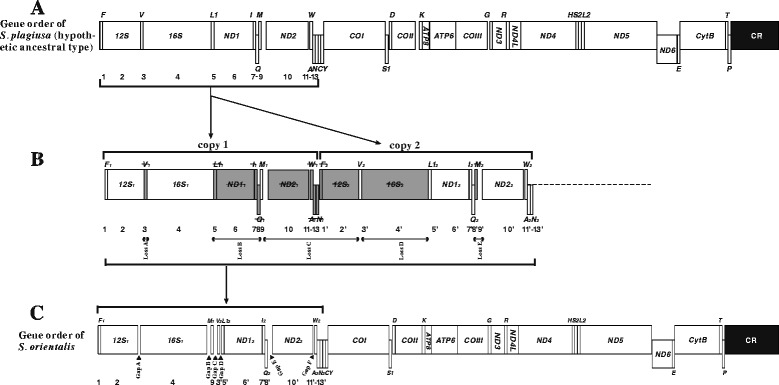
Inferred intermediate steps between the S. plagiusa mitogenome (typical ancestral gene order) and the S. orientalis mitogenome. Protein-coding genes and CRs are indicated with boxes, and tRNA genes are indicated with columns. Genes labeled above the diagram are encoded by the H-strand and those below the diagram by the L-strand. (A) Gene order in the S. plagiusa mitogenome; (B) the genes between CR and N were duplicated and then randomly lost; (C) gene order in the S. orientalis mitogenome.
Intergenic spacers provide evidence supporting the model
In the speculated process, after the five duplicated genes or gene clusters lost their functions, as shown in Figure 3B, they would have degraded to form five successive pseudogene fragments or shorter intergenic spacers (Figure 3B, gray boxes). In general, intergenic spacers vanish quickly because the degradation rate of non-functional genes is high to maintain the parsimony of mitogenomes. Interestingly, the speculated intergenic spacers did not disappear, and the positions of the five degraded gene fragments occur in one-to-one correspondence with those of the five residual intergenic spacers in the S. orientalis mitogenome: loss A in Figure 3B between 12S and 16S, which corresponds to Gap A in Figure 3C; loss B between 16S and tRNA-M, which corresponds to Gap B; loss C between tRNA-M and tRNA-V, which corresponds to Gap C; loss D between tRNA-V and tRNA-L1, which corresponds to Gap D; and loss E between tRNA-Q and ND2, which corresponds to Gap E. The one-to-one correspondence between the loss-of-function fragments and the residual intergenic spacers offers strong evidence supporting the speculated steps of gene duplication and loss in our model (Figure 3).
Conclusions
In summary, we determined the complete mitochondrial genomes of two Symphurus fishes, S. plagiusa and S. orientalis. The gene order of the S. plagiusa mitogenome is the same as that of a typical vertebrate, while that of S. orientalis features large-scale gene rearrangements. In the rearranged fragment from the CR to WANCY in the S. orientalis mitogenome, six large intergenic spacers more than 20 bp in length remain. The positions of these intergenic spacers occur at a one-to-one correspondence with the loss-of-function fragments in our speculated gene rearrangement model based on TDRL, providing strong evidence for both our model and the TDRL.
The two Symphurus fish mitogenomes share another special characteristic: they lack the typical OL region that is conserved in most vertebrates. In addition, in the S. plagiusa mitogenome, there are two regions with the potential ability to form OL-like structures, which means that both could initiate replication of the L-strand. Accordingly, we speculate that the ancestral mitogenome also possessed these two OL-like structures and that during an ancient mitochondrial replication event, both OL-like structures initiated DNA synthesis and induced the doubled replication of the L-strand, leading to the duplication of the region between the two structures. The findings of this study are based only on the primary sequence structures of mitogenomes. To substantiate the speculations presented herein, it will be necessary to observe replication intermediates via electron microscopy or 2D-gel analysis.
Data accessibility
Acknowledgments
This work was supported by the Natural Science Foundation of China (grant numbers 31471979, 31071890 and 41206134). We thank Dr. Richard Paperno and his colleagues of the Florida Fish and Wildlife Commission and Dr. Kwang-Tsao Shao and Dr. Maoying Li, who provided the samples.
Abbreviations
- CR
Control region
- WANCY
tRNA cluster of tRNA-W, tRNA-A, tRNA-N, tRNA-C and tRNA-Y
- rRNA
Ribosomal ribonucleic acid
- OL
Origin for light-strand replication
- tRNA
Transfer ribonucleic acid
- Mitogenome
mitochondrial genome
- TDRL
Tandem Duplication and Random Loss
- TDNL
Tandem Duplication and Non-random Loss
- DMNR
Dimer-Mitogenome and Non-Random Loss
- mtDNA
Mitochondrial DNA
- PCR
Polymerase chain reaction
- H-strand
Heavy strand
- L-strand
Light strand
Additional files
The primers used for fragment amplification of the Symphurus plagiusa mitogenome.
The primers used for fragment amplification of the Symphurus orientalis mitogenome.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WS collected the datasets, carried out a portion of the experiments, and drafted the manuscript. LG, SYW and XGM carried out a portion of the experiments. XYK directed the entire research project. All authors read and approved the final manuscript. All authors read and approved the final manuscript.
Contributor Information
Wei Shi, Email: shiwei@scsio.ac.cn.
Li Gong, Email: gongli1027@163.com.
Shu-Ying Wang, Email: czwangshuying@163.com.
Xian-Guang Miao, Email: miaoxianguang2008@163.com.
Xiao-Yu Kong, Email: xykong@scsio.ac.cn.
References
- 1.Boore J. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schirtzinger EE, Tavares ES, Gonzales LA, Eberhard JR, Miyaki CY, Sanchez JJ, et al. Multiple independent origins of mitochondrial control region duplications in the order Psittaciformes. Mol Phylogenet Evol. 2012;64(2):342–356. doi: 10.1016/j.ympev.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bensch S, Harlid A. Mitochondrial genomic rearrangements in songbirds. Mol Biol Evol. 2000;17(1):107–113. doi: 10.1093/oxfordjournals.molbev.a026223. [DOI] [PubMed] [Google Scholar]
- 4.Verkuil YI, Piersma T, Baker AJ. A novel mitochondrial gene order in shorebirds (Scolopacidae, Charadriiformes) Mol Phylogenet Evol. 2010;57(1):411–416. doi: 10.1016/j.ympev.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 5.Mueller RL, Boore JL. Molecular mechanisms of extensive mitochondrial gene rearrangement in plethodontid salamanders. Mol Biol Evol. 2005;22(10):2104–2112. doi: 10.1093/molbev/msi204. [DOI] [PubMed] [Google Scholar]
- 6.Okajima Y, Kumazawa Y. Mitochondrial genomes of acrodont lizards: timing of gene rearrangements and phylogenetic and biogeographic implications. BMC Evol Biol. 2010;10:141. doi: 10.1186/1471-2148-10-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Macey JR, Larson A, Ananjeva NB, Fang Z, Papenfuss TJ. Two novel gene orders and the role of light-strand replication in rearrangement of the vertebrate mitochondrial genome. Mol Biol Evol. 1997;14(1):91–104. doi: 10.1093/oxfordjournals.molbev.a025706. [DOI] [PubMed] [Google Scholar]
- 8.Sano N, Kurabayashi A, Fujii T, Yonekawa H, Sumida M. Complete nucleotide sequence of the mitochondrial genome of Schlegel's tree frog Rhacophorus schlegelii (family Rhacophoridae): duplicated control regions and gene rearrangements. Genes Genet Syst. 2005;80(3):213–224. doi: 10.1266/ggs.80.213. [DOI] [PubMed] [Google Scholar]
- 9.Inoue JG, Miya M, Tsukamoto K, Nishida M. Evolution of the deep sea gulper eel mitochondrial genomes: large-scale gene rearrangements originated within the eels. Mol Biol Evol. 2003;20(11):1917–1924. doi: 10.1093/molbev/msg206. [DOI] [PubMed] [Google Scholar]
- 10.Kong XY, Dong XL, Zhang YC, Shi W, Wang ZM, Yu ZY. A novel rearrangement in the mitochondrial genome of tongue sole, Cynoglossus semilaevis: control region translocation and a tRNA gene inversion. Genome. 2009;52(12):975–984. doi: 10.1139/G09-069. [DOI] [PubMed] [Google Scholar]
- 11.Ponce M, Infante C, Jimenez-Cantizano RM, Perez L, Manchado M. Complete mitochondrial genome of the blackspot seabream, Pagellus bogaraveo (Perciformes: Sparidae), with high levels of length heteroplasmy in the WANCY region. Gene. 2008;409(1–2):44–52. doi: 10.1016/j.gene.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 12.Mabuchi K, Miya M, Satoh TP, Westneat MW, Nishida M. Gene rearrangements and evolution of tRNA pseudogenes in the mitochondrial genome of the parrotfish (Teleostei: Perciformes: Scaridae) J Mol Evol. 2004;59(3):287–297. doi: 10.1007/s00239-004-2621-z. [DOI] [PubMed] [Google Scholar]
- 13.Ki JS, Jung SO, Hwang DS, Lee YM, Lee JS. Unusual mitochondrial genome structure of the freshwater goby Odontobutis platycephala: rearrangement of tRNAs and an additional non-coding region. J Fish Biol. 2008;73(2):414–428. doi: 10.1111/j.1095-8649.2008.01911.x. [DOI] [Google Scholar]
- 14.Gong L, Shi W, Si LZ, Kong XY. Rearrangement of mitochondrial genome in fishes. Zool Res. 2013;34(6):666–673. [PubMed] [Google Scholar]
- 15.Shi W, Jiang JX, Miao XG, Kong XY. The complete mitochondrial genome sequence of Heteromycteris japonicus (Pleuronectiformes: Soleidae) Mitochondrial DNA. 2013;1:1–2. doi: 10.3109/19401736.2013.800493. [DOI] [PubMed] [Google Scholar]
- 16.Manchado M, Catanese G, Ponce M, Funes V, Infante C. The complete mitochondrial genome of the Senegal sole, Solea senegalensis Kaup. Comparative analysis of tandem repeats in the control region among soles. DNA Seq. 2007;18(3):169–175. doi: 10.1080/10425170701308956. [DOI] [PubMed] [Google Scholar]
- 17.Wang SY, Shi W, Wang ZM, Gong L, Kong XY. The complete mitochondrial genome sequence of Aesopia cornuta (Pleuronectiformes: Soleidae) Mitochondrial DNA. 2013;21:1–2. doi: 10.3109/19401736.2013.803544. [DOI] [PubMed] [Google Scholar]
- 18.Fonseca MM, Posada D, Harris DJ. Inverted replication of vertebrate mitochondria. Mol Biol Evol. 2008;25(5):805–808. doi: 10.1093/molbev/msn050. [DOI] [PubMed] [Google Scholar]
- 19.Fonseca MM, Harris DJ, Posada D. The Inversion of the Control Region in Three Mitogenomes Provides Further Evidence for an Asymmetric Model of Vertebrate mtDNA Replication. PLoS One. 2014;9(9) doi: 10.1371/journal.pone.0106654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi W, Dong XL, Wang Z-M, Miao X-G, Wang S-Y, Kong X-Y. Complete mitogenome sequences of four flatfishes (Pleuronectiformes) reveal a novel gene arrangement of L-strand coding genes. BMC Evol Biol. 2013;13:173. doi: 10.1186/1471-2148-13-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shi W, Miao X-G, Kong X-Y. A novel model of double replications and random loss accounts for rearrangements in the Mitogenome of Samariscus latus (Teleostei: Pleuronectiformes) BMC Genomics. 2014;15(1):352. doi: 10.1186/1471-2164-15-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lunt DH, Hyman BC. Animal mitochondrial DNA recombination. Nature. 1997;387(6630):247. doi: 10.1038/387247a0. [DOI] [PubMed] [Google Scholar]
- 23.Arndt A, Smith MJ. Mitochondrial gene rearrangement in the sea cucumber genus Cucumaria. Mol Biol Evol. 1998;15(8):1009–1016. doi: 10.1093/oxfordjournals.molbev.a025999. [DOI] [PubMed] [Google Scholar]
- 24.Moritz C, Dowling TE, Brown WM. Evolution of Animal Mitochondrial-DNA - Relevance for Population Biology and Systematics. Annu Rev Ecol Syst. 1987;18:269–292. doi: 10.1146/annurev.es.18.110187.001413. [DOI] [Google Scholar]
- 25.Lavrov DV, Boore JL, Brown WM. Complete mtDNA sequences of two millipedes suggest a new model for mitochondrial gene rearrangements: duplication and nonrandom loss. Mol Biol Evol. 2002;19(2):163–169. doi: 10.1093/oxfordjournals.molbev.a004068. [DOI] [PubMed] [Google Scholar]
- 26.Cantatore P, Gadaleta MN, Roberti M, Saccone C, Wilson AC. Duplication and remoulding of tRNA genes during the evolutionary rearrangement of mitochondrial genomes. Nature. 1987;329(6142):853–855. doi: 10.1038/329853a0. [DOI] [PubMed] [Google Scholar]
- 27.Jacobs HT, Herbert ER, Rankine J. Sea urchin egg mitochondrial DNA contains a short displacement loop (D-loop) in the replication origin region. Nucleic Acids Res. 1989;17(22):8949–8965. doi: 10.1093/nar/17.22.8949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beckenbach AT. Mitochondrial Genome Sequences of Nematocera (Lower Diptera): Evidence of Rearrangement following a Complete Genome Duplication in a Winter Crane Fly. Genome Biol Evol. 2012;4(2):89–101. doi: 10.1093/gbe/evr131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.San Mauro D, Gower DJ, Zardoya R, Wilkinson M. A hotspot of gene order rearrangement by tandem duplication and random loss in the vertebrate mitochondrial genome. Mol Biol Evol. 2006;23(1):227–234. doi: 10.1093/molbev/msj025. [DOI] [PubMed] [Google Scholar]
- 30.Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic acids symposium series. 1999;41:95–98. [Google Scholar]
- 31.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo XH, Liu SJ, Liu Y. Comparative analysis of the mitochondrial DNA control region in cyprinids with different ploidy level. Aquaculture. 2003;224(1–4):25–38. doi: 10.1016/S0044-8486(03)00168-6. [DOI] [Google Scholar]
- 33.Shi W, Kong XY, Wang ZM, Jiang JX. Utility of tRNA genes from the complete mitochondrial genome of Psetta maxima for implying a possible sister-group relationship to the Pleuronectiformes. Zool Stud. 2011;50(5):665–681. [Google Scholar]
- 34.Miya M, Pietsch TW, Orr JW, Arnold RJ, Satoh TP, Shedlock AM, et al. Evolutionary history of anglerfishes (Teleostei: Lophiiformes): a mitogenomic perspective. BMC Evol Biol. 2010;10:58. doi: 10.1186/1471-2148-10-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yagishita N, Miya M, Yamanoue Y, Shirai SM, Nakayama K, Suzuki N, et al. Mitogenomic evaluation of the unique facial nerve pattern as a phylogenetic marker within the percifom fishes (Teleostei: Percomorpha) Mol Phylogenet Evol. 2009;53(1):258–266. doi: 10.1016/j.ympev.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 36.Wang Z-M, Shi W, Jiang J-X, Wang S-Y, Miao X-G, Huang L-M, et al. The complete mitochondrial genome of a striped sole Zebrias zebrinus (Pleuronectiformes: Soleidae) Mitochondrial DNA. 2013;24(6):633–635. doi: 10.3109/19401736.2013.772160. [DOI] [PubMed] [Google Scholar]
- 37.Fujita MK, Boore JL, Moritz C. Multiple origins and rapid evolution of duplicated mitochondrial genes in parthenogenetic geckos (Heteronotia binoei; Squamata, Gekkonidae) Mol Biol Evol. 2007;24(12):2775–2786. doi: 10.1093/molbev/msm212. [DOI] [PubMed] [Google Scholar]
- 38.Desjardins P, Morais R. Sequence and gene organization of the chicken mitochondrial genome. A novel gene order in higher vertebrates. J Mol Biol. 1990;212(4):599–634. doi: 10.1016/0022-2836(90)90225-B. [DOI] [PubMed] [Google Scholar]
- 39.Seligmann H, Krishnan NM, Rao BJ. Possible multiple origins of replication in primate mitochondria: Alternative role of tRNA sequences. J Theor Biol. 2006;241(2):321–332. doi: 10.1016/j.jtbi.2005.11.035. [DOI] [PubMed] [Google Scholar]
- 40.Seligmann H, Krishnan NM. Mitochondrial replication origin stability and propensity of adjacent tRNA genes to form putative replication origins increase developmental stability in lizards. J Exp Zool B Mol Dev Evol. 2006;306(5):433–449. doi: 10.1002/jez.b.21095. [DOI] [PubMed] [Google Scholar]
- 41.Seligmann H, Labra A. The relation between hairpin formation by mitochondrial WANCY tRNAs and the occurrence of the light strand replication origin in Lepidosauria. Gene. 2014;542(2):248–257. doi: 10.1016/j.gene.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 42.Seligmann H. Hybridization between mitochondrial heavy strand tDNA and expressed light strand tRNA modulates the function of heavy strand tDNA as light strand replication origin. J Mol Biol. 2008;379(1):188–199. doi: 10.1016/j.jmb.2008.03.066. [DOI] [PubMed] [Google Scholar]
- 43.Clayton DA, Brown TA. Replication of mammalian mitochondrial DNA occurs by strand displacement with alternative light-strand origins. FASEB J. 2006;20(4):A453–3. doi: 10.1101/gad.1352105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yasukawa T, Reyes A, Cluett TJ, Yang MY, Bowmaker M, Jacobs HT, et al. Replication of vertebrate mitochondrial DNA entails transient ribonucleotide incorporation throughout the lagging strand. EMBO J. 2006;25(22):5358–5371. doi: 10.1038/sj.emboj.7601392. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.