

Abstract
Elevated blood pressure is an important global health problem, and in-utero under-nutrition may be an important factor in the pathogenesis of hypertension. In the present study, we tested the hypothesis that antenatal maternal low protein diet (MLPD) leads to sexually dimorphic developmental programming of the components of the pulmonary renin-angiotensin system. This may be important in the antenatal MLPD-associated development of hypertension. In pregnant mice, we administered normal (control) and isocaloric 50 % protein restricted diet, commencing one week before mating and continuing until delivery of the pups. From the 18th to 24th week postnatal, we measured blood pressure in the offspring by use of a non-invasive tail-cuff method. In the same mice, we examined the mRNA and protein expression of the key components of the pulmonary renin-angiotensin system. Also, we examined microRNA complementary to angiotensin converting enzymes (ACE) 2 in the offspring lungs. Our results demonstrate that as a consequence of antenatal MLPD: 1) pup birthweight was significantly reduced in both sexes. 2) female offspring developed hypertension, but males did not. 3) In female offspring, ACE-2 protein expression was significantly reduced without any change in the mRNA levels. 4) miRNA 429, which has a binding site on ACE-2 - 3’ UTR was significantly upregulated in the female antenatal MLPD offspring. 5) In males, ACE-2 mRNA and protein expression were unaltered. We conclude that in the mouse, antenatal MLPD-induced reduction of ACE-2 in the female offspring lung may be an important mechanisms in sexually dimorphic programming of hypertension.
Electronic supplementary material
The online version of this article (doi:10.1186/s12899-015-0016-6) contains supplementary material, which is available to authorized users.
Keywords: Angiotensin converting enzyme, microRNA, Epigenetics, DOHaD, Fetal programming, Developmental origins, Barker hypothesis
Background
More than 150 years after its first description, high blood pressure has been listed as the primary cause of death of 61,005 Americans in 2008 (www.heart.org/statistics). In 90 to 95% of these cases, the cause of hypertension is not known (AHA, 2008). Importantly, a family history of hypertension is an important risk factor for its development [1]; which suggests a strong genetic or familial environmental factor in its genesis. Moreover, during recent years, accumulating evidence indicates that to a significant degree, hypertension may have a developmental origin. Epidemiological data from human studies [2], and experiments in several species of laboratory animals including mice [3–6], rats [7], guinea pigs [8], and sheep [9], have demonstrated the importance of maternal nutrition during gestation in the genesis of hypertension in the adult offspring.
In humans, evidence supports sexually dimorphic trends in the occurrence of hypertension [10], with males being more affected than females before the latter’s menopause; and males and females being affected equally following female menopause [11]. Notably, with increasing age the blood pressure in females increases more rapidly than that of males [12]. The mechanisms of these changes are not known, however.
Following the discovery of renin as a pressor molecule [13], the renin-angiotensin system (RAS) has been established as one of the major pathways involved in both the development of hypertension and in fluid homeostasis. RAS has received increased attention, and our understanding of this system has changed radically over the past several decades. The cascade starts with an α2-globulin angiotensinogen (AGT), which is produced constitutively and released into the circulation, chiefly by the liver. A substrate for renin (secreted by kidneys), AGT is converted into the decapeptide angiotensin I (Ang I), and subsequently by angiotensin converting enzyme 1 (ACE 1) to the octapeptide Ang II. By the action of ACE-2, Ang II activity is terminated by its conversion to Ang 1–7. Of note, both ACE-1 and ACE-2 are secreted by the lung [14]. Importantly, a number of drugs (enalapril, lisinopril, etc.) used clinically [15–17] inhibit ACE1 and underscore the role of lungs in hypertension. Furthermore, studies have demonstrated that modulation of ACE from the lungs have important implications in the development of hypertension [18, 19]. Studies from our laboratory [3–5] and others [20] have demonstrated significant changes in the RAS in response to antenatal MLPD. However, the extent to which antenatal MLPD can lead to sexual dimorphic programing of the RAS in association with hypertension is not known. Also, the specific components of the RAS pathway that may be important in this programing are not well understood. Thus, we tested the hypothesis that antenatal MLPD leads to sexually dimorphic developmental programming of the components of the pulmonary renin-angiotensin system, which may be important in the antenatal MLPD-associated development of hypertension.
Methods
Experimental animal and tissues
The present study was in compliance with the Animal Welfare Act, guidelines of the American Physiological Society, and was approved by the Institutional Animal Care and Use Committee (IACUC) of Loma Linda University. We have described all these methods in our previous publications [3–6]. Briefly, we obtained FVB/NJ mice (~8 weeks of age) from Jackson Laboratories (Bar Harbor, ME), and housed them in the Animal Research Facility, Loma Linda University under conditions of 14 h light, 10 h darkness, ambient temperature of 20 °C, and relative humidity of 30 to 60 %. At 16 weeks of age, the mice were bred, by keeping the males and females together for 12 h (overnight). In the morning, mating was confirmed by examination of vaginal plugs, and considered 0.5 day post coitum (DPC). We started the study with 16 animals in each group. However, following overnight mating, the mice dams without vaginal plug and significant weight gain on day 7 post coitum were excluded from the study. This reduced the number of mice dams to 8 to 10 dams in each group. Following birth, 8 pups per group from 8 different mice dams were used to conduct the blood pressure measurement study. Molecular biology study (real-time PCR) were conducted on 4 animals from each group.
Protein deprivation
Protein restricted chow was obtained from Newco Distributors Inc. (Rancho Cucamonga, CA). Mice dams were divided in two groups: control protein diet (18 g/100 g, 100 % protein content), and antenatal MLPD (9 g/100 g, 50 % protein diet). To avoid the stress of food change during gestation and to include the peri-conceptual period, diet administration was started one week before mating. To maintain an isocaloric diet with low protein, we replaced the proteins with carbohydrates. The normal diet contained 18 g protein/100 g food, as described (additional file 1) [3–6]. We measured the amount of food (g/day) consumed by the mice on both normal and isocaloric low protein diets; and these were similar, [being 3 ± 0.5 g for females (n = 16) and 4 ± 0.3 g for males (n = 16) in the control group and 3 ± 0.4 (n = 16) for females and 4 ± 0.3 g for males (n = 16) in the antenatal MLPD group, respectively].
Physiological Measurement
We measured body weight on the morning after the pups were delivered (0.5 Days). Intact placentas were collected from the cage and placental weight was determined. After birth, all the mice dams were maintained on the control diet to examine the effect of antenatal developmental programming per se. Blood pressure was measured non-invasively, weekly by determining the tail blood volume, flow, and pressure with a volume pressure recording sensor and an occlusion tail-cuff (CODA System, Kent Scientific, Torrington, CT). This system is significantly different from the plethysmographic based tail-cuff measurement system, which measures only systolic blood pressure [21]. This is a highly accurate system with the capability of measuring systolic and diastolic blood pressures with the heart rate simultaneously and non-invasively [22, 23]. Moreover, in numerous other studies, the tail cuff method has been shown to measure the blood pressure non-invasively and accurately in mice and rats [24, 25]. Before commencing our studies, mice were placed on a warming plate at 37° centigrade until the temperature of the tail-region measured 37° by an infrared thermometer. Following the warming, the mice were trained for three 15-min sessions each day for three days, or until we obtained stable blood pressure recordings. Blood pressure was measured 20 times on a fixed day (Tuesday) and time (~11:30 AM), once weekly from 18 to 24 weeks of age.
mRNA and protein quantification
At 32 weeks of age the mice were euthanized by cervical dislocation and the lungs were isolated. The isolated lungs were snap frozen in liquid nitrogen and stored at −80° centigrade for later analysis. For each experiment lung tissue from 4 different mice from different mothers were used. Real-time PCR and western immunoblot assays were conducted, as described and validated previously in our laboratory [3–5]. We isolated and quantified RNA and protein by Allprep DNA/RNA Mini Kit according to the manufacturer’s instructions (Qiagen Inc, Valencia, CA Cat # 80204). Isolated mRNA was analyzed using a NanoDrop1000 Spectrophotometer (Thermo Scientific, Waltham, MA) at 260/280 wavelength UV rays to check for quality and quantity. The 260/280 ratio of 1.8 to 2 was accepted for quantification with real-time PCR. Real-time PCR was performed on Light Cycler 1.5 (Roche Inc., Indianapolis, IN) using hydrolysis Taqman probes and primers, designed using the Universal Probe Library, a web based software (Roche Inc.), and the Quantfast Real-Time PCR Kit (Qiagen). Total RNA (1 ug per reaction) was reverse transcribed using Quantitect reverse transcriptase kit (Qiagen, Valencia, CA). Relative expression was normalized to 18S RNA and fold-changes were calculated using the ΔΔCt method with normalization of individual PCR efficiencies [26].
Western-immunoblot experiments were conducted as described previously [4, 5]. Briefly, frozen samples were homogenized in the 1 x cell lysing buffer (Cell Signaling Technology, Beverly, MA) containing a 1× phosphatase and protease inhibitor cocktail (Sigma). Nuclei and debris were pelleted by centrifugation at 1000 × g for 10 min. The supernatant was collected and stored at −80 °C. SDS-gel and western blot were performed by using appropriate antibodies (Table 1). All secondary antibodies were obtained from Abcam (Cambridge, MA). Twenty μg protein from each sample was loaded on a SDS-gel and electrophoresed at 100 V for 3 h. Proteins were transferred to a nitrocellulose membrane, and subjected to immunoblotting with antibodies. Bands were detected with enhanced chemiluminescence using a ChemiImager (Alpha-Innotech, San Leandro, CA). The results are expressed as fraction of control. We performed control experiments with glyceraldehyde-3-phosphate dehydrogenase (GAPDH), beta-tubulin, alpha-actin, and extracellular regulated signal kinase1/2 (ERK1/2). Our results demonstrated total ERK1/2 protein expression to be the most uniform among all the four study groups. Moreover, ERK1/2 integrated density (arbitrary unit) on densitometry analysis correlated well with different amounts of protein loaded, and was used as an internal control to account for uniform protein loading.
Table 1.
Antibodies used in the study
| No. | Name | Company | Cat. No. | Dilution |
|---|---|---|---|---|
| 1 | Angiotensinogen | Swant Inc. Switzerland | 138 | 1:1000 |
| 2 | Renin | Santa Cruz Biotechnologies | sc-27320 | 1:300 |
| 3 | ACE-1 | Abcam Inc. | ab39172 | 1:1000 |
| 4 | ACE-2 | Abcam Inc. | ab59351 | 1:1000 |
| 5 | AT-1 | Abcam Inc. | ab59018 | 1:250 |
| 6 | AT-2 | Abcam Inc. | ab19134 | 1:250 |
| 7 | GAPDH | Abcam Inc. | ab9485 | 1:2000 |
| 8 | Alpha-Actin | Abcam Inc. | Ab1801 | 1:1000 |
| 9 | ERK1/2 | Cell Signaling Technology | #9102 | 1:1000 |
| 10 | Beta-tubulin | Santa Cruz Biotechnology | sc-5274 | 1:1000 |
MicroRNA studies
MicroRNAs (miRNA) were identified for the 3’ UTR of the ACE-2 mRNAs, using the web based bio-informatics software – TargetScan 4.2 (http://www.targetscan.org). Of 10 miRNA suggested by the bio-informatics software, we chose mmu-mir-429 miRNA for the present study, by the use of the Context Score of more than 90th percentile, as described by Grimson et al. [27]. The identified miRNA levels were measured by the use of Real-Time Taqman microRNA PCR assays, according to manufacturer’s instructions (Life Technologies, Grand Island, NY).
Statistical analysis
We analyzed the data using repeated measure (RM) two-way Univariate Analysis of Variance (ANOVA) with Bonferroni’s and Tukey’s post-hoc analysis as well as linear and non-linear regression to determine statistically significant differences between groups, by the use of GraphPad Prism software (GraphPad Software Inc., San Diego, CA) and IBM SPSS (IBM Corp., NY). The hypothesis was accepted at P < 0.05. For the measurement of blood pressure one male or female offspring was included from one mice dam and considered n = 1.
Results
Offspring mice weight in response to antenatal protein restriction
As a consequence of antenatal protein restriction during the entire gestation, we observed a significant reduction in birth weight in mice offspring. Both male and female antenatal MLPD were associated with a significant reduction in the birth weight (Fig. 1A), being (g) for control male 1.5 ± 0.04 (n = 8), antenatal MLPD male 1.3 ± 0.04 (n = 11), control female 1.6 ± 0.05 (n = 8), and antenatal MLPD female 1.2 ± 0.04 (n = 11). Of importance, the antenatal MLPD offspring demonstrated catch-up growth, so that by 32 weeks of age, female from the antenatal MLPD group were significantly over-weight. Males were affected to a significantly lesser degree, however (Fig. 1).
Fig. 1.
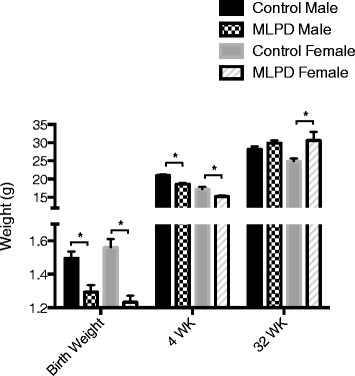
Weight in grams (g) of mice offspring at birth, 4 weeks, and 32 weeks of age from control and antenatal MLPD group. *Denotes P < 0.05. N = 16 for each group. MLPD - Mice offspring from the maternal low protein diet group
Litter size and placental weight
Neither litter size nor placental weights differ significantly. The average litter size was 7 ± 2 and 6 ± 3 in the control and antenatal MLPD group, respectively (P > 0.05). The pups sex distribution was between 40 to 60 % per mice dam between control and MLPD group (P > 0.05).
Blood pressure
We measured mean arterial blood pressure from the 18th week to 24th postnatal week (n = 8 mice in each group). “A sexually dimorphic trend of hypertension was observed in males and females as observed by a significant interaction of Sex*Diet by two-way RM ANOVA”. As shown in Fig. 2, the data demonstrates that antenatal MLPD female offspring had significantly higher mean arterial blood pressure, compared to the offspring from dams on the normal diet. In males, however, there was a significantly less increase in mean arterial blood pressure in response to antenatal MLPD, compared to females (Fig. 2).
Fig. 2.
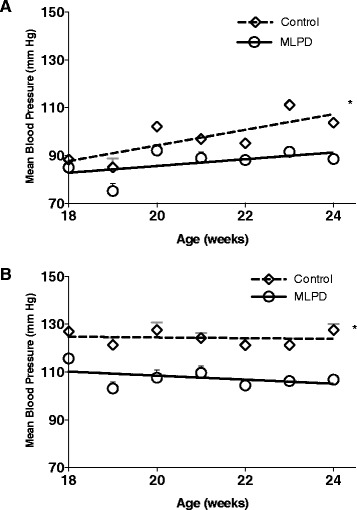
Mean arterial blood pressure for 7 weeks (18 weeks to 24 weeks of age) of male mice offspring (A) and female mice offspring (B) from control and antenatal MLPD group. *Denotes P < 0.05. N = 8 for each group. MLPD - Mice offspring from the maternal low protein diet group
Renin angiotensin system expression
To examine the underlying molecular alterations in association with hypertension, we examined several key components of the pulmonary renin-angiotensin system. With antenatal MLPD, we observed no significant changes in the protein expression of renin, ACE1, or angiotensin II type 1 and 2 receptors protein expression (Table 2). Notably, as shown in the Fig. 3A, in female antenatal MLPD offspring, ACE-2 protein expression was significantly reduced; whereas, no change was observed in the male offspring. To examine, the extent to which the alteration in the ACE-2 expression occurs at the transcriptional level, we conducted real-time PCR analysis of ACE-2 mRNA (Fig. 3B). No significant changes were observed in ACE-2 mRNA levels in antenatal MLPD lungs from either males or females.
Table 2.
Protein levels of un-altered components of the pulmonary renin-angiotensin system in response to antenatal maternal low-protein diet
| Gene Name | Males | Females | ||
|---|---|---|---|---|
| Protein Levels | Protein Levels | |||
| Control | MLPD | Control | MLPD | |
| Renin | 0.82 ± 0.07 | 0.92 ± 0.05 | 0.7 ± 0.1 | 0.7 ± 0.15 |
| ACE1 | 0.94 ± 0.04 | 0.81 ± 0.03 | 0.28 ± 0.15 | 0.16 ± 0.01 |
| AT1 | 0.15 ± 0.01 | 0.14 ± 0.03 | 0.19 ± 0.01 | 0.21 ± 0.03 |
| AT2 | 0.08 ± 0.01 | 0.08 ± 0.02 | 0.1 ± 0.2 | 0.9 ± 0.02 |
Protein levels are shown as integrated density (arbitrary measurement) relative to total-ERK. N = 4, no statistically significant difference (P < 0.05) was identified by one way ANOVA and post-hoc Bonferroni’s in any of the measurement.
Fig. 3.
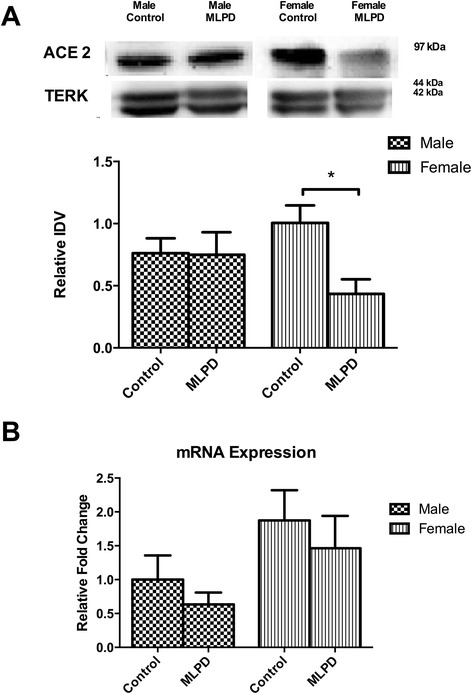
Expression levels of ACE2. (A) Western blot analysis (B) mRNA analysis of mice offspring from control and antenatal MLPD groups. IDV - integrated density value, an arbitrary unit. *Denotes P < 0.05. N = 4 for each group. MLPD - Mice Offspring from the maternal low protein diet group
ACE-2 complementary miRNA expression
To examine, whether the reduction in ACE-2 protein levels in the female antenatal MLPD group is as a consequence of reduced translation, we examined miRNA miR-429, which has a complementary binding site on 3’ UTR of ACE-2. Of importance, as shown in Fig. 4 miR-429 levels were significantly increased in the female and decreased in maleantenatal MLPD lungs, compared to controls.
Fig. 4.
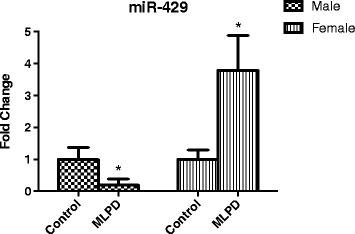
Expression levels of miR-429 from control and antenatal MLPD groups. *Denotes P < 0.05. N = 4 for each group. MLPD - Mice Offspring from the maternal low protein diet group
Discussion
Accumulating evidence suggests that antenatal maternal protein deprivation can lead to the development of hypertension in the offspring [6, 7, 9, 28, 29]. Of note, in a manner similar to humans, mice also show an increase in blood pressure with aging [30]. The mechanisms of this phenomenon are not known, however. In the present study, we identified that in females, antenatal MLPD-induced hypertension is associated with a reduction in the level of ACE-2 enzyme (Fig. 3). Importantly, female mice offspring were affected with hypertension to a significantly greater extent than males (Fig. 2), which may relate to the ACE-2 gene being located on the X chromosome [31]. Further, studies in three different rat strains have demonstrated that reduced ACE-2 expression is associated with hypertension [32]. In addition, ACE-2 gene maps to a defined quantitative trait locus (QTL) for increased blood pressure on the X chromosome in these hypertensive rats [32].
Several other reports also have suggested an important role of ACE-2 in hypertension [33, 34]. In the present study, as a consequence of antenatal MLPD, we observed that ACE-2 was reduced only in female, and not male offspring (Fig. 3A). Importantly, only female offspring from the antenatal MLPD group were significantly hypertensive (Fig. 2). It appears that, as a consequence of antenatal MLPD, in females ACE-2 is differentially regulated, and may play an important role in the developmental programming of hypertension. Being located on an X chromosome, ACE-2 gene is susceptible for sexually dimorphic programming, as the chromosome is well known for other sexually dimorphic gene expression and disorders [35]. Additionally, ACE-2 is known to be regulated in a sexually dimorphic manner by estrogen [36]. Also, a high fat diet in rats is known to cause sexually dimorphic regulation of ACE-2 and the development of hypertension [37]. The present study, adds to the accumulating evidence that ACE-2 can be developmentally programmed by antenatal stressors during gestation, and may be a critical factor in the development of hypertension.
Of vital importance, in the present study, we identified that in response to antenatal MLPD, ACE-2 is not regulated at the transcriptional level, but rather programming occurs at the translational level. The ACE-2 mRNA levels were not significantly altered as a consequence of antenatal maternal protein deprivation (Fig. 3B); however, the ACE-2 protein levels were significantly lower in the antenatal MLPD females. Recent studies have demonstrated that miRNA are important in post-translational gene regulation, and play an important role in the developmental programming of hypertension. miRNA can bind to the 3’ UTR of a near-complementary sequence of mRNA and lead to reduced translation. As noted in Fig. 4, we observed that miRNA 429, which is complementary to ACE-2 was significantly upregulated, which agrees with other studies [14, 38, 39]. Importantly, there was no increase in ACE2 proteins and associated miRNA-429 in lungs from the male MLPD group. As shown in the summary Fig. 5 and previous studies, we speculate that antenatal maternal protein restriction leads to increased miR-429, which causes reduced production of ACE-2 protein. Reduced levels of ACE-2 lead to reduced degradation of the potent vasoconstrictor angiotensin II and thus increased blood pressure.
Fig. 5.
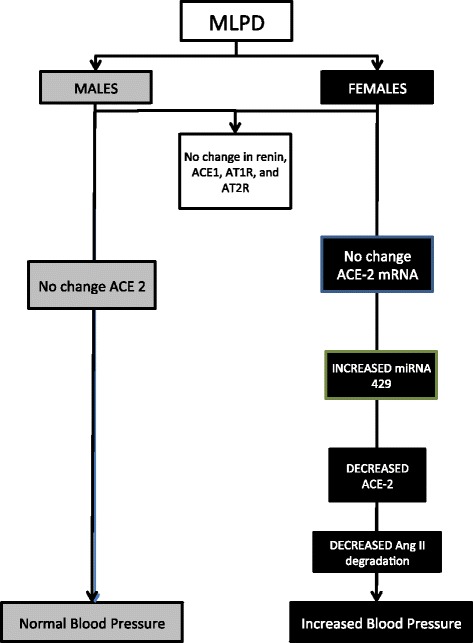
Summarized findings of the study
Conclusion
Developmental programming of adult health and disease is an important factor in the genesis of hypertension. The present study, demonstrates that sexual dimorphism of the development of hypertension may be mediated through miRNA-mediated mechanisms and alterations in the ACE-2 gene. The questions remain, by what mechanism miRNA expression is regulated and programmed by in-utero environment? What is the role of estrogen in this signaling? We shall pursue thesein future studies.
Additional files
Composition of the diet administered to control group of mice dams. Table S2 Composition of the diet administered to the antenatal maternal low protein group of mice dams.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conception and Design: RG and LDL. Acquisition of Data and Analysis/Interpretation of Data: RG, JVW, DG. Drafting of the manuscript and revising it critically: RG and LDL. Final Approval: RG and LDL. All authors read and approved the final manuscript.
Contributor Information
Ravi Goyal, Phone: 909-558-4325, Email: rgoyal@llu.edu.
Jonathan Van-Wickle, Email: jonvanwick@gmail.com.
Dipali Goyal, Email: dipaligoyal@llu.edu.
Lawrence D. Longo, Email: llongo@llu.edu
References
- 1.Rodríguez-Moran M, et al. Family history of hypertension and cardiovascular risk factors in prepubertal children. Am J Hypertens. 2010;23:299–304. doi: 10.1038/ajh.2009.257. [DOI] [PubMed] [Google Scholar]
- 2.Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ. 1989;298:564–7. doi: 10.1136/bmj.298.6673.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goyal R, Wong C, Van Wickle J, Longo LD. Antenatal maternal protein deprivation: sexually dimorphic programming of the pancreatic renin-angiotensin system. J Renin Angiotensin Aldosterone Syst. 2013;14:137–45. doi: 10.1177/1470320312456329. [DOI] [PubMed] [Google Scholar]
- 4.Goyal R, Goyal D, Leitzke A, Gheorghe CP, Longo LD. Brain renin-angiotensin system: fetal epigenetic programming by maternal protein restriction during pregnancy. Reprod Sci. 2010;17:227–38. doi: 10.1177/1933719109351935. [DOI] [PubMed] [Google Scholar]
- 5.Goyal R, Galffy A, Field SA, Gheorghe CP, Mittal A, Longo LD. Maternal protein deprivation: changes in systemic renin-angiotensin system of the mouse fetus. Reprod Sci. 2009;16:894–904. doi: 10.1177/1933719109337260. [DOI] [PubMed] [Google Scholar]
- 6.Goyal R, Longo LD. Maternal protein deprivation: sexually dimorphic programming of hypertension in the mouse. Hypertens Res. 2013;36:29–35. doi: 10.1038/hr.2012.129. [DOI] [PubMed] [Google Scholar]
- 7.Brawley L, Poston L, Hanson MA. Mechanisms underlying the programming of small artery dysfunction: review of the model using low protein diet in pregnancy in the rat. Arch Physiol Biochem. 2003;111:23–35 [DOI] [PubMed]
- 8.Persson E, Jansson T. Low birth weight is associated with elevated adult blood pressure in the chronically catheterized guinea-pig. Acta Physiol Scand. 1992;145:195–6 [DOI] [PubMed]
- 9.Gilbert JS, Lang AL, Grant AR, Nijland MJ. Maternal nutrient restriction in sheep: hypertension and decreased nephron number in offspring at 9 months of age. J Physiol. 2005;565:137–47. [DOI] [PMC free article] [PubMed]
- 10.Sandberg K, Ji H. Sex differences in primary hypertension. Biol sex dif. 2012;3:7. doi: 10.1186/2042-6410-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kannel WB. The Framingham Study: historical insight on the impact of cardiovascular risk factors in men versus women. J Gend Specif Med. 2002;5:27–37. [PubMed] [Google Scholar]
- 12.Burt VL, et al. Prevalence of hypertension in the US adult population. Results from the Third National Health and Nutrition Examination Survey, 1988–1991. Hypertension. 1995;25:305–13. doi: 10.1161/01.HYP.25.3.305. [DOI] [PubMed] [Google Scholar]
- 13.Marks LS, Maxwell MH. Tigerstedt and the discovery of renin. An historical note. Hypertension. 1979;1:364–88. doi: 10.1161/01.HYP.1.4.384. [DOI] [PubMed] [Google Scholar]
- 14.Goyal R, Leitzke A, Goyal D, Gheorghe CP, Longo LD. Antenatal maternal hypoxic stress: adaptations in fetal lung Renin-Angiotensin system. Reprod Sci. 2011;18:180–9. doi: 10.1177/1933719110385134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laragh JH, Mancia G. A symposium: systemic hypertension: contribution of trandolapril, a new angiotensin-converting enzyme inhibitor, toward patient protection. Introduction. Am J Cardiol. 1992;70:1D–3. doi: 10.1016/0002-9149(92)90264-Y. [DOI] [PubMed] [Google Scholar]
- 16.Messerli FH, Oparil S, Feng Z. Comparison of efficacy and side effects of combination therapy of angiotensin-converting enzyme inhibitor (benazepril) with calcium antagonist (either nifedipine or amlodipine) versus high-dose calcium antagonist monotherapy for systemic hypertension. Am J Cardiol. 2000;86:1182–7. doi: 10.1016/S0002-9149(00)01199-1. [DOI] [PubMed] [Google Scholar]
- 17.Schnaper HW. Angiotensin-converting enzyme inhibitors for systemic hypertension in young and elderly patients. Am J Cardiol. 1992;69:54C–8. doi: 10.1016/0002-9149(92)90282-4. [DOI] [PubMed] [Google Scholar]
- 18.Forslund T, Tikkanen I, Gronhagen-Riska C, Fyhrquist F. Dissociation of the effect of captopril on blood pressure and angiotensin converting enzyme in serum and lungs of spontaneously hypertensive rats. Acta Pharmacol Toxicol (Copenh). 1981;49:416–21. doi: 10.1111/j.1600-0773.1981.tb00925.x. [DOI] [PubMed] [Google Scholar]
- 19.Wallace KB, Bailie MD, Hook JB. Development of angiotensin-converting enzyme in fetal rat lungs. Am J Physiol. 1979;236:R57–60. doi: 10.1152/ajpregu.1979.236.1.R57. [DOI] [PubMed] [Google Scholar]
- 20.Bogdarina I, Welham S, King PJ, Burns SP, Clark AJL. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circulation Research. 2007;100:520–6. doi: 10.1161/01.RES.0000258855.60637.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whitesall SE, Hoff JB, Vollmer AP, D’Alecy LG. Comparison of simultaneous measurement of mouse systolic arterial blood pressure by radiotelemetry and tail-cuff methods. Am J Physiol Heart Circ Physiol. 2004;286:H2408–15. doi: 10.1152/ajpheart.01089.2003. [DOI] [PubMed] [Google Scholar]
- 22.Starr A, et al. A reactive oxygen species-mediated component in neurogenic vasodilatation. Cardiovasc Res. 2008;78:139–47. doi: 10.1093/cvr/cvn012. [DOI] [PubMed] [Google Scholar]
- 23.Shue EH, Carson-Walter EB, Liu Y, Winans BN, Ali ZS, Chen J, et al. Plasmalemmal Vesicle Associated Protein-1 (PV-1) is a marker of blood–brain barrier disruption in rodent models. BMC Neurosci. 2008;9:29. [DOI] [PMC free article] [PubMed]
- 24.Huang BS, White RA, Ahmad M, Jeng AY, Leenen FH. Central infusion of aldosterone synthase inhibitor prevents sympathetic hyperactivity and hypertension by central Na + in Wistar rats. Am J Physiol Regul Integr Comp Physiol. 2008;295:R166–72. doi: 10.1152/ajpregu.90352.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johns C, Gavras I, Handy DE, Salomao A, Gavras H. Models of experimental hypertension in mice. Hypertension. 1996;28:1064–9. doi: 10.1161/01.HYP.28.6.1064. [DOI] [PubMed] [Google Scholar]
- 26.Ramakers C, Ruijter JM, Deprez RHL, Moorman AFM. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci Lett. 2003;339:62–6. doi: 10.1016/S0304-3940(02)01423-4. [DOI] [PubMed] [Google Scholar]
- 27.Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA Targeting Specificity in Mammals: Determinants beyond Seed Pairing. Molecular cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Abeelen AF, et al. The fetal origins of hypertension: a systematic review and meta-analysis of the evidence from animal experiments of maternal undernutrition. J Hypertens. 2012;30:2255–67. doi: 10.1097/HJH.0b013e3283588e0f. [DOI] [PubMed] [Google Scholar]
- 29.Curhan GC, et al. Birth weight and adult hypertension and obesity in women. Circulation. 1996;94:1310–5. doi: 10.1161/01.CIR.94.6.1310. [DOI] [PubMed] [Google Scholar]
- 30.Tsukuda K, et al. Influence of angiotensin II type 1 receptor-associated protein on prenatal development and adult hypertension after maternal dietary protein restriction during pregnancy. J Am Soc Hypertens. 2012;6:324–30. doi: 10.1016/j.jash.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 31.Komatsu T, et al. Molecular cloning, mRNA expression and chromosomal localization of mouse angiotensin-converting enzyme-related carboxypeptidase (mACE2) DNA Seq. 2002;13:217–20. doi: 10.1080/1042517021000021608. [DOI] [PubMed] [Google Scholar]
- 32.Crackower MA, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–8. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 33.Ingelfinger JR. Angiotensin-converting enzyme 2: implications for blood pressure and kidney disease. Curr Opin Nephrol Hypertens. 2009;18:79–84. doi: 10.1097/MNH.0b013e32831b70ad. [DOI] [PubMed] [Google Scholar]
- 34.Patel SK, et al. Association of ACE2 Genetic Variants With Blood Pressure, Left Ventricular Mass, and Cardiac Function in Caucasians With Type 2 Diabetes. Am J Hypertens. 2011;25:216–22. doi: 10.1038/ajh.2011.188. [DOI] [PubMed] [Google Scholar]
- 35.Dobyns WB, et al. Inheritance of most X-linked traits is not dominant or recessive, just X-linked. Am J Med Genet. 2004;129A:136–43. doi: 10.1002/ajmg.a.30123. [DOI] [PubMed] [Google Scholar]
- 36.Liu J, et al. Sex differences in renal angiotensin converting enzyme 2 (ACE2) activity are 17Œ ≤ −oestradiol-dependent and sex chromosome-independent. Biol sex dif. 2010;1:6. doi: 10.1186/2042-6410-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gupte M, et al. Angiotensin converting enzyme 2 contributes to sex differences in the development of obesity hypertension in C57BL/6 mice. Arterioscler Thromb Vasc Biol. 2012;32:1392–9. doi: 10.1161/ATVBAHA.112.248559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goyal R, Lister R, Leitzke A, Goyal D, Gheorghe CP, Longo LD. Antenatal maternal hypoxic stress: adaptations of the placental renin-angiotensin system in the mouse. Placenta. 2011;32:134–9. doi: 10.1016/j.placenta.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang G, et al. Intrarenal expression of miRNAs in patients with hypertensive nephrosclerosis. Am J Hypertens. 2009;23:78–84. doi: 10.1038/ajh.2009.208. [DOI] [PubMed] [Google Scholar]