

Abstract
The evolutionarily conserved PIKfyve, which synthesizes PtdIns5P from PtdIns, and PtdIns(3,5)P2 from PtdIns3P, requires PtdIns3P as both an enzyme substrate and a membrane recruitment signal. Whereas the PtdIns3P source is undetermined, class III PI3K (Vps34), the only evolutionarily conserved of the eight mammalian PI3Ks, is presumed as a main candidate. A hallmark of PIKfyve deficiency is formation of multiple translucent cytoplasmic vacuoles seen by light microscopy in cells cultured in complete media. Such an aberrant phenotype is often observed in cells from conditional Vps34 knockout (KO) mice. To clarify the mechanism of Vps34KO-triggered vacuolation and the PtdIns3P source for PIKfyve functionality, here we have characterized a podocyte cell type derived from Vps34fl/fl mice, which, upon Cre-mediated gene KO, robustly formed cytoplasmic vacuoles resembling those in PikfyveKO MEFs. Vps34wt, expressed in Vps34KO podocytes restored the normal morphology, but only if the endogenous PIKfyve activity was intact. Conversely, expressed PIKfyvewt rescued completely the vacuolation only in PikfyveKO MEFs but not in Vps34KO podocytes. Analyses of phosphoinositide profiles by HPLC and localization patterns by a PtdIns3P biosensor revealed that Vps34 is the main supplier of localized PtdIns3P not only for PIKfyve activity but also for membrane recruitment. Concordantly, Vps34KO podocytes had severely reduced steady-state levels of both PtdIns(3,5)P2 and PtdIns5P, along with PtdIns3P. We further present evidence for a plausible physiologically-relevant Vps34-independent PtdIns3P supply for PIKfyve, operating through activated class I PI3Ks. Our data provide the first evidence that the vacuolation phenotype in Vps34KO podocytes is due to PIKfyve dysfunction and that Vps34 is a main PtdIns3P source for constitutive PIKfyve functionality.
Keywords: PIKfyve, class III PI 3-kinase, phosphatidylinositol phosphates, endomembrane homeostasis, Vps34 knockout podocytes, class I PI 3-kinase
1. Introduction
PIKfyve is an evolutionarily conserved kinase that is a product of a single gene in metazoans. Mammalian PIKfyve, synthesizing phosphatidylinositol (PtdIns)1 5P from PtdIns, and PtdIns(3,5)P2 from PtdIns3P, represents a major node in PI signaling and membrane trafficking downstream of PtdIns3P (1). PIKfyve controls both constitutive and regulated endocytic trafficking as evidenced by the presence of its lipid products in quiescent cells and their elevation upon activation of endosome dynamics by cues such as insulin, EGF or viral infection (2–8). A hallmark of PIKfyve cellular inactivation, achieved by means of PIKfyve dominant-negative kinase-deficient mutants (9,10), pharmacological inhibition (5,11,12), siRNA-mediated silencing (13) or Cre-mediated gene disruption (14,15), is progressive dilation of endosomal membranes, culminating in the appearance of multiple translucent cytoplasmic vacuoles readily seen by light microscopy in cells grown in complete media. Because the vacuoles are selectively rescued by cytoplasmic microinjections of PtdIns(3,5)P2 but not PtdIns5P (10), the aberrant phenotype has been widely used as a sensitive intracellular indicator for selective PIKfyve-dependent PtdIns(3,5)P2 deficiency [reviewed in (1,2)]. These and other morphological and biochemical data have suggested a rate-limiting function of the PIKfyve-catalyzed PtdIns3P to PtdIns(3,5)P2 conversion in the course of early endosome maturation, controlling cargo exit from early endosomes and progression of endocytic trafficking (1,2).
The PtdIns3P-producing enzyme(s) supplying the PtdIns3P substrate for the PIKfyve lipid kinase activity is (are) not yet delineated. Likewise, whereas it is well established that PtdIns3P is a major targeting signal for PIKfyve recruitment to Rab5GTP-organized endosome platforms (16,17), whether it is generated by the same enzyme supplying the substrate awaits clarification. Based on the fact that, like PIKfyve, human Vps34, a class III phosphatidylinositol 3-kinase (gene symbol PIK3C3) is evolutionary conserved, with the orthologous Vps34 being the only PtdIns3P-synthesizing enzyme in S. cerevisiae (18), its role as a main PtdIns3P source for both PIKfyve activity and localization has been presumed but never studied. Indeed, the vesicular localization pattern of PIKfyve is rendered diffuse in several cell types treated with low concentrations of wortmannin, a powerful catalytic inhibitor of Vps34 (13,16,17). However, in metazoans, PtdIns3P is also produced by Vps34-independent mechanisms operating through class I and class II PI3Ks. For example, PtdIns(3,4,5)P3, made at the internal leaflet of the plasma membrane by activated wortmannin-sensitive class I PI3Ks (19,20), might serve as a source of endosomal PtdIns3P subsequent to two consecutive dephosphorylation reactions by the PI 5- and 4-phosphatases (21,22). Likewise, class II PI3Ks produce PtdIns3P directly from PtdIns and, indirectly, by 3-phosphorylation of PtdIns4P followed by 4-dephosphorylation of PtdIns(3,4)P2 (23–25), with class II PI3Kβ, also being wortmannin-sensitive (26).
Strikingly, Vps34 perturbation by siRNA silencing or delivery of inhibitory antibodies causes cytoplasmic vacuolation resembling that seen upon PIKfyve dysfunction (27–29). Similarly, primary cells derived from recently developed mouse models with conditional Vps34 gene disruption also demonstrate the aberrant phenotype of translucent cytoplasmic vacuoles, which often occurs in the absence of autophagic abnormalities (30–32). Hence, an outstanding question awaiting clarification is whether the aberrant endomembrane phenotype in cells with ablated Vps34-catalyzed PtdIns3P pool is due to arrested downstream signaling by PIKfyve.
Here we used podocytes with Vps34 Cre recombinase-mediated gene disruption in combination with a rescue approach to unravel the functional relationship between PIKfyve and Vps34 lipid kinases and its role in endomembrane homeostasis. MEFs with Pikfyve Cre recombinase-mediated gene disruption were used as a reference. We identified that PtdIns3P, generated by Vps34, serves as both a major precursor for PtdIns(3,5)P2 synthesis and a membrane localization target of PIKfyve in resting cells. We clarified that disrupted endomembrane homeostasis in Vps34 KO podocytes is due to an ablated Vps34-controlled PtdIns3P pool and consequent abrogation of PIKfyve-catalyzed PtdIns(3,5)P2 and PtdIns5P production. We further revealed that activated class I PI3Ks could contribute to some extent to the regulation of endomembrane homeostasis.
2. Materials and methods
2.1. Cell lines
Pikfyve and Vps34 (fl/fl or wt/wt) MEFs were isolated from mouse E12.5 embryos derived from the transgenic models (14,33) following previously described procedures (14). Glomerular cells were isolated from kidneys of Vps34fl/fl mice (33). Briefly, removed kidneys were chopped on ice into fine pieces and then passed through sieves in a laminar flow hood. Small pieces were first passed through a 70 μm sieve (BD Falcon) and collected in a 50 ml tube. A sterile syringe plunger was used to push the small pieces through the sieve. Following thorough rinses with sterile PBS, the material was collected and then passed through a second 50 μm sieve, followed by rinses with PBS. The material on top of the 50 μm sieve was transferred to a 50 ml tube and then spun down to form a pellet. Cells, resuspended in RPMI supplemented with insulin, transferrin and selenium (each at 1%), and containing 50 U/ml Penicillin/50 μg/ml Streptomycin, were seeded on 10 cm tissue-culture plates. Plates were incubated at 37°C for 10–15 days with media change every 3–4 days. Cellular outgrowths were visible at day 6–8 and predominantly consisted of podocytes, with only a minor presence of mesangial and endothelial cells (34,35). Hence, the glomerular cells are hereafter referred to as podocytes. Cells were then trypsinized and seeded on fresh culture plates for 24–48 hours. Cells were then immortalized using SV40 T antigen, packaged in a Retro Viral vector (MMLV-SV40-VSVG), and serum-free DMEM/RPMI media containing polybrene (8μg/ml) as previously described (34). Primary cells or transiently transfected cells stopped dividing after a few rounds of sub-culturing and therefore further selection was not required. Podocytes used in this study were derived from two Vps34fl/fl mice (#3176 and #3061) and showed similar results. Control podocytes were from the Vps34wt/wt mouse (#3175). All experiments were performed with cells between 3 and 5 passages. Unless indicated, all cell types were maintained in complete DMEM, containing 10% FBS, 50 U/ml penicillin and 50 μg/ml streptomycin sulfate.
2.2. Adenoviral transduction and transient transfection
Cells were treated with recombinant adenovirus expressing Cre recombinase (Ad-Cre) or empty Ad (titer 1.3×1011; Vector Core, University of Michigan, Ann Arbor), dissolved in complete media at MOI=200 and a minimal volume covering the cells. Four h later, complete media was added to a volume optimal for the dish size. Media was replaced 14 h later with fresh complete media. Pikfyve KO MEFs (100 mm confluent dishes) were transfected on day 4 post Ad-Cre treatment by electroporation using 50 μg of pEGFP-2xFYVEPIKfyve (14) or pTag-YFP-Vps34wt cDNAs under a single pulse of 975 μF and 0.16 kV (Biorad-Gene pulser II) as described previously (36). Vps34 KO podocytes were transfected with the above cDNAs by Lipofectamine 3000 transfection kit (Invitrogen) following manufacturer protocols. Where indicated, 24 h post transfection, Vps34 KO podocytes were treated (60 min; 37°C) with 1 μM of the YM201636 inhibitor (Symansis NZ) dissolved in DMSO or only with vehicle.
Recombinant adenoviruses expressing HA-PIKfyvewt and GFP from separate CMV promoters (AdGFP-PIKfyvewt) or only GFP (AdGFP) were used at MOI=10, with MOI=1, being the lowest dilution reaching 100% infection efficiency in HEK293 cells for 14 h (9). The chosen MOI delivered infection in ~20–25% of the total Pikfyve KO or Vps34 KO cells, the non-infected cells on the same dish thereby serving as controls.
2.3. Light, fluorescence and electron microscopy analyses
Cells, live or fixed in 4% formaldehyde for 30 min were viewed at 200x or 400x magnification with a Nikon Eclipse TE200 inverted fluorescence microscope as specified in the figure legends. Images were captured with a SPOT RT Slider charge-coupled device camera (Diagnostic Instruments). Cells expressing eGFP-2xFYVEPIKfyve or YFP-Vps34wt were visualized by phase-contrast and fluorescence microscopy using a standard green channel (14,17). Cells exhibiting at least 5 perinuclear translucent vacuoles were considered as vacuolated. Under each experimental design, 20–100 cells were counted in 3–4 separate experiments. For electron microscopy analyses, Ad or Ad-Cre-treated Vps34fl/fl podocytes grown in complete media (10 cm dish) were trypsinized and pelleted at 4000 g for 5 minutes. Cell pellets were washed with PBS and fixed in a Sorenson’s buffer (0.1M), containing 2% paraformaldehyde and 2.5% glutaraldehyde, again washed with PBS and fixed using 1% osmium tetroxide. Cell pellets were dehydrated in different strengths of ethanol followed by propylene oxide. After embedding in a resin, 70–100 nm thick sections were collected over copper grids and stained with 2% aqueous uranyl acetate and lead citrate solutions. For imaging, a Philips CM-100 transmission electron microscope equipped with compustage and high resolution (2K × 2K) digital camera was used at the University of Michigan core imaging facility.
2.4. Myo-[2-3H] inositol cell labeling, lipid extraction and HPLC analyses
Vps34fl/fl podocytes (35 mm dishes) were treated with Ad-Cre or empty Ad viruses as specified above. On day 4, cells were washed two times with inositol/glucose-free DMEM, containing 5% dialyzed FBS, 0.5% BSA, 5 μg/ml insulin and 5 μg/ml transferrin, 2 mM pyruvate, 25 mM HEPES, pH 7.4, and 100 units/ml penicillin and 100 μg/ml streptomycin for 24 h. Cells were then labeled with 25 μCi/ml myo-[2-3H]inositol (Perkin Elmer) in a fresh media, as detailed above, for 26 h. Cellular lipids were extracted, deacylated, and analyzed by HPLC (Waters 5215) on a 5-micron Partisphere SAX column (Whatman) as we described previously (5,7,14,37). [32P]GroPIns5P, [32P]GroPIns3P, [32P]GroPIns(4,5)P2 and [32P]GroPIns(3,5)P2 prepared by enzymatic synthesis with [γ-32P]-ATP were used as HPLC standards as described elsewhere (5,7,14,37). Fractions were collected every 0.25 min and analyzed for [3H] and [32P] radioactivity after addition of scintillation mixture. Data evaluation and documentation was performed by Microsoft EXCEL software. Individual peak radioactivity was quantified by area integration and presented as a percentage of the summed radioactivity from the [3H]-GroPIns3P, -4P, -5P, -(3,5)P2, and -(4,5)P2 peaks (“total radioactivity”).
2.5. Western blot analysis
Cells were scraped in RIPA+ buffer [composed of 50 mM Tris HCl, pH 8.0, 150 mM NaCl, 1% Nonidet P-40 and 0.5% Na deoxycholate), supplemented with 1 x protease inhibitors (14)]. Proteins (60–150 μg/lane) were separated by PAGE, transferred onto nitrocellulose membrane and probed with previously characterized polyclonal antibodies against Vps34 [a gift from Jon Backer (38)], Sac3 (37), PIKfyve [R7069 (39)] and anti-GDI-1 (40).
2.6. Immunoprecipitation and in vitro PI3K activity assay
Fresh lysates from Vps34 KO podocytes, treated with or without Ad-GFP for 16h, were collected in RIPA++ buffer containing 1x protease and 1x phosphatase inhibitor cocktails as described (41). Precleared lysates (20,000 × g, 15 min, 4°C) were subjected to immunoprecipitation with rabbit polyclonal anti-p85 or mouse monoclonal anti-phosphotyrosine antibodies (4G10), both from EMD Millipore. The in vitro PI3K activity assay was conducted essentially as we described previously (42) and detailed in Supplementary Experimental Procedures.
2.7. Other assays and statistics
Protein concentration was determined by bicinchoninic acid protein assay. Protein and lipid levels on the blots and TLC plates, respectively, were quantified by laser scanning densitometry (Epson V700) and UN-SCAN-IT software (Silk Scientific). Several films of different exposure times were analyzed to assure that the signals were within the linear range of detection sensitivity. Data are presented as mean +/− SE. Statistical analysis of variance was performed by t test for independent samples and a one-tail t test for paired samples, with p<0.05 considered statistically significant.
3. Results
3.1. Identification of Vps34 KO cells forming robustly cytoplasmic vacuoles in complete culture media
Along with PIKfyve protein loss, the excision of the floxed alleles in MEFs isolated from Pikfyvefl/fl mice triggered progressively exacerbating cytoplasmic vacuolation 3–5 d post transfection with GFP-Cre recombinase, readily visible by light microscopy of cells cultured in complete (serum-containing) media, as we have previously reported (14). This result was extended herein for cells transduced with adenovirus expressing Cre recombinase, whereby only Pikfyvefl/fl but not the Pikfyvewt/wt MEFs showed a profound vacuolation phenotype, reaching ~90% on the 5th day post transduction (Fig. 1A and B). PIKfyve protein levels were undetectable under these conditions (Fig. 1D). By contrast, control Pikfyvefl/fl MEFs infected with empty adenovirus displayed only a very low % of vacuolating cells (<2%, Fig. 1A, panel b) until the end of the 7-day observation.
Fig. 1.



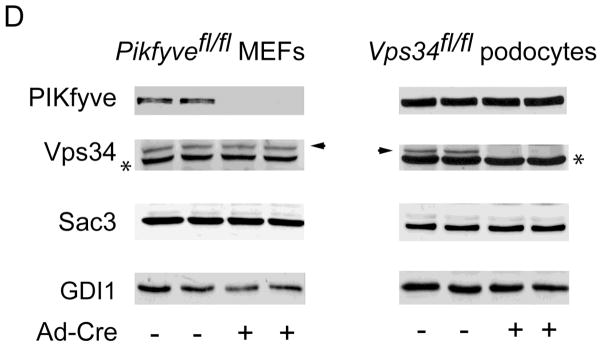
Gene disruption of Vps34 in podocytes and of Pikfyve in MEFs triggers cytoplasmic vacuolation at a similar pace. Pikfyvefl/fl or Pikfyvewt/wt mouse embryonic fibroblasts (MEFs), Vps34fl/fl or Vps34wt/wt podocytes and Vps34fl/fl MEFs cultured in complete media were treated with adenovirus, either empty (Ad) or expressing full-length Cre recombinase (Ad-Cre). A, representative phase-contrast images, taken 5 days post-treatment depicting similar multiple translucent cytoplasmic vacuoles in nearly all Ad-Cre-treated Pikfyvefl/fl and Vps34fl/fl cells (panels a and c). Bar, 10 μm. B, time-course quantification of cytoplasmic vacuolation subsequent to Ad-Cre treatment showing similar precipitous vacuolation progression in Pikfyvefl/fl MEFs or Vps34fl/fl podocytes, reaching ~90% of total cells at day 5–6, weak cytoplasmic vacuolation (~25% of total cells at day 7 post Ad-Cre, but increasing thereafter) in Ad-Cre-treated Vps34fl/fl MEFs and no vacuoles in Ad-Cre-treated Pikfyvewt/wt or Vps34wt/wt cells. Quantification of the vacuolating cells was obtained by counting >100 cells per condition per cell type in 3 separate experiments (mean ± SE). C, Vps34fl/fl podocytes maintained in complete media and treated with Ad or Ad-Cre (6 days) were observed by electron microscopy. Large-sized dilated empty vacuoles surrounded by a single membrane, containing only few intraluminal vesicles, are observed only in Vps34 KO podocytes. In contrast, in control Vps34fl/fl podocytes, single-membraned MVBs are visible (black arrowheads) containing multilamellar membrane whorls and intraluminal vesicles. Micrographs are representative of 30–60 images out of 2 independent experiments with similar results. m, mitochondria (white arrows), n, nucleus. Bar, 0.5 μm; D, Western blot analysis of lysates derived from indicated cells on day 5 post infection using antibodies against the indicated proteins confirms complete and selective ablation of only the targeted gene products by Ad-Cre. Sac3 phosphatase levels are unaltered by Pikfyve KO or Vps34 KO. Shown are representative blots out of three to six independent experiments with similar results. GDI1, loading control; *, unspecific bands recognized by the anti-Vps34 antibodies, also indicating equal loading; arrowhead, the Vps34 protein band.
To begin elucidating the functional relationship between the enzymatic products of Vps34 and PIKfyve as well as their role in endomembrane homeostasis, we aimed at identifying a Vps34fl/fl cell type that, upon Ad-Cre treatment would exhibit a robust time-course of cell vacuolation as seen in Pikfyve fl/fl - Ad-Cre-treated MEFs. Surprisingly, examination of primary Vps34fl/fl MEFs showed only 10 and 25% vacuolating cells on day 5 and 7 post transduction, respectively (Fig. 1B). Consistently, an independent study (43) in MEFs, derived from the same Vps34fl/fl mouse model used herein (33), also revealed aberrant vacuolation only upon prolonged treatment with Cre recombinase. Conversely, Ad-Cre transduction of immortalized Vps34fl/fl podocytes resulted in formation of translucent cytoplasmic vacuoles at a pace similar to that observed in Ad-Cre-treated Pikfyvefl/fl MEFs (Fig. 1A and B). Indeed, as high as 90% of the Vps34fl/fl podocytes cultured in complete media exhibited cytoplasmic vacuoles at day 5 post-Ad-Cre treatment, which were readily seen by light microscopy (Fig. 1A, panel c and B). Our observation for robust cytoplasmic vacuolation in podocytes with Vps34 ablation confirms and extends previous findings in primary podocytes isolated from podocyte-specific KO mice (31), generated from the same conditional Vps34fl/fl mouse model used herein (33). Of note, control Vps34fl/fl or Vps34wt/wt podocytes transduced with empty Ad or Ad-Cre, respectively, demonstrated a negligible number of vacuolating cells (<2%; Fig. 1A, panel d, and Fig. 1B).
In addition to a similar pace of vacuole appearance in the two cell types, electron microscopy revealed that, similarly to HEK293 cells with impaired PIKfyve activity (44), the cytoplasmic vacuoles formed in Vps34fl/fl podocytes at 5–6 day post-Cre-infection were surrounded by a single-membrane and largely empty inside (Fig. 1C). This indicates that the swollen vacuoles in podocytes with Vps34 ablation, as in the case with PIKfyve (44), are distinct from autophagosome structures induced by serum starvation. Moreover, even under conditions of 24 h serum starvation, the morphology in Vps34fl/fl podocytes remained normal by light microscopy (Fig. S1). Finally, the exclusion of the Oil red O lipid marker from vacuoles in both Pikfyvefl/fl MEFs and Vps34fl/fl podocytes at 5–6 days post-Ad-Cre treatment indicated that the translucent vacuoles were not lipid droplets (Fig. S2).
Ad-Cre transduction led to drastic and selective decreases of Vps34 protein levels in Vps34fl/fl podocytes 5–6 day post-infection, thus confirming the expected complete gene disruption (Fig. 1D). Importantly, after day 7 post Ad-Cre treatment, vacuolated Vps34fl/fl podocytes showed progressively reduced viability, with most of them demising by day 10 likely through apoptosis, as determined in 9-day primary podocytes in culture isolated from the podocyte-specific Vps34 KO mouse model (31).
Considering these similarities and limitations, detailed above, we focused our study on Vps34fl/fl podocytes and Pikfyvefl/fl MEFs at days 5–6 after Ad-Cre treatment (referred to further as Vps34 KO and Pikfyve KO). Cross examination of protein levels under these conditions revealed that PIKfyve was not significantly affected in Vps34 KO podocytes, nor was Sac3 (Fig. 1D), the 5-phosphatase that associates with PIKfyve and is potentially able to modulate the PtdIns3P pool (1). Similarly, Vps34 protein levels were unaltered in Pikfyve KO MEFs (Fig. 1D). Likewise, in Pikfyve KO MEFs, the Sac3 phosphatase was at the control protein levels (Fig. 1D), in agreement with our previous studies for unaltered Sac3 protein levels under PIKfyve perturbations (14,37).
3.2. The cytoplasmic vacuolation in Vps34 KO podocytes is due to PIKfyve dysfunction
Notably, the morphological defect caused by Vps34 KO in podocytes was phenocopied by inhibition of the PIKfyve activity in this cell type, which was achieved by treatment with the cell permeable PIKfyve inhibitor YM2013616 (5,12) (Fig. S3). This raised the question as to whether PtdIns3P ablation in the Vps34 KO podocytes disrupted endomembrane homeostasis indirectly, i.e., by depleting the substrate for PIKfyve activity and, hence, PtdIns(3,5)P2 synthesis. We reasoned that if this were the case, then heterologous expression of Vps34 should prevent the endomembrane defect in Vps34 KO podocytes, but only if the latter harbor an intact PIKfyve activity. Likewise, increased PIKfyve protein should restore the aberrant phenotype back to normal only in Pikfyve KO MEFs but not in Vps34 KO podocytes. As illustrated in Fig. 2A, panels a and b, morphological inspection of Vps34 KO podocytes positive for YFP-Vps34wt fluorescence signals from three independent experiments revealed the absence of vacuoles in practically all transfected cells (Fig. 2B), consistent with the restored PtdIns3P pool and the downstream PIKfyve function. However, if cells were acutely treated with YM201636 expressed YFP-Vps34wt failed to rescue the vacuolation (Fig. 2A, panels c and d), which was observed in ~98% of transfected cells (Fig. 2B). Likewise, similar expression of YFP-Vps34wt in Pikfyve KO MEFs was ineffective as evidenced by sustained aberrant vacuolation in all transfected cells 24–48 h post-transfection (Fig. 2A, panels e and f). We next increased levels of PIKfyve in Vps34 KO and Pikfyve KO cells by adenovirus-mediated gene delivery of PIKfyvewt. This approach was fully effective only in Pikfyve KO cells (see below). These data, together with other published observations (see discussion), suggest that under basal conditions, the cytoplasmic vacuolation in the Vps34 KO podocytes is due to PIKfyve-dependent loss of PtdIns(3,5)P2 synthesis and that the Vps34-catalyzed PtdIns3P pool is the main source for sustaining PIKfyve function in the maintenance of endomembrane homeostasis in podocytes.
Fig. 2.

PIKfyve dysfunction in Vps34 KO podocytes underlies the aberrant vacuolation. On day 4 post-Ad-Cre-treatment, Vps34 KO podocytes (panels a–d) and Pikfyve KO MEFs (panels e and f) were transfected with pTag-YFP-Vps34wt cDNA. Twenty-four h post-transfection, cells were treated with vehicle (panels a, b, e and f) or 1 μm YM201636 for 1 hour (panels c, d and not shown). A, Live cells were observed by fluorescence microscopy and phase-contrast. Shown are representative images of transfected cells (panels a, c and e) and the overlay with the phase-contrast images (panels b, d and f). Rescue of the vacuolation phenotype occurs in Vps34 KO podocytes upon YFP-Vps34wt expression (panels a and b) in practically all transfected cells (see B). If endogenous PIKfyve activity is inhibited by YM201636 such rescue is not observed (panels c and d). As expected, YFP-Vps34wt expression in Pikfyve KO MEFs did not prevent the cytoplasmic vacuolation (panels e and f). Bar, 10 μm. B, Quantification of vacuolated Vps34wt-expressing cells in the presence or absence of the inhibitor was obtained by inspecting >20 transfected cells per condition per experiment in three separate experiments and presented as percentage of the total number of transfected cells. *, different vs. (−) inhibitor, P<0.001
3.3. Vps34-produced PtdIns3P is a major membrane recruitment signal for PIKfyve
In addition to serving as an enzyme substrate, endosomal PtdIns3P represents a major determinant for PIKfyve membrane localization that is mediated through the PI3P-binding FYVE domain in PIKfyve (16). Membrane recruitment is critical for PIKfyve functionality as evidenced by the inability of the kinase-deficient dominant-negative point mutant PIKfyveK1831 harboring a disrupted FYVE domain to trigger cytoplasmic vacuolation (9). To determine whether Vps34 catalytic activity is also supplying the PIKfyve localization signal, we used the 2xFYVEPIKfyve biosensor that avidly localized to PtdIns3P/EEA1-enriched vesicular structures in Pikfyvewt/wt MEFs (14). In agreement with our previous data, transient transfection of eGFP-2xFYVEPIKfyve in Pikfyve KO MEFs or Vps34fl/fl podocytes showed a similar vesicular localization (Fig. 3, panels c and d, and not shown). By contrast, the eGFP-2xFYVEPIKfyve fluorescent signals in Vps34 KO podocytes were mostly diffuse (Fig. 3, panels a and b), although vesicle association could still be observed in some vacuolated cells (~25% of the cells). These data corroborate the notion that PtdIns3P produced by Vps34 is also a major membrane recruitment signal for PIKfyve. The sparse eGFP-2xFYVEPIKfyve-positive vesicles seen in some cells support the idea for additional sources of localized PtdIns3P production, as also has been recently concluded in Vps34 KO MEFs by usage of an enhanced FYVE domain probe (43).
Fig. 3.

2xFYVE vesicular pattern is rendered diffuse in Vps34 KO podocytes. On day 4 post-Ad-Cre-treatment, Vps34 KO podocytes and Pikfyve KO MEFs were transfected with pEGFP-2xFYVEPIKfyve cDNA for visualizing PtdIns3P-enriched endosomal structures. Twenty-four h post-transfection cells were fixed for fluorescence microscopy (panels a and c) and phase-contrast inspection (panels b and d). White arrows denote transfected cells. The 2xFYVEPIKfyve fluorescence signals in the transfected cells (and hence, the PtdIns3P-stained vesicles) were diffuse in Vps34 KO podocytes (panels a and b), which was observed in 77 ± 4% of transfected Vps34 KO podocytes. The appearance of the 2xFYVEPIKfyve in Pikfyve KO MEFs was vesicular (panels c and d), seen in 93 ± 3% of transfected cells. Quantitation was obtained by counting >20 transfected cells per condition per experiment in 3 separate experiments. Bar, 10 μm.
3.4. Commensurate decreases in steady-state levels of PtdIns3P, PtdIns(3,5)P2 and PtdIns5P in Vps34 KO podocytes
The data presented above indicate that PIKfyve is a major downstream effector of Vps34-catalyzed localized PtdIns3P production in the maintenance of endomembrane homeostasis. However, a Vps34-independent PtdIns3P pool, generated directly and indirectly by class I and class II PI3Ks could be reportedly quite substantial (up to 65% of total PtdIns3P) (6,22,23,43,45). To reveal the extent of Vps34 contribution to the PtdIns3P pool in Vps34 podocytes and the proportion of Vps34-catalyzed PtdIns3P used by the PIKfyve catalytic activity, we measured steady-state levels of PIs by HPLC inositol head-group analysis subsequent to cell labeling with myo-[2-3H]inositol. As illustrated in Fig. 4, selective and significant changes were observed only in steady-state levels of PtdIns3P, PtdIns5P and PtdIns(3,5)P2, consistent with the key role of Vps34 in PIKfyve functionality. Steady-state levels of PtdIns(4,5)P2 and PtdIns4P remained unaltered (Fig. 4). In agreement with the HPLC profiles of PIs in other quiescent cells (5,14), PtdIns(3,4)P2 and PtdIns(3,4,5)P3 were not detected. Data from three separate labeling experiments in Vps34 KO vs. Vps34fl/fl control podocytes revealed that steady-state levels of PtdIns3P were reduced to ~45% of the control levels, with comparable diminution in PtdIns(3,5)P2 (~40%) and PtdIns5P (~41%) (Fig. 4). Of note, during the labeling procedure, a relative increase (up to 20–25%) of non-vacuolated cells (presumably, expressing some Vps34) in parallel with preferential death of vacuolated Vps34 KO podocytes has occurred. Therefore, if we normalize for the total number of vacuolating cells seen at the end of cell labeling, the values of the residual amounts of these lipids are overestimated by ~20–25%.
Fig. 4.

Reduced PIKfyve lipid products in Vps34 KO podocytes. Vps34fl/fl podocytes, seeded on 35 mm dishes, were treated with Ad-Cre or Ad in complete culture media, as described under “Materials and Methods”. On day 4 post-treatment media was replaced with inositol/glucose-free media for 24 h, followed by myo-[2-3H]inositol labeling for 26 h. Lipids were extracted, deacylated and fractionated by HPLC. Shown is quantitation from three independent experiments, calculated as a percentage of the total PI radioactivity and presented as mean ± SE. The total PI radioactivity was obtained by summing the counts within the elution times corresponding to the indicated [3H]GroPIns peaks. *, PtdIns(3,5)P2, PtdIns5P and PtdIns3P in Vps34 KO vs. those in Vps34fl/fl podocytes, P<0.05.
Consistent with the failure of PIKfyve membrane recruitment under localized depletion of PtdIns3P, a step critical for formation of productive complex of PIKfyve with its protein partners and, hence, activation of PIKfyve lipid kinase (1,46), we observed similar reduction in both PIKfyve lipid products PtdIns5P and PtdIns(3,5)P2 (Fig. 4). Taken together, the data for residual PtdIns3P levels even after normalization and roughly commensurate residual levels of PtdIns(3,5)P2 and PtdIns5P by Vps34 KO suggest that: 1) in podocytes, a small proportion of basal PtdIns3P could be synthesized through Vps34-independent pathway(s) and, 2) this alternatively synthesized Ptdins3P pool could, at least in part, serve as a catalytic precursor and localization signal for PIKfyve.
3.5. Sustained activation of class I PI3Ks delays the vacuolation phenotype in Vps34 KO
Our findings that the vacuolation phenotype in Vps34 KO podocytes is mainly due to PIKfyve dysfunction provided a paradigm to address the question as to whether PIKfyve could utilize PtdIns3P produced by class I PI3Ks, found previously to indirectly feed into the endosomal PtdIns3P pool (47). We took advantage of the fact that activation of class I PI3Ks is a common cellular survival response to diverse viral infections (48), with the non-replicating E1/E3-deleted adenoviral vector type 5 causing sustained activation, beginning 10 min subsequent to virus binding to its receptors and persisting for over 24 hours (49–52). We reasoned that if class I PI3Ks were to provide PtdIns3P for PIKfyve function, then adenovirus infection of Vps34 KO should mitigate the susceptibility of cytoplasmic vacuole formation. To this end, we used E1/E3-deleted adenoviral vector type 5 harboring GFP (AdGFP) to facilitate detection of infected cells by fluorescent microscopy (Fig. 5). Remarkably, AdGFP infection of Vps34 KO cells on day 4 post Ad-Cre treatment, a time point when the vacuolated cells were ~50% (see Fig. 1), led to a statistically significant time delay of vacuole formation at the 16-h, and less so, at the 36-h post-infection vs. non-infected neighboring Vps34 KO cells on the same dish (Fig. 5A and C). Intriguingly, if wortmannin was present during the 16 h AdGFP infection such a delay was not apparent (Fig. S4). Direct measurement of class I PI3K activity in Vps34 KO podocytes subsequent to the 16 h AdGFP infection revealed on average 15 and 50% elevation in PtdIns3P product in p85 and phospho-tyrosine immunoprecipitates, respectively, in comparison to non-treated Vps34 KO podocytes (Fig. S5), consistent with activated class I PI3K underlying the vacuolation delay.
Fig. 5.



Transient activation of class I PI3Ks selectively delays the cytoplasmic vacuolation only in Vps34 KO podocytes. A and B, on day 4 post-Ad-Cre treatment Vps34 KO podocytes and Pikfyve KO MEFs were infected with either AdGFP (A) or AdGFP-PIKfyvewt (B) at MOI=10, as described under “Materials and methods”. 16–36 hours post-infection, cells were observed live by fluorescence and phase-contrast microscopy monitoring the GFP signals and the vacuolation phenotype in both infected [GFP(+)] and noninfected [GFP(−)] cells. A, fluorescence and phase-contrast images of representative cells at 16 h post-infection showing AdGFP -infected Vps34 KO podocytes (panels a and b) that did not exhibit cytoplasmic vacuoles in contrast to the GFP(−) neighboring cells and AdGFP-infected Pikfyve KO MEFs, in which the vacuolation phenotype was unaltered (panels c and d). See C for quantification. Bar, 10 μm. B, fluorescence and phase-contrast images of representative cells at 16 h post-infection with AdGFP-PIKfyvewt showing the absence of vacuolation in AdGFP-PIKfyvewt-expressing Vps34 KO podocytes (panels a and b) and Pikfyve KO MEFs (panels c and d). In both cell lines the aberrant vacuoles persisted in the GFP(−) neighboring cells. See D for quantification. Bar, 10 μm. C and D, Quantification of vacuolation in the non-infected GFP(−) control cells (white bars) and GFP(+)-infected condition (grey bars) as percentage of total GFP(−) control cells and total GFP(+) cells, respectively by counting >100 cells per condition in three separate experiments. *, different vs. GFP(−) cells at the same time point; #, different vs. the 16h time point, P<0.001.
Contrary to Vps34 KO, similar AdGFP infection of Pikfyve KO cells failed to delay the vacuolation phenotype (Fig. 5A and C), consistent with lack of PtdIns(3,5)P2 production in PIKfyve’s absence. Conversely, introduction of AdGFP that simultaneously expresses PIKfyvewt from an independent promoter (AdGFP-PIKfyvewt) on day 4 of the KO resulted in nearly complete and sustained rescue of the vacuolation phenotype in Pikfyve KO MEFs (Fig. 5B and D), in agreement with studies utilizing other approaches for PIKfyve perturbation/vacuolation rescue (9,11). However, AdGFP-PIKfyvewt infection of Vps34 KO podocytes produced only a transient delay of cytoplasmic vacuolation, quantitatively similar to that observed by the AdGFP infection (Fig. 5C and D), further reinforcing the conclusion from Fig. 2 that PtdIns3P substrate deprivation and reduced PtdIns(3,5)P2 synthesis by PIKfyve is causing the endomembrane defect. Combined data support the notion that under induced conditions, class I PI3Ks could deliver to some extent the PtdIns3P precursor for PIKfyve function.
4. Discussion
The observation that perturbations of the PIKfyve kinase trigger aberrant cell morphology in the form of translucent cytoplasmic vacuoles, readily reversed by exogenous PtdIns(3,5)P2 delivery, together with unavailability of sufficiently specific probes for PtdIns(3,5)P2 cellular detection, has promoted the cytoplasmic vacuolation phenotype as a sensitive functional measure of localized PtdIns(3,5)P2 reduction (1,2). Intriguingly, similar translucent vacuoles accompanied by endocytic trafficking defects and severe in vivo phenotypes have been recently reported in cells derived from mouse models with conditional Vps34 gene disruption (30–32). Importantly, in podocytes-specific Vps34 KO mice, the aberrant vacuolation phenotype and disrupted endocytic trafficking precede the early-age severe proteinuria and glomerulosclerosis that lead to premature death (31,53). As deficient autophagy is reportedly not the primary cause of the severe defects in the podocytes or in other cells and tissues with Vps34-deficiency (30–32), elucidating the molecular mechanism(s) causing the faulty performance of the endosomal system is of critical importance. The current study was undertaken to determine, first, if the Vps34 KO-triggered endomembrane defect in podocytes is secondary to PIKfyve dysfunction and hence, loss of PtdIns(3,5)P2 production and, second, the degree to which Vps34 activity is responsible for providing PtdIns3P as a catalytic substrate and membrane localization signal for the constitutive PIKfyve function in this cell type. The key finding we presented here is that in podocytes, Vps34 is the main supplier of PtdIns3P that serves as both a substrate for PIKfyve lipid kinase activity and an endosome targeting signal for PIKfyve membrane recruitment. We further revealed that the cytoplasmic vacuolation seen upon Vps34 KO in podocytes results from loss of PIKfyve downstream function due to substrate deprivation and enzyme mislocalization. We also demonstrated that induction of class I PI3Ks in the background of Vps34 KO in podocytes can transiently restore the PIKfyve function. Our results provide the first experimental evidence that the Vps34 catalytic activity is the main PtdIns3P supplier for PIKfyve functionality, at least in podocytes, and that the aberrant cytoplasmic vacuolation seen by Vps34 disruption is secondary to PIKfyve dysfunction.
Several lines of evidence in the current study support the conclusion that dysregulated endomembrane homeostasis in Vps34-deficient cells is due to deficiency of PtdIns(3,5)P2 synthesis by PIKfyve. Thus, unlike PIKfyve, increased protein levels of Vps34 were able to restore the normal morphology in Vps34 KO podocytes, but only if the endogenous PIKfyve activity was intact (Fig. 2). Conversely, increased protein levels of PIKfyve but not of Vps34 in Pikfyve KO MEFs produced a complete and sustained restoration of the normal endomembrane morphology (Fig. 5). Concordantly, PtdIns(3,5)P2 levels were profoundly reduced in Vps34 KO podocytes (Fig. 4). Additional support that reduced PtdIns(3,5)P2, rather than PtdIns3P synthesis, causes the cytoplasmic vacuolation is provided by findings for marked decreases of PtdIns3P under increased PIKfyve activity and elevated PtdIns(3,5)P2 synthesis, yet vacuole formation is not apparent as seen in HEK293 cells infected with Ad-PIKfyvewt (9). In the same vein, unlike with PtdIns(3,5)P2, delivery of exogenous PtdIns3P in COS cells, vacuolated due to ectopically expressed PIKfyve dominant-negative kinase-deficient point mutant K1831, did not rescue the phenotype (10).
Our observations for similar and substantial reduction in steady-state levels of PtdIns3P and PtdIns(3,5)P2 in Vps34 KO podocytes (Fig. 4) allow the conclusion that Vps34 provides a considerable subfraction of PtdIns3P as a substrate for PIKfyve enzymatic activity (~65–70% after normalization for total Vps34 KO cell number, see text to Fig. 4). The observed diffuse cytosolic pattern of the PtdIns3P reporter 2xFYVEPIKfyve (Fig. 3) suggests that Vps34-generated PtdIns3P also serves as a major targeting signal for PIKfyve membrane recruitment. The ramification of the lost PIKfyve endosome localization is a commensurate reduction of PtdIns5P production (Fig. 4), a finding consistent with direct PIKfyve-catalyzed synthesis of PtdIns5P (46,54). The concomitant PtdIns5P diminution suggests that the cytosolic PIKfyve fails to gain access to the PtdIns substrate on membranes and/or form a productive heterooligomeric complex (PAS complex) with the two partner proteins, i.e., the scaffolding regulator ArPIKfyve and the antagonistic PtdIns(3,5)P2 phosphatase Sac3 (1,37,55). Both membrane recruitment and the PAS complex formation are crucial steps for PIKfyve activity, as evidenced by the inability of the dominant-negative kinase-deficient point mutant PIKfyveK1831 to trigger the vacuolation phenotype if incapacitated for PtdIns3P binding or ArPIKfyve-Sac3 association through truncated mutations in the FYVE domain or ArPIKfyve-Sac3 interaction sites, respectively (9,56). Concordantly, gene disruption of ArPIKfyve or Sac3 impairs PtdIns5P synthesis, further reinforcing the prerequisite of intact PAS complex in PIKfyve-catalyzed PtdIns5P production [reviewed in (46,54)].
Previous studies in cells of kidney origin have indicated the key role of PIKfyve and Vps34 in the progression of endocytic trafficking, with fluid-phase endocytosis being particularly sensitive to PIKfyve or Vps34 dysfunction (31,44). In addition to similar endomembrane vacuolation and endocytic defects upon Vps34 or PIKfyve perturbations, cells with genetic Vps34 KO may exhibit defects in autophagy. Reportedly, the latter could precede or follow the endomembrane dysfunction, with differences likely related to the cell type, the strategy in the Vps34 gene targeting and/or the mouse genetic background. For example, MEFs isolated from two independent conditional Vps34 KO mouse models (30,33) exhibit different phenotypes. Indeed, whereas MEFs from the model used by Jaber et al form severe cytoplasmic vacuoles at early stage of excision with Cre recombinase (5–6 days) (30), those derived from the model of Zhou et al (33) vacuolate only after prolonged ablation of Vps34 [herein, Fig. 1 and (43), 14–15 days]. Intriguingly, in the latter case, the MEFs exhibit predominant autophagic defects (43). By contrast, Vps34 disruption causes predominantly endocytic dysfunction in sensory neurons, podocytes (Fig. 1) or lens cells (31–33). Consistent with this, our observations in Vps34 KO podocytes at the ultrastructural level failed to reveal double-membrane autophagic structures (Fig. 1C). Instead, large swollen empty vacuoles were the most pronounced defect in these cells. These data suggest that the autophagic and endocytic phenotypes seen upon Vps34 KO have separate molecular mechanisms and are consistent with the Vps34 endocytic functions being delivered predominantly by PIKfyve. Whereas our data indicate that the latter mechanism operates in podocytes, it is conceivable that it has a more general significance. Future studies should define plausible cell specificity in the requirement of Vps34-dependent PtdIns3P synthesis for PIKfyve function in maintaining endomembrane homeostasis.
Class II PI3Ks that regulate key processes in different cell types, including autophagy, hormonal and immune responses, cilia movements and glomerular filtration, also supply the constitutive PtdIns3P pool (6,23,43,45,57). The observations for residual PtdIns3P, as documented in Vps34 KO podocytes (Fig. 4) or MEFs (43), derived from the same Vps34fl/fl mouse model, together with the ubiquitous distribution of class II PI3K-C2α/β, are consistent with the latter feeding endosomal PtdIns3P for PIKfyve function. Although this has not been addressed in our study, evidence for such a pathway has been recently provided by findings for concurrent, though small, decreases in PtdIns3P and PtdIns(3,5)P2 upon PI3K-C2α knock-down in 3T3L1 adipocytes (6).
Whereas the degree of PtdIns3P contribution by class II PI3Ks is a matter of future investigation, in the framework of this study we have provided evidence for a Vps34-independent pathway, comprised of activated class I PI3Ks, supplying the substrate for PIKfyve in Vps34 KO podocytes. Intriguingly, a Rab5-regulated enzyme cascade, comprised of class I PI3Kβ and two phosphatases (PI 5- and PI 4-phosphatase), has been suggested to generate as much as 30% of the endosomal PtdIns3P pool (22). Under our experimental paradigm of temporary activation of class I PI3Ks, we observed a transient delay of the endomembrane defects in Vps34 KO podocytes in parallel with elevated class I PI3K activity (Fig. 5 and Fig. S5), corroborating the notion of a class I PI3K-related endosomal PtdIns3P pool being used for PtdIns(3,5)P2 production. That this pathway could be physiologically significant is inferred by the comparable phenotypes between mice with muscle-specific KO of Pikfyve (MPIfKO) and those with muscle-specific inactivation of Pik3r1 combined with global Pik3r2 (58,59) (note that both models are generated by the same muscle-creatine kinase (MCK) promoter-driven Cre recombinase and on C57Bl6 background). The two models share remarkably similar metabolic abnormalities, including systemic glucose intolerance, muscle insulin resistance and increased adiposity, suggesting that the metabolic defects in these mice might be related, in part, to ablation of a class I PI3K-controlled PtdIns3P pool that serves as a precursor for PIKfyve function. These data become more prominent in light of findings for distinct phenotypes in mice with muscle-specific Vps34 KO (30,60) and MPIfKO mice (59) (also controlled by the same MCK promoter-driven Cre recombinase and on Bl6 background). Indeed, MPIfKO mice have normal life span and size of heart or striated muscles (59), whereas muscle-specific Vps34 KO causes premature death at 2–4 months of age, enlarged heart and striated muscular dystrophy (30,60). The more severe phenotypes upon muscle-specific Vps34 KO suggests that if the Vps34-controlled PtdIns3P pool is also a main substrate supplier for PIKfyve functionality in muscle cells, as observed herein for podocytes, only a subfraction of this pool might undergo the PAS complex-dependent PtdIns3P-PtdIns(3,5)P2 cycling.
In conclusion, our study demonstrates that podocytes lacking Vps34 produce an abnormal vacuolation phenotype that is due to inadequate Vps34-dependent PtdIns3P synthesis on endosomes and consequent PtdIns(3,5)P2/PtdIns5P reduction due to PIKfyve dysfunction. These data are consistent with an important physiological relationship of Vps34 and PIKfyve in podocyte endomembrane homeostasis and maintenance of glomerular filtration.
Supplementary Material
Highlights.
PIKfyve is critical for endomembrane homeostasis
PIKfyve uses PtdIns3P as both an enzyme substrate and membrane recruitment signal
We find that Vps34 is a main PtdIns3P supplier for constitutive PIKfyve functionality
Cytoplasmic vacuoles due to Vps34 KO in podocytes are secondary to PIKfyve dysfunction
Podocyte endomembrane homeostasis requires a functional link between Vps34 and PIKfyve
Acknowledgments
This project was supported by National Institutes of Health (DK58058), American Diabetes Association (BS-161), Wayne State University and WSU School of Medicine Research Offices (to AS); National Institutes of Health (DK096053 and DK097027, to PG) and (GM068813, to ET). We thank Dr. Jon Backer, for the Vps34 antibodies, and Linda McCraw and Rochelle LaMacchio, for the outstanding secretarial assistance. The senior author expresses gratitude to the late Violeta Shisheva for her many years of support.
Footnotes
Abbreviations: PtdIns, phosphatidylinositol; PI, phosphoinositide; PIKfyve, PhosphoInositide Kinase for position five containing a fyve domain; PAS, PIKfyve-ArPIKfyve-Sac3; Ad-Cre, adenovirus with Cre-recombinase; MEFs, mouse embryonic fibroblasts; MOI, multiplicity of infection; eGFP, enhanced green fluorescence protein; GroPIns, glycerophosphorylinositol; wt, wild-type; KO, knockout; RIPA, radioimmune precipitation assay buffer.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shisheva A. PIKfyve and its lipid products in health and in sickness. Curr Topics in Microbiology and Immunology. 2012;362:127–162. doi: 10.1007/978-94-007-5025-8_7. [DOI] [PubMed] [Google Scholar]
- 2.Shisheva A. PIKfyve: Partners, significance, debates and paradoxes. Cell Biol Int. 2008;32:591–604. doi: 10.1016/j.cellbi.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sarkes D, Rameh LE. A novel HPLC-based approach makes possible the spatial characterization of cellular PtdIns5P and other phosphoinositides. Biochem J. 2010;428:375–384. doi: 10.1042/BJ20100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sbrissa D, Ikonomov OC, Strakova J, Shisheva A. Role for a novel signaling intermediate, phosphatidylinositol 5-phosphate, in insulin-regulated F-actin stress fiber breakdown and GLUT4 translocation. Endocrinology. 2004;145:4853–4865. doi: 10.1210/en.2004-0489. [DOI] [PubMed] [Google Scholar]
- 5.Sbrissa D, Ikonomov OC, Filios C, Delvecchio K, Shisheva A. Functional dissociation between PIKfyve-synthesized PtdIns5P and PtdIns(3,5)P2 by means of the PIKfyve inhibitor YM201636. Am J Physiol Cell Physiol. 2012;303:C436–446. doi: 10.1152/ajpcell.00105.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bridges D, Ma JT, Park S, Inoki K, Weisman LS, Saltiel AR. Phosphatidylinositol 3,5-bisphosphate plays a role in the activation and subcellular localization of mechanistic target of rapamycin 1. Mol Biol Cell. 2012;23:2955–2962. doi: 10.1091/mbc.E11-12-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ikonomov OC, Sbrissa D, Ijuin T, Takenawa T, Shisheva A. Sac3 is an insulin-regulated phosphatidylinositol 3,5-bisphosphate phosphatase: gain in insulin responsiveness through Sac3 down-regulation in adipocytes. J Biol Chem. 2009;284:23961–23971. doi: 10.1074/jbc.M109.025361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawasaki T, Takemura N, Standley DM, Akira S, Kawai T. The second messenger phosphatidylinositol-5-phosphate facilitates antiviral innate immune signaling. Cell host & microbe. 2013;14:148–158. doi: 10.1016/j.chom.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 9.Ikonomov OC, Sbrissa D, Shisheva A. Mammalian cell morphology and endocytic membrane homeostasis require enzymatically active phosphoinositide 5-kinase PIKfyve. J Biol Chem. 2001;276:26141–26147. doi: 10.1074/jbc.M101722200. [DOI] [PubMed] [Google Scholar]
- 10.Ikonomov OC, Sbrissa D, Mlak K, Kanzaki M, Pessin J, Shisheva A. Functional dissection of lipid and protein kinase signals of PIKfyve reveals the role of PtdIns 3,5-P2 production for endomembrane integrity. J Biol Chem. 2002;277:9206–9211. doi: 10.1074/jbc.M108750200. [DOI] [PubMed] [Google Scholar]
- 11.Jefferies HB, Cooke FT, Jat P, Boucheron C, Koizumi T, Hayakawa M, Kaizawa H, Ohishi T, Workman P, Waterfield MD, Parker PJ. A selective PIKfyve inhibitor blocks PtdIns(3,5)P(2) production and disrupts endomembrane transport and retroviral budding. EMBO Rep. 2008;9:164–170. doi: 10.1038/sj.embor.7401155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikonomov OC, Sbrissa D, Shisheva A. YM201636, an inhibitor of retroviral budding and PIKfyve-catalyzed PtdIns(3,5)P2 synthesis, halts glucose entry by insulin in adipocytes. Biochem Biophys Res Commun. 2009;382:566–570. doi: 10.1016/j.bbrc.2009.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rutherford AC, Traer C, Wassmer T, Pattni K, Bujny MV, Carlton JG, Stenmark H, Cullen PJ. The mammalian phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) regulates endosome-to-TGN retrograde transport. J Cell Sci. 2006;119:3944–3957. doi: 10.1242/jcs.03153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikonomov OC, Sbrissa D, Delvecchio K, Xie Y, Jin JP, Rappolee D, Shisheva A. The Phosphoinositide Kinase PIKfyve Is Vital in Early Embryonic Development: PREIMPLANTATION LETHALITY OF PIKfyve−/− EMBRYOS BUT NORMALITY OF PIKfyve+/− MICE. J Biol Chem. 2011;286:13404–13413. doi: 10.1074/jbc.M111.222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takasuga S, Horie Y, Sasaki J, Sun-Wada GH, Kawamura N, Iizuka R, Mizuno K, Eguchi S, Kofuji S, Kimura H, Yamazaki M, Horie C, Odanaga E, Sato Y, Chida S, Kontani K, Harada A, Katada T, Suzuki A, Wada Y, Ohnishi H, Sasaki T. Critical roles of type III phosphatidylinositol phosphate kinase in murine embryonic visceral endoderm and adult intestine. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1726–1731. doi: 10.1073/pnas.1213212110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sbrissa D, Ikonomov OC, Shisheva A. Phosphatidylinositol 3-phosphate-interacting domains in PIKfyve. Binding specificity and role in PIKfyve. Endomenbrane localization. J Biol Chem. 2002;277:6073–6079. doi: 10.1074/jbc.M110194200. [DOI] [PubMed] [Google Scholar]
- 17.Ikonomov OC, Sbrissa D, Shisheva A. Localized PtdIns 3,5-P2 synthesis to regulate early endosome dynamics and fusion. Am J Physiol Cell Physiol. 2006;291:C393–404. doi: 10.1152/ajpcell.00019.2006. [DOI] [PubMed] [Google Scholar]
- 18.Herman PK, Emr SD. Characterization of VPS34, a gene required for vacuolar protein sorting and vacuole segregation in Saccharomyces cerevisiae. Mol Cell Biol. 1990;10:6742–6754. doi: 10.1128/mcb.10.12.6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 20.Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 21.Munday AD, Norris FA, Caldwell KK, Brown S, Majerus PW, Mitchell CA. The inositol polyphosphate 4-phosphatase forms a complex with phosphatidylinositol 3-kinase in human platelet cytosol. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:3640–3645. doi: 10.1073/pnas.96.7.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shin HW, Hayashi M, Christoforidis S, Lacas-Gervais S, Hoepfner S, Wenk MR, Modregger J, Uttenweiler-Joseph S, Wilm M, Nystuen A, Frankel WN, Solimena M, De Camilli P, Zerial M. An enzymatic cascade of Rab5 effectors regulates phosphoinositide turnover in the endocytic pathway. J Cell Biol. 2005;170:607–618. doi: 10.1083/jcb.200505128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falasca M, Maffucci T. Regulation and cellular functions of class II phosphoinositide 3-kinases. Biochem J. 2012;443:587–601. doi: 10.1042/BJ20120008. [DOI] [PubMed] [Google Scholar]
- 24.Posor Y, Eichhorn-Gruenig M, Puchkov D, Schoneberg J, Ullrich A, Lampe A, Muller R, Zarbakhsh S, Gulluni F, Hirsch E, Krauss M, Schultz C, Schmoranzer J, Noe F, Haucke V. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature. 2013;499:233–237. doi: 10.1038/nature12360. [DOI] [PubMed] [Google Scholar]
- 25.Ivetac I, Munday AD, Kisseleva MV, Zhang XM, Luff S, Tiganis T, Whisstock JC, Rowe T, Majerus PW, Mitchell CA. The type Ialpha inositol polyphosphate 4-phosphatase generates and terminates phosphoinositide 3-kinase signals on endosomes and the plasma membrane. Mol Biol Cell. 2005;16:2218–2233. doi: 10.1091/mbc.E04-09-0799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maffucci T, Cooke FT, Foster FM, Traer CJ, Fry MJ, Falasca M. Class II phosphoinositide 3-kinase defines a novel signaling pathway in cell migration. J Cell Biol. 2005;169:789–799. doi: 10.1083/jcb.200408005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eskelinen EL, Prescott AR, Cooper J, Brachmann SM, Wang L, Tang X, Backer JM, Lucocq JM. Inhibition of autophagy in mitotic animal cells. Traffic. 2002;3:878–893. doi: 10.1034/j.1600-0854.2002.31204.x. [DOI] [PubMed] [Google Scholar]
- 28.Futter CE, Collinson LM, Backer JM, Hopkins CR. Human VPS34 is required for internal vesicle formation within multivesicular endosomes. J Cell Biol. 2001;155:1251–1264. doi: 10.1083/jcb.200108152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson EE, Overmeyer JH, Gunning WT, Maltese WA. Gene silencing reveals a specific function of hVps34 phosphatidylinositol 3-kinase in late versus early endosomes. J Cell Sci. 2006;119:1219–1232. doi: 10.1242/jcs.02833. [DOI] [PubMed] [Google Scholar]
- 30.Jaber N, Dou Z, Chen JS, Catanzaro J, Jiang YP, Ballou LM, Selinger E, Ouyang X, Lin RZ, Zhang J, Zong WX. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2003–2008. doi: 10.1073/pnas.1112848109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bechtel W, Helmstadter M, Balica J, Hartleben B, Kiefer B, Hrnjic F, Schell C, Kretz O, Liu S, Geist F, Kerjaschki D, Walz G, Huber TB. Vps34 deficiency reveals the importance of endocytosis for podocyte homeostasis. Journal of the American Society of Nephrology : JASN. 2013;24:727–743. doi: 10.1681/ASN.2012070700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morishita H, Eguchi S, Kimura H, Sasaki J, Sakamaki Y, Robinson ML, Sasaki T, Mizushima N. Deletion of autophagy-related 5 (Atg5) and Pik3c3 genes in the lens causes cataract independent of programmed organelle degradation. J Biol Chem. 2013;288:11436–11447. doi: 10.1074/jbc.M112.437103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou X, Wang L, Hasegawa H, Amin P, Han BX, Kaneko S, He Y, Wang F. Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:9424–9429. doi: 10.1073/pnas.0914725107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ni L, Saleem M, Mathieson PW. Podocyte culture: tricks of the trade. Nephrology. 2012;17:525–531. doi: 10.1111/j.1440-1797.2012.01619.x. [DOI] [PubMed] [Google Scholar]
- 35.Garg P, Holzman LB. Podocytes: gaining a foothold. Exp Cell Res. 2012;318:955–963. doi: 10.1016/j.yexcr.2012.02.030. [DOI] [PubMed] [Google Scholar]
- 36.Ikonomov OC, Sbrissa D, Mlak K, Shisheva A. Requirement for PIKfyve enzymatic activity in acute and long-term insulin cellular effects. Endocrinology. 2002;143:4742–4754. doi: 10.1210/en.2002-220615. [DOI] [PubMed] [Google Scholar]
- 37.Sbrissa D, Ikonomov OC, Fu Z, Ijuin T, Gruenberg J, Takenawa T, Shisheva A. Core protein machinery for mammalian phosphatidylinositol 3,5-bisphosphate synthesis and turnover that regulates the progression of endosomal transport. Novel Sac phosphatase joins the ArPIKfyve-PIKfyve complex. J Biol Chem. 2007;282:23878–23891. doi: 10.1074/jbc.M611678200. [DOI] [PubMed] [Google Scholar]
- 38.Siddhanta U, McIlroy J, Shah A, Zhang Y, Backer JM. Distinct roles for the p110alpha and hVPS34 phosphatidylinositol 3′-kinases in vesicular trafficking, regulation of the actin cytoskeleton, and mitogenesis. J Cell Biol. 1998;143:1647–1659. doi: 10.1083/jcb.143.6.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sbrissa D, Ikonomov OC, Shisheva A. PIKfyve, a mammalian ortholog of yeast Fab1p lipid kinase, synthesizes 5-phosphoinositides. Effect of insulin. J Biol Chem. 1999;274:21589–21597. doi: 10.1074/jbc.274.31.21589. [DOI] [PubMed] [Google Scholar]
- 40.Ikonomov OC, Filios C, Sbrissa D, Chen X, Shisheva A. The PIKfyve-ArPIKfyve-Sac3 triad in human breast cancer: Functional link between elevated Sac3 phosphatase and enhanced proliferation of triple negative cell lines. Biochem Biophys Res Commun. 2013;440:342–347. doi: 10.1016/j.bbrc.2013.09.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shisheva A, Rusin B, Ikonomov OC, DeMarco C, Sbrissa D. Localization and insulin-regulated relocation of phosphoinositide 5-kinase PIKfyve in 3T3-L1 adipocytes. J Biol Chem. 2001;276:11859–11869. doi: 10.1074/jbc.M008437200. [DOI] [PubMed] [Google Scholar]
- 42.Sbrissa D, Ikonomov O, Shisheva A. Selective insulin-induced activation of class I(A) phosphoinositide 3-kinase in PIKfyve immune complexes from 3T3-L1 adipocytes. Mol Cell Endocrinol. 2001;181:35–46. doi: 10.1016/s0303-7207(01)00539-1. [DOI] [PubMed] [Google Scholar]
- 43.Devereaux K, Dall’Armi C, Alcazar-Roman A, Ogasawara Y, Zhou X, Wang F, Yamamoto A, De Camilli P, Di Paolo G. Regulation of mammalian autophagy by class II and III PI 3-kinases through PI3P synthesis. PloS one. 2013;8:e76405. doi: 10.1371/journal.pone.0076405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ikonomov OC, Sbrissa D, Foti M, Carpentier JL, Shisheva A. PIKfyve controls fluid phase endocytosis but not recycling/degradation of endocytosed receptors or sorting of procathepsin D by regulating multivesicular body morphogenesis. Mol Biol Cell. 2003;14:4581–4591. doi: 10.1091/mbc.E03-04-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Franco I, Gulluni F, Campa CC, Costa C, Margaria JP, Ciraolo E, Martini M, Monteyne D, De Luca E, Germena G, Posor Y, Maffucci T, Marengo S, Haucke V, Falasca M, Perez-Morga D, Boletta A, Merlo GR, Hirsch E. PI3K class II alpha controls spatially restricted endosomal PtdIns3P and Rab11 activation to promote primary cilium function. Dev Cell. 2014;28:647–658. doi: 10.1016/j.devcel.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shisheva A. PtdIns5P: news and views of its appearance, disappearance and deeds. Arch Biochem Biophys. 2013;538:171–180. doi: 10.1016/j.abb.2013.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schink KO, Raiborg C, Stenmark H. Phosphatidylinositol 3-phosphate, a lipid that regulates membrane dynamics, protein sorting and cell signalling. Bioessays. 2013;35:900–912. doi: 10.1002/bies.201300064. [DOI] [PubMed] [Google Scholar]
- 48.Diehl N, Schaal H. Make yourself at home: viral hijacking of the PI3K/Akt signaling pathway. Viruses. 2013;5:3192–3212. doi: 10.3390/v5123192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li E, Stupack D, Klemke R, Cheresh DA, Nemerow GR. Adenovirus endocytosis via alpha(v) integrins requires phosphoinositide-3-OH kinase. J Virol. 1998;72:2055–2061. doi: 10.1128/jvi.72.3.2055-2061.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller-Jensen K, Janes KA, Wong YL, Griffith LG, Lauffenburger DA. Adenoviral vector saturates Akt pro-survival signaling and blocks insulin-mediated rescue of tumor necrosis-factor-induced apoptosis. J Cell Sci. 2006;119:3788–3798. doi: 10.1242/jcs.03102. [DOI] [PubMed] [Google Scholar]
- 51.Frese KK, Lee SS, Thomas DL, Latorre IJ, Weiss RS, Glaunsinger BA, Javier RT. Selective PDZ protein-dependent stimulation of phosphatidylinositol 3-kinase by the adenovirus E4-ORF1 oncoprotein. Oncogene. 2003;22:710–721. doi: 10.1038/sj.onc.1206151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kong K, Kumar M, Taruishi M, Javier RT. The human adenovirus E4-ORF1 protein subverts discs large 1 to mediate membrane recruitment and dysregulation of phosphatidylinositol 3-kinase. PLoS Pathog. 2014;10:e1004102. doi: 10.1371/journal.ppat.1004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen J, Chen MX, Fogo AB, Harris RC, Chen JK. mVps34 deletion in podocytes causes glomerulosclerosis by disrupting intracellular vesicle trafficking. Journal of the American Society of Nephrology : JASN. 2013;24:198–207. doi: 10.1681/ASN.2012010101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shisheva A, Sbrissa D, Ikonomov O. Plentiful PtdIns5P from scanty PtdIns(3,5)P or from ample PtdIns? PIKfyve-dependent models: Evidence and speculation (response to: DOI 10.1002/bies.201300012) Bioessays. 2014 doi: 10.1002/bies.201400129. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sbrissa D, Ikonomov OC, Fenner H, Shisheva A. ArPIKfyve homomeric and heteromeric interactions scaffold PIKfyve and Sac3 in a complex to promote PIKfyve activity and functionality. J Mol Biol. 2008;384:766–779. doi: 10.1016/j.jmb.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ikonomov OC, Sbrissa D, Fenner H, Shisheva A. PIKfyve-ArPIKfyve-Sac3 core complex: contact sites and their consequence for Sac3 phosphatase activity and endocytic membrane homeostasis. J Biol Chem. 2009;284:35794–35806. doi: 10.1074/jbc.M109.037515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harris DP, Vogel P, Wims M, Moberg K, Humphries J, Jhaver KG, DaCosta CM, Shadoan MK, Xu N, Hansen GM, Balakrishnan S, Domin J, Powell DR, Oravecz T. Requirement for class II phosphoinositide 3-kinase C2alpha in maintenance of glomerular structure and function. Mol Cell Biol. 2011;31:63–80. doi: 10.1128/MCB.00468-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luo J, Sobkiw CL, Hirshman MF, Logsdon MN, Li TQ, Goodyear LJ, Cantley LC. Loss of class IA PI3K signaling in muscle leads to impaired muscle growth, insulin response, and hyperlipidemia. Cell Metab. 2006;3:355–366. doi: 10.1016/j.cmet.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 59.Ikonomov OC, Sbrissa D, Delvecchio K, Feng HZ, Cartee GD, Jin JP, Shisheva A. Muscle-specific Pikfyve gene disruption causes glucose intolerance, insulin resistance, adiposity, and hyperinsulinemia but not muscle fiber-type switching. Am J Physiol Endocrinol Metab. 2013;305:E119–131. doi: 10.1152/ajpendo.00030.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reifler A, Li X, Archambeau AJ, McDade JR, Sabha N, Michele DE, Dowling JJ. Conditional knockout of pik3c3 causes a murine muscular dystrophy. Am J Pathol. 2014;184:1819–1830. doi: 10.1016/j.ajpath.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.