

Abstract
The human brain contains more than 100 trillion (1014) synaptic connections, which form all of its neural circuits. Neuroscientists have long been interested in how this complex synaptic web is weaved during development and remodelled during learning and disease. Recent studies have uncovered that glial cells are important regulators of synaptic connectivity. These cells are far more active than was previously thought and are powerful controllers of synapse formation, function, plasticity and elimination, both in health and disease. Understanding how signalling between glia and neurons regulates synaptic development will offer new insight into how the nervous system works and provide new targets for the treatment of neurological diseases.
Glial cells occupy more than half of the volume of the human brain. There are several types in the central nervous system (CNS), including astrocytes, oligodendrocytes and microglia1. In addition, a new class of glial cell, oligodendrocyte precursor cells (OPCs) that express the proteoglycan NG2, has recently been identified and has morphological and physiological features that are distinct from those of other glia2, 3. Astrocytes and oligodendrocyte lineage cells are derived from neural stem cells, whereas microglia originate from the immune system4. In the peripheral nervous system, there are two classes of Schwann cell (myelinating and non-myelinating), which functionally and antigenically resemble the glia of the CNS5.
Glia are vital for the survival and function of neurons. Oligodendrocytes and Schwann cells myelinate axons to ensure fast, saltatory movement of action potentials. Astrocytes regulate blood flow, provide much-needed energy to neurons, and supply the building blocks of neurotransmitters, which fuel synapse function. But the functions of glia are not restricted to supporting neuronal function4, 6.
In this Review, we describe the numerous recent findings that illustrate the importance of glia in the formation, function, plasticity and elimination of synapses in the nervous system. We also discuss how these findings provide new insight into the pathophysiology of chronic pain, neurological diseases such as epilepsy, and neurodegenerative disorders such as Alzheimer’s disease and glaucoma.
Glia are intimately associated with synapses
In the peripheral nervous system, synapses are ensheathed by non-myelinating Schwann cells, and in the CNS by astrocytes (Fig. 1a). The CNS also contains two forms of elongated, radial glial cell: Bergmann glia in the cerebellum, and Müller cells in the retina. These have many features in common with astrocytes and are closely associated with synapses7. Such an association of glia with synapses seems to have been conserved across evolution, because a class of synapse-ensheathing glial cell is also found in the brain of the fruitfly Drosophila melanogaster and has surprisingly similar morphology to rodent astrocytes8. This structural association extends to function.
Figure 1. The tri-partite synapse.
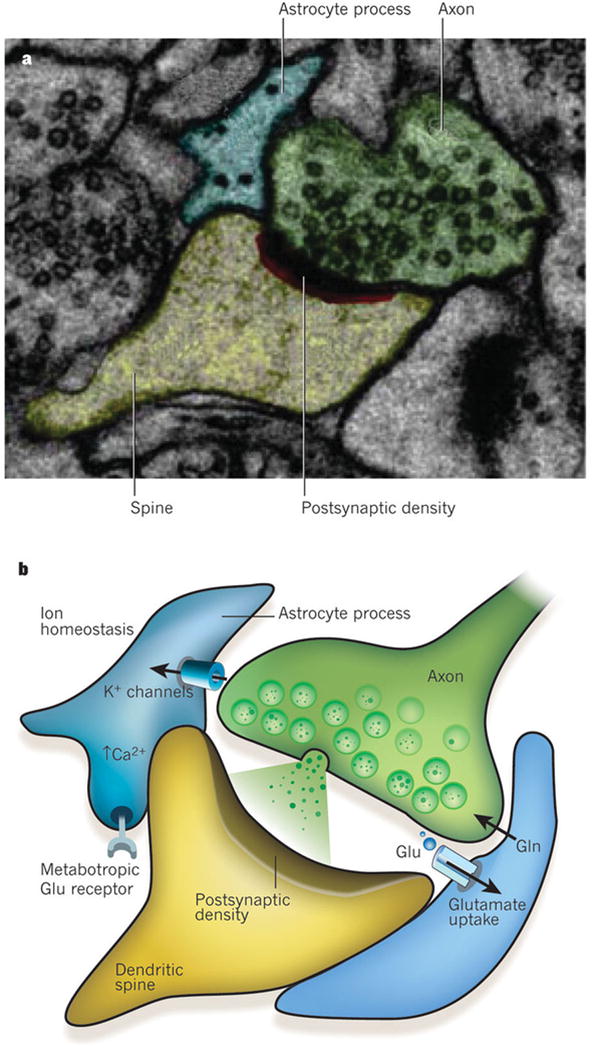
The processes of astrocytes are intimately associated with synapses. This association is both structural and functional. a, Electron micrograph showing a tripartite synapse in the hippocampus. The astrocyte process (blue) ensheaths the perisynaptic area. The axon of the neuron is shown in green, with the dendritic spine in yellow and the postsynaptic density in red and black. Reproduced, with permission, from ref. 22. b, Schematic representation of a tripartite synapse. Perisynaptic astrocyte processes contain transporters that take up glutamate (Glu, green circles) that has been released into the synapse and return it to neurons in the form of glutamine (Gln). Glutamate receptors on astrocytes (such as metabotropic glutamate receptors) sense synaptic glutamate release, which in turn induces a rise in Ca2+ concentration in the astrocytes. One of the main functions of glia at the synapse is to maintain ion homeostasis, for example regulating extracellular K+ concentrations and pH.
Perisynaptic glia ensure potassium ion homeostasis and regulate extracellular pH (Fig. 1b). Moreover, these cells express several receptors for neurotransmitters, enabling them to ‘listen’ to synapse function and respond to synapse activity by making localized and global changes in intracellular calcium ion concentrations9, 10 (Fig. 1b). In addition, glia modulate the properties of synapses by releasing neurologically active substances such as ATP and D-serine4. The extensive structural and functional association of perisynaptic glia with the synapse gave rise to the concept of the ‘tripartite synapse’, in which synapses are defined as comprising the presynaptic and postsynaptic specializations of the neurons and the glial process that ensheaths them11 (Fig. 1b).
In the past decade surprising new findings showed that one glial subtype receives synaptic inputs. Fast synaptic transmission occurs between OPCs and axons, both in the hippocampus and the cerebellum12, 13. These OPCs can receive input mediated by the neurotransmitters glutamate and GABA (γ-aminobutyric acid)14, 15. The functional significance of these neuron-to-glia synapses is not known. Although a matter of speculation at present, it is possible that neuronal activity regulates the differentiation of OPCs into myelinating oligodendrocytes according to the requirements of the neural circuit.
Astrocytes are the main population of glia in the brain. Rodent astrocytes have been classified into two groups on the basis of their morphology and location. One group contains the protoplasmic astrocytes of the grey matter, which are highly ramified. These cells ensheath synapses and are in contact with blood vessels. The other group is the fibrous astrocytes of the white matter, cells that are in contact with the nodes of Ranvier1. Recent physiological and gene expression profiling studies indicate that astrocytes, like neurons, are a diverse cell population, with distinct properties in different brain regions and at different periods of development. So this classification into two groups might not be adequate to appreciate the full extent of astrocyte diversity16. Moreover, the number and size of astrocytes in the brain varies between species and increases with species brain size and cognitive capability. For example, the human brain contains several more populations of astrocytes than the rodent brain, and human astrocytes are up to threefold larger and more ramified than their rodent counterparts17.
The processes of protoplasmic astrocytes infiltrate into the neuropil and wrap themselves around synapses18, 19, 20. In the hippocampus, individual astrocytes parcel out the neuropil in a non-overlapping manner to form separate anatomical domains18. A similar tile-like organization of glia is characteristic of the anatomical distribution of cortical astrocytes19 and also of cerebellar Bergmann glia, which ensheath most Purkinje cell synapses21.
Astrocytes do not readily wrap around all synapses and the presence and extent of astrocyte coverage might be regulated20. In the hippocampus, protoplasmic astrocytes have been found to ensheath 57% of the synapses, most of which are excitatory synapses22. What directs astrocyte processes to synapses? It has been postulated that astrocytes extend processes towards regions where the glutamate concentration is higher. In agreement with this hypothesis, in the hippocampus of adult rats, large perforated synapses with higher probabilities of glutamate release were found to be more likely to be ensheathed by astrocytes22. It is equally possible, however, that synapses that are wrapped by an astrocyte are stabilized and receive nurturing signals that allow them to mature further. It is important to point out that astrocyte processes do not fully insulate a synapse from the surrounding environment but, instead, allow some flow into and out of the synaptic cleft. For example, at hippocampal synapses, regardless of their size, there are astrocyte-free perimeters, where substances could escape from or enter the synaptic clefts. These regions might be important for the trans-synaptic activation of glutamate receptors20.
Association of synapses and astrocyte processes might be a dynamic process. In agreement with this possibility, astrocyte coverage can be altered during development in response to injury and in various physiological conditions23, 24, 25, 26. Live imaging of organotypic hippocampal slices shows that astrocytes rapidly extend and retract their processes to engage and disengage from postsynaptic dendritic spines27, 28. Astrocytes are more motile than their dendritic counterpart, and their movement is regulated by actin dynamics. Interestingly, dendritic protrusions that had contacts with astrocytes survived longer and were morphologically more mature than those without such contacts28. These observations suggest that astrocyte processes might control the stabilization of individual dendritic protrusions and their subsequent maturation into spines, and they present the possibility that astrocytes could have a crucial role in the experience-dependent structural synaptic changes that underlie learning and memory.
A series of studies in the adult rodent hippocampus suggested that the receptor tyrosine kinase EphA4 on the dendritic spines of pyramidal neurons regulates spine morphology by interacting with its ligand ephrin-A3, which is located in the perisynaptic processes of astrocytes29. Perturbing ephrin–EphA signalling led to increased numbers of glutamate transporters on astrocytes, aberrant morphology of dendritic spines, and defects in hippocampal learning30 and long-term potentiation31.
A remarkable interplay between astrocyte processes and synapses occurs in the hypothalamo-hypophyseal system at the hypothalamic supraoptic nucleus. In physiological conditions such as parturition, lactation and chronic dehydration, most of the astrocyte processes in contact with the soma and dendrites of magnocellular oxytocin neurons denervate these neurons in an oxytocin-dependent manner32. Concurrently, new inhibitory and excitatory synapses are formed onto these neurons, and these disappear when stimulation ends and the astrocyte processes reinnervate these oxytocinergic neurons33.
Taken together, these findings show that different glial cell types regulate different aspects of nervous-system architecture, function and plasticity, through dynamic and often bidirectional structural interactions with synapses.
Glia control synapse formation
Establishment of the correct number and type of synapses is crucial for the proper development and function of the human brain. Many presynaptic and postsynaptic ‘target-derived’ factors have been implicated as important regulators of synapse formation and specificity34. Because astrocytes are an integral part of synapses, they probably contribute to the establishment of synapses. In addition, gliogenesis and synaptogenesis occur concurrently in the brain, and glial-cell maturation marks the end of the synaptogenic and plastic periods35, 36.
In vitro studies using purified neurons and astrocytes paved the way for understanding the extent to which astrocytes influence the shaping of synapse formation. They showed that synaptogenesis is not solely controlled by neurons but, instead, can be instructed by glia-derived signals. Retinal ganglion cells (RGCs), unlike many other CNS neurons, can be isolated from rodent retina as a purified population and can then be cultured in serum-free media of known composition37. Under these conditions, RGCs show little spontaneous synapse activity and form few synapses, whereas RGCs cultured in the presence of a feeding layer of astrocytes or astrocyte-conditioned medium show about tenfold more excitatory synapse activity and a fivefold to sevenfold increase in the number of synapses38, 39. In addition to rat RGCs, astrocytes have been shown to be necessary for inducing synapse formation by human neurons generated from embryonic stem cells40. Astrocyte-induced synapses are ultrastructurally normal with functional excitatory synaptic events mediated by AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) receptors39. Several studies using this system uncovered the presence of at least three classes of factor secreted by astrocytes: those that induce the formation of structurally normal but postsynaptically silent synapses; those that facilitate presynaptic activity and enhance the probability of neurotransmitter release; and those that stimulate the insertion of glutamate receptors, which are stored in intracellular vesicles to the postsynaptic membranes, thus facilitating postsynaptic function1 (Fig. 2).
Figure 2. Glial regulation of synaptic development.
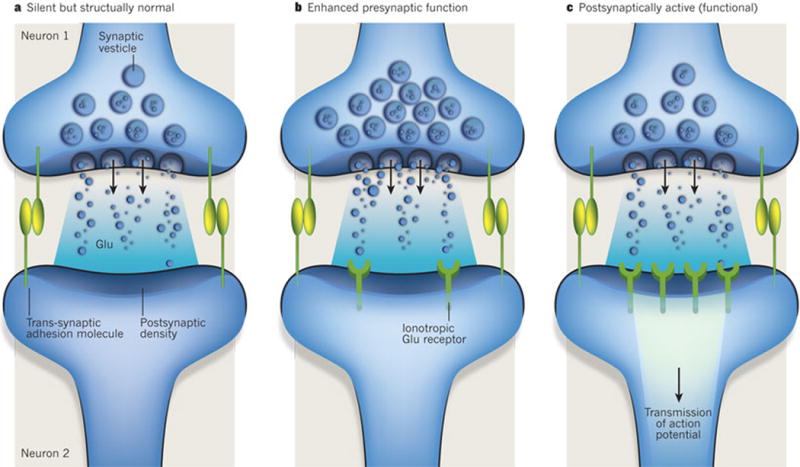
Several studies using the retinal ganglion cell culture system have shown that there are at least three classes of factor secreted by astrocytes. These factors control different aspects of the development of glutamate-mediated synapses. a, One type induces the formation of structurally normal but postsynaptically silent synapses. Thrombospondins are an example of this type of factor. b, Another type facilitates presynaptic activity and increases the probability of neurotransmitter release. Cholesterol functions in this way. c, A third type induces the formation of functional synapses or converts silent synapses into active ones by facilitating the insertion of glutamate receptors into postsynaptic sites. These factors have yet to be identified.
The synapse-formation-inducing signals from astrocytes were identified to be a family of extracellular matrix proteins called thrombospondins (TSPs)41. Purified TSPs alone have been found to increase synapse number in RGC cultures to a level comparable to that induced by culturing in astrocyte-conditioned medium. TSP-induced synapses are ultrastructurally normal, presynaptically active but postsynaptically silent because of a lack of AMPA receptors on the postsynaptic side. Removal of TSPs from astrocyte-conditioned medium diminishes the synaptogenic activity of the medium. These results show that TSPs are the necessary and sufficient synaptogenic factors in astrocyte-conditioned medium for inducing the formation of structural synapses. In vivo, TSP1 and TSP2 are expressed by developing astrocytes at early postnatal stages, when most excitatory synapses are forming, and their expression is downregulated in adults. In addition, mice lacking both TSP1 and TSP2 have significantly fewer excitatory synapses in the cortex, showing that TSPs are important for excitatory synapse formation in vivo41.
Using molecular and biochemical techniques, the α2δ-1 subunit (encoded by the geneCACNA2D1) of the voltage-dependent Ca2+ channel complex was identified as the relevant neuronal receptor for TSPs42. All five mammalian TSPs can induce synapse formation by binding through their type 2 epidermal-growth-factor-like repeats to the von Willebrand factor type A (vWFA) domain of neuronal α2δ-1. It is postulated that the interaction between TSPs and α2δ-1 triggers cellular events that lead to synapse formation by activating a synaptogenic signalling complex, which might include Ca2+ channels42. TSPs also bind to integrins, which are important regulators of synapse structure and function43, 44. Moreover, TSP1 was recently identified to be a ligand for the neuroligin family of synaptic adhesion proteins45. Given its ability to interact with multiple molecule types, it is possible that TSPs can not only induce synapse formation but can also modulate presynaptic and postsynaptic function. Understanding how TSP–α2δ-1 signalling in neurons induces synapse formation has the potential to provide new insight into the molecular basis of this process, which has long been mysterious.
The α2δ-1 subunit is also the receptor for the drug gabapentin (Neurontin), which is used to treat neuropathic pain and epilepsy and has an unknown mechanism of action46. Gabapentin blocks TSP-induced excitatory synaptogenesis in vitro and markedly inhibits excitatory synapse formation between neurons throughout the entire developing brain. It prevents this type of synapse formation by blocking the ability of TSPs to bind to α2δ-1, thus inhibiting synaptogenic signalling initiated by the TSP–α2δ-1 interaction without dissolving previously formed synapses42. These findings provide an additional line of evidence highlighting the ability of astrocytes as powerful promoters of synapse formation in vivo. They also suggest that TSP–α2δ-1 signalling and astrocyte-induced synapse formation might be involved in the pathophysiology of disorders such as neuropathic pain and epilepsy.
As astrocytes can induce both presynaptic and postsynaptic activity, whereas TSPs can only instruct the formation of postsynaptically silent synapses, there must be other signals secreted by astrocytes that facilitate the insertion of glutamate receptors at the postsynaptic site and thus convert silent synapses to active ones. Through these signals, astrocytes can regulate synapse strength and plasticity. The identity of the astrocyte-secreted signal that can induce AMPA receptor insertion into synapses is unknown. Cholesterol in apolipoprotein E particles secreted by astrocytes, however, has been shown to increase the induced synaptic responses substantially in autaptic, cultured RGCs47 by increasing presynaptic function and dendrite differentiation in this system41, 48. The relevance of cholesterol and apolipoprotein E in synapse function in vivo is not yet known.
Factors secreted by astrocytes also regulate the formation of inhibitory synapses between hippocampal neurons in vitro. They do this by modulating inhibitory postsynaptic development, by stimulating neuron–neuron signalling through the neurotrophic receptor tyrosine kinase TrkB49. The exact mechanism is not clear; however, the neurons themselves are the source of the TrkB ligand brain-derived neurotrophic factor (BDNF). TSP1 did not induce inhibitory synapse formation in this culture system, although it facilitated excitatory synapse formation and neurite outgrowth50. The identity of the factor(s) that is secreted by astrocytes and promotes inhibitory synaptogenesis through increasing neuronal BDNF production remains unknown.
Schwann cells of the peripheral nervous system, similar to astrocytes in the CNS, also secrete synaptogenic factors. These factors include transforming growth factor-β, which controls neuromuscular junction formation51, and unidentified low-molecular-mass molecules that increase synapse function52. In addition, both TSP4 and α2δ-1 are highly enriched at the neuromuscular junction53, 54, suggesting that they might be important in inducing synapse formation in the peripheral nervous system, as well as in the CNS.
Astrocytes also regulate synapse formation through contact-mediated mechanisms. In the retina, there is a developmental switch in which the ability of RGCs to respond to soluble synaptogenic signals from astrocytes is induced by contact with astrocytes themselves55. This finding was made by observing rat RGCs at embryonic day (E) 17 co-cultured with postnatal RGCs in the presence of astrocyte-conditioned medium. Synapses did not form on the dendrites of E17 RGCs, whereas E19 RGCs received synapses. This two-day period (E17–E19) coincides with the developmental window in vivo during which astrocytes migrate along the optic nerve into the retina and populate the inner retinal layer adjacent to the RGCs. Contact with astrocytes but not amacrine cells, which are interneurons in the retina, was sufficient for cultured E17 RGCs to become receptive to synapse formation. Although the exact mechanism is not yet clear, it is known that local contact with astrocytes through integrins activates protein kinase C signalling in individual dissociated neurons from the embryonic hippocampus, and facilitates excitatory synapse formation56. It is possible that this global protein kinase C activation initiates neuronal maturation events that affect major cellular processes in neurons, such as the sorting of synaptic adhesion proteins into the correct subcellular compartments. Consistent with this hypothesis, it has been shown that astrocyte contact with E17 RGCs stimulates the localization of the synaptic adhesion molecule neurexin away from dendrites55, where neurexin functions as an inhibitor of synapse formation by interacting in cis with the postsynaptic adhesion molecule neuroligin57.
Recent gene-expression analysis of astrocytes in vivo showed that astrocytes produce messenger RNAs that encode several synaptic adhesion proteins, such as neurexins, neuroligins and cadherins58, which are known to be important for synapse formation and are thought to function only in neurons34. Astrocyte processes might also use these cell-surface molecules to guide synapse formation or mediate the astrocyte–synapse interactions discussed earlier. In agreement with this, γ-protocadherins, a family of neuronal adhesion molecules encoded by a single gene cluster of 22 genes, are also expressed by astrocytes. These adhesion molecules localize to perisynaptic astrocyte processes, and homophilic astrocyte–neuron γ-protocadherin contacts are crucial for synaptogenesis in vitro. In vivo, restricted mutation of the γ-protocadherin gene cluster in astrocytes significantly delays the formation of both excitatory and inhibitory synapses59. These results suggest that cell adhesion molecules that are important for neuron–neuron interactions can also participate in astrocyte–neuron interactions, thus enabling astrocytes to guide synapse formation and morphology.
There is evidence that the glia–neuron signalling observed to regulate synapse formation in mammals also occurs in invertebrates. For example, in Caenorhabditis elegans, the glia-derived cell-surface receptor UNC-6 (also known as netrin) is used not only for guiding axons but also for defining sites for synaptogenesis60. In addition, the function of sensory neurons in C. elegansdepends on FIG-1, a protein that is secreted by glia and contains TSP type I and type II EGF-like repeats61. Moreover, a Drosophila homologue of α2δ-1, Cacna2d3, is required for presynaptic maturation at the neuromuscular junction62. Together, these findings suggest that the molecular basis of glia-induced synapse formation, as well as synapse formation itself, might be highly evolutionarily conserved. Interestingly, two isoforms of TSP (TSP2 and TSP4) are among the few genes that are substantially upregulated in the human brain compared with the primate and mouse brain63, suggesting that TSPs — and thus astrocytes — might contribute to the greater brain plasticity of humans.
Several recent studies have shown that glia, through their ability to signal to neurons, also have an active role in activity-dependent modulation of synaptic efficacy. This process contributes to neural circuit development and experience-dependent plasticity. Regulation of synapse plasticity by long-term potentiation and long-term depression requires rapid adjustments in the strength of individual synapses in response to patterns of correlated synapse activity. The main mechanisms regulating these processes involved in plasticity are thought to be changes in the cell-surface delivery, retention and Ca2+-channel properties of the postsynaptic ionotropic glutamate receptors, AMPA receptors and NMDA (N-methyl-D-aspartate) receptors64, 65. NMDA receptors contain a glutamate-binding site, as well as a site for binding to glycine, a co-agonist that is required for the opening of the Ca2+ channel. The amino acid D-serine, which is produced exclusively by astrocytes, is a potent agonist for this site66. In hippocampal cultures, neuronal-activity-dependent release of D-serine from astrocytes was found to be necessary for long-term potentiation67. Moreover, in the hypothalamic supraoptic nucleus, a system in which structural changes in astrocyte association with synapses are important for oxytocin-mediated responses at synapses, the endogenous co-agonist of NMDA receptors is D-serine rather than glycine. The degree of astrocyte coverage of neurons governs the level at which D-serine occupies the glycine-binding site on the NMDA receptor, thereby controlling the activity dependence of long-term potentiation and long-term depression68.
In addition to NMDA receptor function, cell-surface delivery of AMPA receptors is also regulated by the pro-inflammatory cytokine tumour-necrosis factor-α (TNF-α), which is released by glia. Glial TNF-α has been shown to improve synaptic efficacy by increasing the cell-surface expression of AMPA receptors. Conversely, blocking TNF-α-mediated signalling was found to have the opposite effect69. Interestingly, even though TNF-α increased the cell-surface expression of AMPA receptors, long-term potentiation and long-term depression were not affected in its absence70. Prolonged changes in the cell’s synaptic activity, such as during blockade of synapse function, lead to adjustments in the strength of all synapses on that cell. This form of synaptic plasticity is called homeostatic synaptic scaling and is thought to be a crucial mechanism for preventing neural networks from becoming unstable and dysfunctional71. Glial TNF-α mediates synaptic scaling in response to prolonged activity blockade70. The exact glial-cell source of TNF-α(astrocytes or microglia) is not yet clear. Because TNF-α is secreted by microglia in response to injury or insult to the CNS, TNF-α-mediated synaptic plasticity might modulate neural responses to injury and neurodegeneration.
In summary, secreted and cell-surface-associated signals from glia coordinate excitatory and inhibitory synapse formation and modulate synapse function and plasticity in the CNS and peripheral nervous system (Table 1).
Table 1.
Glial-cell signals that control synapse development
| Glia type | Signal type | Signal | Action |
|---|---|---|---|
| Rat astrocyte | Secreted | TSP1, TSP2, TSP3, TSP4 and TSP5 | Induce excitatory synapse formation (but synapses are postsynaptically silent, owing to a lack of AMPA receptors)41,42 |
| Unknown | Induces excitatory synapse formation (and synapses are postsynaptically active, given the presence of AMPA receptors at the synapse) | ||
| Unknown | Induces inhibitory synapse formation and neurite outgrowth by activating TrkB signalling in neurons49, 50 | ||
| Cholesterol and apolipoprotein E | Enhance synaptic function47 by facilitating presynaptic release41 and dendritic maturation48 | ||
| D-Serine | Controls activity dependence of long-term potentiation and long-term depression68 | ||
| Cell surface | Unknown, possibility integrins (PKC-signalling dependent) | Regulates synaptic receptivity of embryonic neurons55 in response to contact-dependent astrocyte signals56 | |
| γ-Protocadherins | Facilitate correct execution of excitatory and inhibitory synaptogenesis59 | ||
| Frog Schwann cell | Secreted | TGF-β | Induces neuromuscular junction formation51 |
| Unidentified low-molecular-mass molecule (not ATP or glutamate) | Enhances synaptic activity at the neuromuscular junction52 | ||
| C. elegans glia | Secreted | FIG-1 | Regulates sensory organ formation and function61 |
| Cell surface | UNC-6 (also known as netrin) | Instructs axonal growth and presynaptic assembly60 |
Glia regulate axon pruning and synapse elimination
During development, neurons often extend their axons beyond their intended targets and form an excessive number of synapses. The selective elimination of synapses and the pruning of axons to fine-tune synaptic territories are crucial for the proper development and function of the nervous system. The specific molecular mechanisms that drive synapse elimination and axon pruning remain mostly unknown, although, in the past decade, studies in invertebrate systems have suggested that synapses are removed actively through engulfment and phagocytosis by glia. Recent evidence suggests that vertebrate glia are also actively involved in the process of synapse elimination through mechanisms that are both similar and distinct from those observed in invertebrates.
Developmental axon pruning is widely used in Drosophila metamorphosis, which involves major pruning events that require the breakdown and reconstruction of synaptic networks72. For example, in Drosophila larvae, γ-neurons in the mushroom body initially extend axon branches into both the dorsal and the medial mushroom-body-axon lobes. Axon branches to both lobes degenerate during pupal stages before the formation of adult connections. This pruning is triggered in a developmentally regulated manner by intrinsic molecular mechanisms such as activation of local axon degeneration by the neuronal nuclear hormone ecdysone73. Live imaging studies have shown that during early pupal stages, the processes of the glia that surround the neurons infiltrate bundles of axon branches. Glial processes engulf clusters of axon varicosities, which accumulate in intracellular lysosomal compartments. Selective inhibition of endocytosis by glia or of the ecdysone receptors in mushroom-body neurons suppresses infiltration by glia, as well as elimination of varicosities, and induces a severe delay in axon pruning73, 74. These findings show that glia are activated by mushroom-body axons that are destined for elimination. Activated glia then infiltrate the mass of axon branches to eliminate varicosities and break down axon branches, rather than just scavenging already-degraded axon debris.
Forward genetic screens in Drosophila uncovered two genes — encoding Draper and CED-6, which are essential for the clearance of apoptotic cells in C. elegans — that function in the engulfment of pruned axons by glia during Drosophila metamorphosis (Fig. 3). In flies in which the gene encoding draper is mutated, or in which draper and ced-6 have been knocked down specifically in glia by RNA interference, engulfment by glia is suppressed, resulting in the inhibition of axon pruning during metamorphosis75, 76. These findings suggest that a similar molecular mechanism governs the clearance of apoptotic cells and of degenerating axons of living neurons. But how does Draper recognize modified self proteins? Another Drosophilareceptor involved in phagocytosis, Six-microns-under (SIMU; also known as NIMC4), was recently reported to be required for efficient clearance of apoptotic cells by glia in the nervous system and by macrophages elsewhere77. SIMU is highly expressed by phagocytic cell types during development and is part of a conserved family of proteins that includes Draper. SIMU functions upstream of Draper in the same pathway and affects the recognition and engulfment of apoptotic cells, but only Draper affects their subsequent degradation77 (Fig. 3). These results suggested that, by strongly binding to apoptotic cells or axons that are to be pruned, SIMU couples debris from degenerating axons to Draper signalling, which induces glia to phagocytose the debris.
Figure 3. Molecular pathways known to regulate axon pruning and synapse elimination by glia in invertebrates.
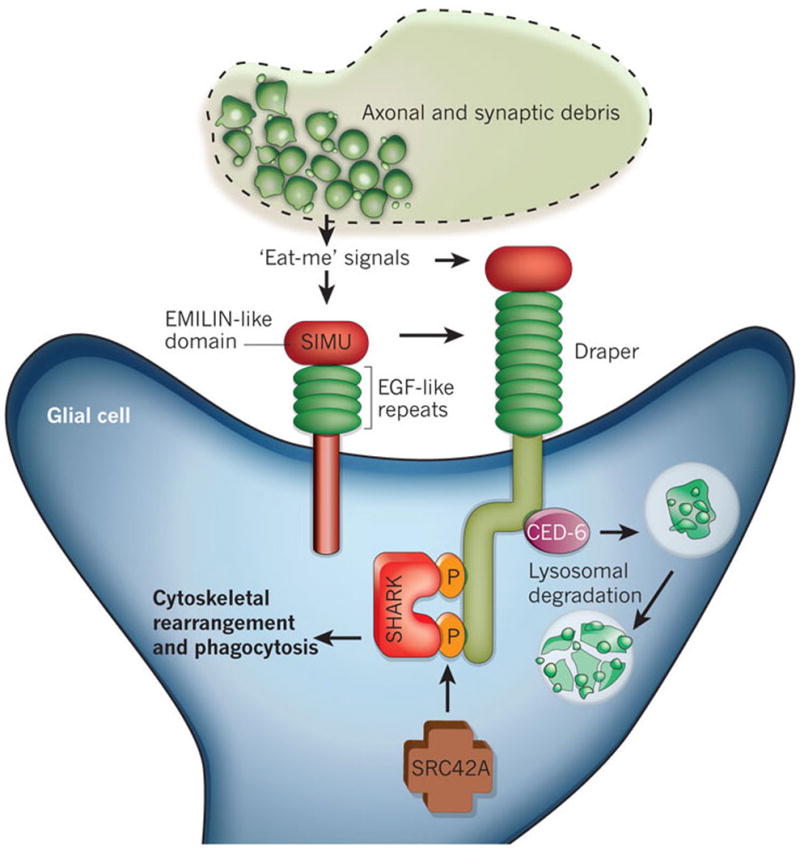
Two transmembrane proteins, Six-microns-under (SIMU) and Draper, regulate the engulfment and phagocytosis of axosomes by glia in Drosophila. These proteins are homologues and have large extracellular regions with multiple epidermal growth factor (EGF)-like repeats and an EMILIN-like domain. SIMU and Draper function in the same pathway, most probably recognizing synaptic debris by binding to unidentified ‘eat-me’ signals on degenerating axons. SIMU seems to be involved in the initial recognition and uptake steps of engulfment, but it lacks an intracellular signalling domain. By contrast, Draper is capable of intracellular signalling and operates downstream of SIMU. The cytoplasmic adaptor protein CED-6 functions downstream of Draper, mediating the internalization and lysosomal degradation of debris. In addition, Draper triggers cytoskeletal rearrangements and phagocytosis through an interaction with the non-receptor tyrosine kinase SHARK. The kinase SRC42A facilitates the SHARK–Draper interaction by phosphorylating Draper.
Signalling of neural injury in Drosophila uses the same molecular and cellular processes observed in developmental axon pruning. Upon axonal injury, glia upregulate the expression of draper, undergo marked changes in morphology and rapidly extend fine processes towards severed axons78. In draper mutants, glia fail to respond to axon injury, and severed axons are not cleared from the CNS. The Drosophila protein SHARK, a non-receptor tyrosine kinase similar to mammalian SYK, has been identified as a downstream signalling molecule that binds to the intracellular domain of Draper (Fig. 3). SHARK activity is essential for Draper signalling events, including the recruitment of glial membranes to severed axons and the phagocytosis of axon debris and neuronal-cell corpses by glia. Another signalling molecule, the SRC-family kinase SRC42A, functions in the same pathway by increasing Draper phosphorylation, thus stimulating the phagocytic activity of glia78 (Fig. 3). These Draper–SRC42A–SHARK interactions are also likely to govern developmental axon pruning and have remarkable similarities to immunoreceptor–SRC-family-kinase–SYK signalling in mammalian immune cells.
Glia also play important roles in synapse elimination in the mammalian nervous system. One of the classic examples of activity-dependent synapse elimination occurs at the mammalian neuromuscular junction. At birth, postsynaptic muscle cells are innervated by multiple motor axons. By the second week after birth, activity-dependent competition permanently eliminates immature inputs, whereas the sole remaining input is maintained and strengthened. Eliminated connections detach from the neuromuscular junction and, as they retract, pieces of axon are shed. In the mammalian peripheral nervous system, similar to the active engulfment and clearance by glia that is observed in Drosophila metamorphosis, Schwann cells break up retracting axons and remove the synaptic debris. Time-lapse imaging and serial electron microscopy demonstrated that as inappropriate axons disappeared, they shed small membrane particles that contained intact presynaptic structures79. These debris structures, termed axosomes, were formed by engulfment of the tips of retreating axons by neighbouring Schwann cells. Further time-lapse imaging was carried out in transgenic mice that differentially express fluorescent proteins in Schwann cells and axons. This experiment directly showed that axosome shedding occurs entirely within the confines of Schwann cells and depends on glial lysosome function80. The molecules that drive Schwann cells to phagocytose retracting axons are unknown. However, a recent study showed that, similar to mammals, during neuromuscular junction development in Drosophila, there is activity-dependent elimination of immature synaptic boutons and widespread appearance of presynaptic debris81. Moreover, like Schwann cells, Drosophilaglia invade the neuromuscular junction and, together with muscle cells, phagocytose the synaptic debris. The Draper signalling pathway is important for this process because suppression of Draper function results in accumulation of presynaptic debris and compromised synapse growth81. These results show that the activity-dependent elimination of presynaptic inputs in the neuromuscular junction through phagocytosis by glia is an evolutionarily conserved phenomenon. The Draper–CED-6 pathway might also be important as a regulator of glia-mediated axon pruning at the mammalian neuromuscular junction because peripheral glia express a similar or identical pathway82.
A recent study showed that the classical complement cascade, which is part of the innate immune system, helps to mediate synapse elimination in the developing CNS83. During development, immature astrocytes produce an unidentified signal that alters gene expression in neurons, leading to the upregulated expression of C1q, the protein that initiates the complement cascade. Early in development, C1q is localized to synapses that are destined for elimination (Fig. 4). In support of this model, mice deficient in either C1q or the downstream complement protein C3 fail to execute synapse elimination properly in the CNS, as determined by the absence of anatomical refinement of retinogeniculate connections and by the retention of excess retinal innervations by lateral geniculate neurons. Moreover, this failure to eliminate synapses in C1q-deficient mice led to enhanced synaptic connectivity and epileptic activity in the adult mice84, suggesting that defects in this pathway could be involved in seizure disorders observed in humans.
Figure 4. Regulation of synapse elimination in the mammalian CNS by the complement cascade.
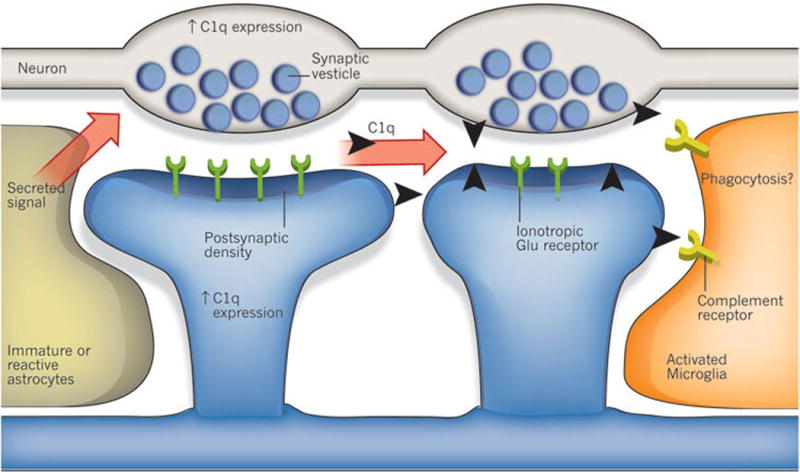
An unidentified secreted signal from immature and reactive astrocytes upregulates expression of the complement component C1q in neurons. It is proposed that C1q binds to weaker synapses and tags them for elimination. This elimination might occur through phagocytosis by microglia, mediated by complement receptors at the surface of microglia. Other complement-cascade components such as C3 are also produced by glia in normal and disease conditions, and another possibility is that synapse elimination is triggered by C1q and C3 not only during development but also during the early stages of neurodegenerative diseases.
These observations beg the question, how does C1q control synapse elimination? Looking to the innate immune system, C1q functions as a molecular tag to mark unwanted cells or debris for removal85. So it is possible that C1q tags weak synapses for elimination (Fig. 4). The molecular interactions that localize C1q specifically to weak synapses are unknown; however, microglia, the resident immune cells of the brain, produce large amounts of the receptors for C1q and C3 and thus are likely to be responsible for the removal of unwanted synapses. Microglia also phagocytose the synaptic terminals of motor neurons after injury, through a process known as synaptic stripping (discussed in the next section)86, and an exciting question is whether synaptic stripping is also complement dependent. Interestingly, homologues of C1q, such as precerebellins87 and C1q-like protein 1 (C1QL1), C1QL2 and C1QL3 (ref. 88), are synaptic molecules that are expressed by neurons and are involved in activity-dependent synapse plasticity and formation. It is thus likely that these molecules all participate in a common mechanism that controls different aspects of synapse remodelling. Perhaps these synaptic C1q-like molecules compete for a common synaptic receptor89, and their interaction with this receptor might be antagonized by C1q. The recent finding that the synaptic pentraxins, which bind to C1q, mediate silent to active synapse conversion in the developing visual system90, potentially provides an exciting link between activity-dependent control of synapse elimination and regulation of synapse function, given that neuronal pentraxins are implicated in postsynaptic glutamate-receptor recruitment.
Glia control synaptic connectivity in disease
Glia rapidly respond to injury in the nervous system. The activation of microglia is triggered by injury-mediated signals, such as ATP released from dead cells and serum factors leaking into the extracellular environment as a result of the breakdown of the blood–brain barrier. Similar signalling events also trigger a marked change in astrocyte behaviour. Both cell types lose their ramified structures. Microglia migrate to the injury site and start to divide, and astrocytes direct their processes towards the damaged region86, 91. This phenomenon is known as reactive gliosis. The mechanisms that affect the behaviour of glia after insult to the CNS are an active area of research. Whether reactive glia aid in CNS recovery after injury or permanently impair CNS regeneration is not clear.
When injured, reactive astrocytes revert to an immature state and express molecules that affect synapse formation. For example, the production of TSP1 and TSP2 increases after traumatic brain injury, ischaemia or stroke92. Purinoceptor signalling and mechanical stimulation have been shown to mediate the upregulation of TSP production in cultured astrocytes, and these proteins could be upregulated in a similar manner in vivo after injury93. TSP1 and TSP2 have been shown to be required for functional recovery after stroke because mice deficient in both TSP1 and TSP2 (TSP1/2-nulls) showed impaired recovery of motor function, synaptic density and axon sprouting94. Another line of evidence suggests that the TSP–α2δ-1 interaction, and thus astrocyte-induced synapse formation, is necessary for the correct execution of injury-mediated developmental plasticity in the mouse brain42. This was determined by using a well-characterized model of barrel-cortex plasticity. Inhibition of TSP-mediated synapse formation, either by injection of gabapentin or in TSP1/2-null mice, strongly perturbed the stereotypical reorganization of the barrel cortex.
TSP–α2δ-1 signalling after injury might not always be beneficial. The fact that the anti-analgesic, anti-epileptic drug gabapentin blocks the interaction between α2δ-1 and TSP suggests that under certain conditions this signalling might be pathological. In agreement with this idea, both Cacna2d1 and TSP4 levels increase in the spinal-nerve ligation model of neuropathic pain95, 96. The increase in the expression levels of Cacna2d-1 and TSPs could be an essential part of synapse remodelling in response to synapse loss after injury. However, aberrant synapse formation — or dysregulation of another cellular process that is also regulated by the TSP–α2δ-1 interaction (for example, increased trafficking of Ca2+ channels to the cell surface) — may lead to outcomes such as epilepsy and neuropathic pain. If this is the case, then drugs that efficiently target the TSP–α2δ-1 interaction may help to alleviate neuropathic pain or even to prevent the development of epilepsy following traumatic brain injury.
Similar to the engulfing glia of Drosophila that were discussed earlier, microglia and possibly astrocytes physically remove synaptic inputs after injury. For example, after axotomy, activated microglia adhere to damaged motor neurons, spread across their soma and dendrites, and actively denervate (remove neuron–neuron (nerve) connections in a way that breaks down damaged synapses) the glutamate-containing presynaptic boutons86. This process, generally referred to as synaptic stripping, is thought to protect neurons from excitotoxicity and increase the overall inhibition of the damaged neuron. There are strong indications that signalling between neurons and glia through receptors that are classically involved in immune responses underlies this stripping response. Inhibition of classical MHC class I molecule signalling led to an aberrant stripping response by microglia such that not only excitatory synapses were removed in response to injury but also a subclass of inhibitory synapses97.
Synaptic stripping could also be harmful. Emerging evidence suggests that the loss of synapses is an early hallmark of neurodegenerative diseases, including Alzheimer’s disease, glaucoma and prion disease85, 86, 91. Interestingly, components of the complement system, such as C1q, the C1 complex and activated C3, have been shown to localize to senile plaques in patients with Alzheimer’s disease but were not present in the brains of a control group of non-demented, elderly individuals. Moreover, activated microglia and astrocytes are present in the vicinity of these plaques, indicating that excessive synaptic stripping might be occurring at these locations. Early in the course of a disease process, it might be useful to strip away dying synapses so that healthy synapses can take over this synaptic territory. It has been postulated that C1q and C3, which may initially be protective, are differentially synthesized early in neurodegenerative diseases. However, as the insult to the CNS persists, the remainder of the components of the complement cascade are synthesized, resulting in synapse loss and cell death85 (Fig. 4). In agreement with this idea, C1q has been shown to be synthesized in response to injury or during the early stages of neurodegeneration in the adult CNS. For example, in the mouse model of glaucoma, C1q production was found to be upregulated, and the protein was localized to RGC synapses in the inner plexiform layer of the retina; however, this occurred only during the early stages of the disease, preceding the substantial synapse loss and eventual RGC loss that is observed in this model83. These findings suggest that C1q-dependent synapse loss may be driving the neurodegenerative process in glaucoma and other neurodegenerative disorders. This exciting finding could be further investigated by determining whether C1q deficiency is protective against neurodegenerative disease in animal models.
Future directions and outstanding questions
The exciting new findings highlighted here have vastly advanced the understanding of the cellular and molecular mechanisms that glia use to achieve correct synaptic connectivity in the nervous system. But many questions and mysteries remain unsolved. For example, even though the importance and diversity of astrocytes is now recognized, the full extent of their properties and the specific roles they have in the formation and function of the CNS are still unclear. Why are astrocytes tiled into non-overlapping anatomical domains? What are the functional consequences of localized and global changes in intracellular Ca2+ concentrations in these cells in response to synapse function? Could astrocytes and other glia actively influence neural circuits to control information processing? Do microglia have an active role in synapse elimination during development? Furthermore, the precise function of OPCs in the adult brain still needs to be uncovered, as well as the purpose and consequences of their direct synaptic contacts with neurons. The good news is that many of the technical difficulties that impeded glial research in the past are, to a great extent, solved. Recent advances in methods to carry out cell-type-specific genetic manipulations in invertebrate and vertebrate model organisms, as well as powerful in vivo imaging techniques, now allow the study and observation of glial function at synapses in great detail. Today, neuroscientists are in a better position than ever before for exploring and uncovering the long-standing mysteries of glia and for gaining new insight into the general workings of the nervous system.
Acknowledgments
We acknowledge all of our colleagues whose important work was not directly cited here because of space limitations. This work is referenced in the review articles cited here. C.E. is supported by the Alfred P. Sloan Foundation and a Klingenstein Fellowship Award in the Neurosciences, from the Esther A. & Joseph Klingenstein Fund.
References
- 1.Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 2.Chittajallu R, Aguirre A, Gallo V. NG2-positive cells in the mouse white and grey matter display distinct physiological properties. J Physiol (Lond) 2004;561:109–122. doi: 10.1113/jphysiol.2004.074252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin SC, Bergles DE. Physiological characteristics of NG2-expressing glial cells. J Neurocytol. 2002;31:537–549. doi: 10.1023/a:1025799816285. [DOI] [PubMed] [Google Scholar]
- 4.Eroglu C, Barres BA, Stevens B. In: Structural and Functional Organization of the Synapse. Hell JW, Ehlers MD, editors. Springer; 2008. pp. 683–714. [Google Scholar]
- 5.Feng Z, Ko CP. Neuronal glia interactions at the vertebrate neuromuscular junction. Curr Opin Pharmacol. 2007;7:316–324. doi: 10.1016/j.coph.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Bolton MM, Eroglu C. Look who is weaving the neural web: glial control of synapse formation. Curr Opin Neurobiol. 2009;19:491–497. doi: 10.1016/j.conb.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 7.Reichenbach A, Derouiche A, Kirchhoff F. Morphology and dynamics of perisynaptic glia. Brain Res Rev. 2010;63:11–25. doi: 10.1016/j.brainresrev.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Doherty J, Logan MA, Tasdemir OE, Freeman MR. Ensheathing glia function as phagocytes in the adult Drosophila brain. J Neurosci. 2009;29:4768–4781. doi: 10.1523/JNEUROSCI.5951-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dani JW, Chernjavsky A, Smith SJ. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron. 1992;8:429–440. doi: 10.1016/0896-6273(92)90271-e. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, et al. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nature Neurosci. 2006;9:816–823. doi: 10.1038/nn1703. This paper shows that the stimulation of whiskers increases the cytosolic Ca2+ concentration in astrocytes in the barrel cortex of adult mice. This is the first in vivoevidence that astrocytes respond to a presynaptic spillover of glutamate by increasing their cytosolic Ca2+ concentrations. [DOI] [PubMed] [Google Scholar]
- 11.Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- 12.Bergles DE, Roberts JD, Somogyi P, Jahr CE. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature. 2000;405:187–191. doi: 10.1038/35012083. This is the first report of glutamate-mediated synaptic contacts and fast synaptic transmission between pyramidal neurons and OPCs in the mammalian hippocampus. [DOI] [PubMed] [Google Scholar]
- 13.Lin SC, et al. Climbing fiber innervation of NG2-expressing glia in the mammalian cerebellum. Neuron. 2005;46:773–785. doi: 10.1016/j.neuron.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 14.De Biase LM, Nishiyama A, Bergles DE. Excitability and synaptic communication within the oligodendrocyte lineage. J Neurosci. 2010;30:3600–3611. doi: 10.1523/JNEUROSCI.6000-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin SC, Bergles DE. Synaptic signaling between GABAergic interneurons and oligodendrocyte precursor cells in the hippocampus. Nature Neurosci. 2004;7:24–32. doi: 10.1038/nn1162. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Barres BA. Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr Opin Neurobiol. 2010;20:588–594. doi: 10.1016/j.conb.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Oberheim NA, et al. Uniquely hominid features of adult human astrocytes. J Neurosci. 2009;29:3276–3287. doi: 10.1523/JNEUROSCI.4707-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG. Synaptic islands defined by the territory of a single astrocyte. J Neurosci. 2007;27:6473–6477. doi: 10.1523/JNEUROSCI.1419-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ventura R, Harris KM. Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci. 1999;19:6897–6906. doi: 10.1523/JNEUROSCI.19-16-06897.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grosche J, et al. Microdomains for neuron–glia interaction: parallel fiber signaling to Bergmann glial cells. Nature Neurosci. 1999;2:139–143. doi: 10.1038/5692. [DOI] [PubMed] [Google Scholar]
- 22.Witcher MR, Kirov SA, Harris KM. Plasticity of perisynaptic astroglia during synaptogenesis in the mature rat hippocampus. Glia. 2007;55:13–23. doi: 10.1002/glia.20415. [DOI] [PubMed] [Google Scholar]
- 23.Genoud C, et al. Plasticity of astrocytic coverage and glutamate transporter expression in adult mouse cortex. PLoS Biol. 2006;4:e343. doi: 10.1371/journal.pbio.0040343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirrlinger J, Hulsmann S, Kirchhoff F. Astroglial processes show spontaneous motility at active synaptic terminals in situ. Eur J Neurosci. 2004;20:2235–2239. doi: 10.1111/j.1460-9568.2004.03689.x. [DOI] [PubMed] [Google Scholar]
- 25.Theodosis DT, et al. Oxytocin and estrogen promote rapid formation of functional GABA synapses in the adult supraoptic nucleus. Mol Cell Neurosci. 2006;31:785–794. doi: 10.1016/j.mcn.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 26.Iino M, et al. Glia–synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science. 2001;292:926–929. doi: 10.1126/science.1058827. This paper shows that Ca2+-permeable AMPA receptors on glia are required for proper structural and functional relationships between Bergmann glia and glutamate-mediated synapses in the cerebellum. [DOI] [PubMed] [Google Scholar]
- 27.Haber M, Zhou L, Murai KK. Cooperative astrocyte and dendritic spine dynamics at hippocampal excitatory synapses. J Neurosci. 2006;26:8881–8891. doi: 10.1523/JNEUROSCI.1302-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishida H, Okabe S. Direct astrocytic contacts regulate local maturation of dendritic spines. J Neurosci. 2007;27:331–340. doi: 10.1523/JNEUROSCI.4466-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murai KK, Nguyen LN, Irie F, Yamaguchi Y, Pasquale EB. Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nature Neurosci. 2003;6:153–160. doi: 10.1038/nn994. [DOI] [PubMed] [Google Scholar]
- 30.Carmona MA, Murai KK, Wang L, Roberts AJ, Pasquale EB. Glial ephrin-A3 regulates hippocampal dendritic spine morphology and glutamate transport. Proc Natl Acad Sci USA. 2009;106:12524–12529. doi: 10.1073/pnas.0903328106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Filosa A, et al. Neuron–glia communication via EphA4/ephrin-A3 modulates LTP through glial glutamate transport. Nature Neurosci. 2009;12:1285–1292. doi: 10.1038/nn.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hatton GI. Function-related plasticity in hypothalamus. Annu Rev Neurosci. 1997;20:375–397. doi: 10.1146/annurev.neuro.20.1.375. [DOI] [PubMed] [Google Scholar]
- 33.Piet R, Poulain DA, Oliet SH. Modulation of synaptic transmission by astrocytes in the rat supraoptic nucleus. J Physiol (Paris) 2002;96:231–236. doi: 10.1016/s0928-4257(02)00010-4. [DOI] [PubMed] [Google Scholar]
- 34.Fox MA, Umemori H. Seeking long-term relationship: axon and target communicate to organize synaptic differentiation. J Neurochem. 2006;97:1215–1231. doi: 10.1111/j.1471-4159.2006.03834.x. [DOI] [PubMed] [Google Scholar]
- 35.Fields RD. Myelination: an overlooked mechanism of synaptic plasticity? Neuroscientist. 2005;11:528–531. doi: 10.1177/1073858405282304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muller CM, Best J. Ocular dominance plasticity in adult cat visual cortex after transplantation of cultured astrocytes. Nature. 1989;342:427–430. doi: 10.1038/342427a0. [DOI] [PubMed] [Google Scholar]
- 37.Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- 38.Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- 39.Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. References 38 and 39 show that synapse formation and function are powerfully affected by astrocytes in culture. This is the first report that these processes are not only controlled by neuronal mechanisms but can also be directed by glia. [DOI] [PubMed] [Google Scholar]
- 40.Wu H, et al. Integrative genomic and functional analyses reveal neuronal subtype differentiation bias in human embryonic stem cell lines. Proc Natl Acad Sci USA. 2007;104:13821–13826. doi: 10.1073/pnas.0706199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Christopherson KS, et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. Following on from the work by Ullian and colleagues39, this paper identifies the extracellular matrix proteins TSPs as the synaptogenic factors that are released by astrocytes. [DOI] [PubMed] [Google Scholar]
- 42.Eroglu C, et al. Gabapentin receptor α2δ-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. In this study, the receptor for TSPs that is involved in synapse formation was identified to be the Ca2+ channel subunit α2δ-1, which is also the receptor for the commonly prescribed pain medication gabapentin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rohrbough J, Grotewiel MS, Davis RL, Broadie K. Integrin-mediated regulation of synaptic morphology, transmission, and plasticity. J Neurosci. 2000;20:6868–6878. doi: 10.1523/JNEUROSCI.20-18-06868.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi Y, Ethell IM. Integrins control dendritic spine plasticity in hippocampal neurons through NMDA receptor and Ca2+/calmodulin-dependent protein kinase II-mediated actin reorganization. J Neurosci. 2006;26:1813–1822. doi: 10.1523/JNEUROSCI.4091-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu J, Xiao N, Xia J. Thrombospondin 1 accelerates synaptogenesis in hippocampal neurons through neuroligin 1. Nature Neurosci. 2010;13:22–24. doi: 10.1038/nn.2459. [DOI] [PubMed] [Google Scholar]
- 46.Taylor CP. Mechanisms of analgesia by gabapentin and pregabalin: calcium channel α2-δ [Cavα2-δ] ligands. Pain. 2009;142:13–16. doi: 10.1016/j.pain.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 47.Mauch DH, et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- 48.Goritz C, Mauch DH, Pfrieger FW. Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol Cell Neurosci. 2005;29:190–201. doi: 10.1016/j.mcn.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 49.Elmariah SB, Hughes EG, Oh EJ, Balice-Gordon RJ. Neurotrophin signaling among neurons and glia during formation of tripartite synapses. Neuron Glia Biol. 2005;1:1–11. doi: 10.1017/S1740925X05000189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hughes EG, Elmariah SB, Balice-Gordon RJ. Astrocyte secreted proteins selectively increase hippocampal GABAergic axon length, branching, and synaptogenesis. Mol Cell Neurosci. 2009;43:136–145. doi: 10.1016/j.mcn.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Feng Z, Ko CP. Schwann cells promote synaptogenesis at the neuromuscular junction via transforming growth factor-β1. J Neurosci. 2008;28:9599–9609. doi: 10.1523/JNEUROSCI.2589-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao G, Ko CP. Schwann cell-derived factors modulate synaptic activities at developing neuromuscular synapses. J Neurosci. 2007;27:6712–6722. doi: 10.1523/JNEUROSCI.1329-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arber S, Caroni P. Thrombospondin-4, an extracellular matrix protein expressed in the developing and adult nervous system, promotes neurite outgrowth. J Cell Biol. 1995;131:1083–1094. doi: 10.1083/jcb.131.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arikkath J, Campbell KP. Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Curr Opin Neurobiol. 2003;13:298–307. doi: 10.1016/s0959-4388(03)00066-7. [DOI] [PubMed] [Google Scholar]
- 55.Barker AJ, Koch SM, Reed J, Barres BA, Ullian EM. Developmental control of synaptic receptivity. J Neurosci. 2008;28:8150–8160. doi: 10.1523/JNEUROSCI.1744-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hama H, Hara C, Yamaguchi K, Miyawaki A. PKC signaling mediates global enhancement of excitatory synaptogenesis in neurons triggered by local contact with astrocytes. Neuron. 2004;41:405–415. doi: 10.1016/s0896-6273(04)00007-8. [DOI] [PubMed] [Google Scholar]
- 57.Taniguchi H, et al. Silencing of neuroligin function by postsynaptic neurexins. J Neurosci. 2007;27:2815–2824. doi: 10.1523/JNEUROSCI.0032-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cahoy JD, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garrett AM, Weiner JA. Control of CNS synapse development by γ-protocadherin-mediated astrocyte–neuron contact. J Neurosci. 2009;29:11723–11731. doi: 10.1523/JNEUROSCI.2818-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Colon-Ramos DA, Margeta MA, Shen K. Glia promote local synaptogenesis through UNC-6 (netrin) signaling in C. elegans. Science. 2007;318:103–106. doi: 10.1126/science.1143762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bacaj T, Tevlin M, Lu Y, Shaham S. Glia are essential for sensory organ function inC. elegans. Science. 2008;322:744–747. doi: 10.1126/science.1163074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kurshan PT, Oztan A, Schwarz TL. Presynaptic α2δ-3 is required for synaptic morphogenesis independent of its Ca2+-channel functions. Nature Neurosci. 2009;12:1415–1423. doi: 10.1038/nn.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caceres M, Suwyn C, Maddox M, Thomas JW, Preuss TM. Increased cortical expression of two synaptogenic thrombospondins in human brain evolution. Cereb Cortex. 2007;17:2312–2321. doi: 10.1093/cercor/bhl140. [DOI] [PubMed] [Google Scholar]
- 64.Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16:825–833. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- 65.Selig DK, Hjelmstad GO, Herron C, Nicoll RA, Malenka RC. Independent mechanisms for long-term depression of AMPA and NMDA responses. Neuron. 1995;15:417–426. doi: 10.1016/0896-6273(95)90045-4. [DOI] [PubMed] [Google Scholar]
- 66.Miller RF. D-Serine as a glial modulator of nerve cells. Glia. 2004;47:275–283. doi: 10.1002/glia.20073. [DOI] [PubMed] [Google Scholar]
- 67.Yang Y, et al. Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc Natl Acad Sci USA. 2003;100:15194–15199. doi: 10.1073/pnas.2431073100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Panatier A, et al. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 69.Beattie EC, et al. Control of synaptic strength by glial TNFα. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 70.Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-α. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- 71.Buckby LE, Jensen TP, Smith PJ, Empson RM. Network stability through homeostatic scaling of excitatory and inhibitory synapses following inactivity in CA3 of rat organotypic hippocampal slice cultures. Mol Cell Neurosci. 2006;31:805–816. doi: 10.1016/j.mcn.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 72.Freeman MR. Sculpting the nervous system: glial control of neuronal development. Curr Opin Neurobiol. 2006;16:119–125. doi: 10.1016/j.conb.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 73.Awasaki T, Ito K. Engulfing action of glial cells is required for programmed axon pruning during Drosophila metamorphosis. Curr Biol. 2004;14:668–677. doi: 10.1016/j.cub.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 74.Watts RJ, Schuldiner O, Perrino J, Larsen C, Luo L. Glia engulf degenerating axons during developmental axon pruning. Curr Biol. 2004;14:678–684. doi: 10.1016/j.cub.2004.03.035. References 73 and 74 are the first reports that the pruning of axons during Drosophila metamorphosis is mediated by their active engulfment by glia. [DOI] [PubMed] [Google Scholar]
- 75.MacDonald JM, et al. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron. 2006;50:869–881. doi: 10.1016/j.neuron.2006.04.028. [DOI] [PubMed] [Google Scholar]
- 76.Awasaki T, et al. Essential role of the apoptotic cell engulfment genes draper and ced-6 in programmed axon pruning during Drosophila metamorphosis. Neuron. 2006;50:855–867. doi: 10.1016/j.neuron.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 77.Kurant E, Axelrod S, Leaman D, Gaul U. Six-microns-under acts upstream of Draper in the glial phagocytosis of apoptotic neurons. Cell. 2008;133:498–509. doi: 10.1016/j.cell.2008.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ziegenfuss JS, et al. Draper-dependent glial phagocytic activity is mediated by Src and Syk family kinase signalling. Nature. 2008;453:935–939. doi: 10.1038/nature06901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bishop DL, Misgeld T, Walsh MK, Gan WB, Lichtman JW. Axon branch removal at developing synapses by axosome shedding. Neuron. 2004;44:651–661. doi: 10.1016/j.neuron.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 80.Song JW, et al. Lysosomal activity associated with developmental axon pruning. J Neurosci. 2008;28:8993–9001. doi: 10.1523/JNEUROSCI.0720-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fuentes-Medel Y, et al. Glia and muscle sculpt neuromuscular arbors by engulfing destabilized synaptic boutons and shed presynaptic debris. PLoS Biol. 2009;7:e1000184. doi: 10.1371/journal.pbio.1000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu HH, et al. Glial precursors clear sensory neuron corpses during development via Jedi-1, an engulfment receptor. Nature Neurosci. 2009;12:1534–1541. doi: 10.1038/nn.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stevens B, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. This study describes an unexpected role for the complement cascade in the elimination of synapses during development and provides evidence that the expression of complement proteins is upregulated and their synaptic localization increases during the early stages of glaucoma. [DOI] [PubMed] [Google Scholar]
- 84.Chu Y, et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci USA. 2010;107:7975–7980. doi: 10.1073/pnas.0913449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alexander JJ, Anderson AJ, Barnum SR, Stevens B, Tenner AJ. The complement cascade: Yin–Yang in neuroinflammation — neuro-protection and -degeneration. J Neurochem. 2008;107:1169–1187. doi: 10.1111/j.1471-4159.2008.05668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cullheim S, Thams S. The microglial networks of the brain and their role in neuronal network plasticity after lesion. Brain Res Rev. 2007;55:89–96. doi: 10.1016/j.brainresrev.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 87.Urade Y, Oberdick J, Molinar-Rode R, Morgan JI. Precerebellin is a cerebellum-specific protein with similarity to the globular domain of complement C1q B chain. Proc Natl Acad Sci USA. 1991;88:1069–1073. doi: 10.1073/pnas.88.3.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Iijima T, Miura E, Watanabe M, Yuzaki M. Distinct expression of C1q-like family mRNAs in mouse brain and biochemical characterization of their encoded proteins. Eur J Neurosci. 2010;31:1606–1615. doi: 10.1111/j.1460-9568.2010.07202.x. [DOI] [PubMed] [Google Scholar]
- 89.Matsuda K, et al. Cbln1 is a ligand for an orphan glutamate receptor δ2, a bidirectional synapse organizer. Science. 2010;328:363–368. doi: 10.1126/science.1185152. [DOI] [PubMed] [Google Scholar]
- 90.Koch SM, Ullian EM. Neuronal pentraxins mediate silent synapse conversion in the developing visual system. J Neurosci. 2010;30:5404–5414. doi: 10.1523/JNEUROSCI.4893-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Johnson EC, Morrison JC. Friend or foe? Resolving the impact of glial responses in glaucoma. J Glaucoma. 2009;18:341–353. doi: 10.1097/IJG.0b013e31818c6ef6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lin TN, et al. Differential regulation of thrombospondin-1 and thrombospondin-2 after focal cerebral ischemia/reperfusion. Stroke. 2003;34:177–186. doi: 10.1161/01.str.0000047100.84604.ba. [DOI] [PubMed] [Google Scholar]
- 93.Tran MD, Neary JT. Purinergic signaling induces thrombospondin-1 expression in astrocytes. Proc Natl Acad Sci USA. 2006;103:9321–9326. doi: 10.1073/pnas.0603146103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liauw J, et al. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab. 2008;28:1722–1732. doi: 10.1038/jcbfm.2008.65. [DOI] [PubMed] [Google Scholar]
- 95.Li CY, Song YH, Higuera ES, Luo ZD. Spinal dorsal horn calcium channel α2δ-1 subunit upregulation contributes to peripheral nerve injury-induced tactile allodynia. J Neurosci. 2004;24:8494–8499. doi: 10.1523/JNEUROSCI.2982-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Valder CR, Liu JJ, Song YH, Luo ZD. Coupling gene chip analyses and rat genetic variances in identifying potential target genes that may contribute to neuropathic allodynia development. J Neurochem. 2003;87:560–573. doi: 10.1046/j.1471-4159.2003.02016.x. [DOI] [PubMed] [Google Scholar]
- 97.Oliveira AL, et al. A role for MHC class I molecules in synaptic plasticity and regeneration of neurons after axotomy. Proc Natl Acad Sci USA. 2004;101:17843–17848. doi: 10.1073/pnas.0408154101. [DOI] [PMC free article] [PubMed] [Google Scholar]