

Abstract
Cyclin-G associated kinase (GAK) emerged as a promising drug target for the treatment of viral infections. However, no potent and selective GAK inhibitors have been reported in the literature to date. This paper describes the discovery of isothiazolo[5,4-b]pyridines as selective GAK inhibitors, with the most potent congeners displaying low nanomolar binding affinity for GAK. Co-crystallization experiments revealed that these compounds behaved as classic type I ATP-competitive kinase inhibitors. In addition, we have demonstrated that these compounds exhibit a potent activity against hepatitis C virus (HCV) by inhibiting two temporally distinct steps in the HCV lifecycle (i.e. viral entry and assembly). Hence, these GAK inhibitors represent chemical probes to study GAK function in different disease areas where GAK has been implicated (including viral infection, cancer and Parkinson's disease).
Introduction
Cyclin G associated kinase (GAK) was first identified in experiments investigating proteins associated with cyclin G, a protein involved in cell cycle regulation.1 GAK (also known as auxillin 2) is a 160 kDa serine/threonine protein kinase that belongs to the numb-associated kinase (NAK) family, which also includes STK16/MPSK1 (Serine/threonine kinase 16/myristoylated and palmitoylated serine/threonine kinase 1), AAK1 (adaptor-associated kinase) and BIKE (BMP-2 inducible kinase).2
GAK is expressed ubiquitously and bears a strong homology (43%) to the neuronal-specific protein auxilin, a heat shock cognate 70 (Hsc70) cochaperone with a role in uncoating clathrin vesicles. GAK is a key regulator of clathrin-mediated trafficking both in the endocytic and secretory pathways. It recruits clathrin and clathrin adaptor protein complex 2 (AP-2) to the plasma membrane3 and phosphorylates a T156 residue within AP2M1, the μ subunit of AP-2, thereby stimulating its binding to cargo proteins and enhancing cargo recruitment, vesicle assembly and efficient internalization.3,4,5,6 Moreover, GAK regulates endocytosis of receptors that is mediated by alternate clathrin adaptors3 and is implicated in later steps of endocytosis, including regulation of clathrin-coated vesicles (CCVs) uncoating, which enables recycling of clathrin back to the cell surface.3,5 GAK is an important regulator of Epidermal Growth Factor Receptor (EGFR); it is known to promote EGF uptake3 and may also function in receptor signaling.7 Last, GAK also plays an important role in regulating clathrin-mediated sorting events in the trans-Golgi network.3,5
Interestingly, GAK-dependent phosphorylation of clathrin adaptor proteins has been implicated in the regulation of viruses. AP2M1 was shown to be recruited to the surface of lipid droplets by the HCV capsid protein, core.8 The interaction between HCV core and AP2M1 was shown to be critical for HCV assembly.8 Notably, either overexpression of an AP2M1 phosphorylation-site mutant or suppression of GAK expression disrupted core-AP2M1 binding and HCV assembly.8 More recently, GAK was shown to regulate HCV entry independently of its effect on HCV assembly, in part by activating AP2M1.11 Hence, GAK represents a cellular host factor essential for regulation of HCV entry and assembly and a potential target for antiviral strategies. Indeed, erlotinib, an approved anticancer drug that potently inhibits GAK (in addition to its known cancer target, EGFR9,10) inhibits HCV entry as well as core-AP2M1 binding, thereby also disrupting HCV assembly, but not HCV RNA replication.8,11
To the best of our knowledge, no potent and selective GAK inhibitors have been reported in the literature to date. Like erlotinib, other approved kinase inhibitors, such as dasatinib, gefitinib, and pelitinib, display a high affinity for GAK with Kd values in the low nanomolar range (Chart 1).12 Similarly, pyridinyl imidazoles, such as SB203580 and SB201290 that have been developed as p38 inhibitors, potently inhibit GAK.13 Nevertheless, since all these compounds were designed to target other kinases, their inhibitory effect on GAK represents an off-target effect, and their use is limited by significant toxicities resulting from lack of selectivity. Moreover, while several compounds that bind GAK with an excellent ligand efficiency (LE) of 0.51 kcal/mol (Figure 1) were discovered by a fragment-based screening using weak affinity chromatography, their binding affinity was low (Kd value of 2 μM).14
Chart 1. Known GAK inhibitors.

Figure 1. Hit compound.

Because of the potential for GAK to serve as an antiviral drug target and the lack of selective small-molecule GAK inhibitors, we embarked on the synthesis and biological evaluation of a novel series of GAK inhibitors. In addition to their potential as lead molecules for the development of a novel antiviral strategy, these compounds represent useful chemical probes to further investigate the function of GAK in aspects of general cell biology and other disease conditions, such as cancer15 and Parkinson's disease,16 where GAK plays an important role.
Screening – Hit discovery
In order to discover novel GAK inhibitors, a drug-like compound library of 150 analogues was screened to identify potential ligands of GAK. This compound collection was made as part of a program to synthesize novel compound libraries based on original and patentable chemistry, which has not been explored before in drug discovery, and hence, represent unexplored chemical space. This library consists mainly of compounds based on a bicyclic, hetero-aromatic flat scaffold. The different scaffolds are unrelated to each other, and, in addition, they differ in their substitution pattern (the nature, as well as the spatial orientation of the substituents). These flat core structures are typical for kinase inhibitors, since they function as a central scaffold that binds to the ATP-binding site of the enzyme, and is therefore predicted to yield a high hit rate in kinase assays. We opted for the KINOMEscan™ screening platform that employs an active site-directed competition binding assay to quantitatively measure interactions between a test compound and a given kinase.12 Compounds that bind the kinase active site and either directly (sterically) or indirectly (allosterically) prevent kinase binding to the immobilized ligand reduce the amount of kinase captured on the solid support. Conversely, test molecules that do not bind the kinase have no effect on the amount of kinase captured on the solid support. Hits are identified by measuring the amount of kinase captured in test versus control samples using a quantitative, precise and sensitive qPCR method that detects the associated DNA label. In the first round of screening, the compound library was tested at a single concentration of 10 μM. The results are reported as the percentage of kinase/phage remaining bound to the ligands/beads, relative to a control. High affinity compounds have % of control values close to zero, while weaker binders have higher % control values. For the most promising compounds, dose-response curves were generated to determine binding constant (Kd) values. This initial screening led to the discovery of a promising hit (Figure 1), with a good potency (%Ctrl = 0.3; Kd = 0.3 μM), moderate lipophilicity (clogP = 2.44) and a favorable ligand efficiency (LE) of 0.494 kcal/mol. Overall, these parameters qualified this hit compound as an optimal starting point for the discovery of novel GAK inhibitors. Notably, this hit compound is based on an isothiazolo[4,3-b]pyridine scaffold, a skeleton that is unexplored in organic chemistry. This is in contrast to the bicyclic hetero-aromatics (such as purines, quinazolines, benzimidazoles and indoles), which are considered ‘privileged structures’ in kinase drug discovery and are widely described in scientific and patent literature, making novelty difficult to achieve. Indeed, SciFinder searches revealed only 19 known isothiazolo[4,3-b]pyridine derivatives,17 and, hence, further elaboration of this scaffold gives access to a largely unexplored chemical space.
Chemistry
The general approach for the synthesis of the isothiazolo[4,3-b]pyridine derivatives is shown in Scheme 1. The synthesis started from 3-nitro-5-bromopyridine-2-carbonitrile 1. Reduction of the nitro group to the corresponding amino group was achieved by treatment with iron under acidic conditions.18 The major compound was the desired compound 2, however, it was always accompanied by formation of the corresponding carboxamide, resulting from acidic hydrolysis of the cyano group. This mixture was used as such in the subsequent reaction. The aromatic thioamide 3 was obtained by treatment of the mixture with phosphorus pentasulfide as thionation reagent.19 An oxidative ring closure using hydrogen peroxide yielded the 3-amino-6-bromo-isothiazolo[4,3-b]pyridine 4.20 Subsequent palladium-catalyzed Suzuki coupling21 with appropriate arylboronic acids afforded a series of 3-amino-6-aryl-isothiazolo[4,3-b]pyridines 5a-e. Alternatively, diazotation of the exocyclic amino group with sodium nitrite, hydrogen bromide and CuBr, furnished the 3,6-dibromo-isothiazolo[4,3-b]pyridine 6.20 Treatment of 6 with sodium methoxide or nitrogen-containing nucleophiles yielded a series of 3-substituted-6-bromo-isothiazolo[4,3-b]pyridine analogues 7. Finally, reaction of 7 with a number of arylboronic acids then yielded target compounds 8-13. These Suzuki reactions were either conducted in a mixture of dioxane/water, using sodium carbonate as base and Pd(dppf)Cl2 as a catalyst, or alternatively, in a mixture of dimethoxyethane/water with potassium carbonate as a base and Pd(PPh3)4 as a catalyst.
Scheme 1. Synthesis of isothiazolo[4,3-b]pyridine analogues.

Reagents and conditions: a) Fe, CH3COOH, 0°C to rt; b) P2S5, EtOH, 75°C; c) 30% aq. H2O2, CH3OH, rt; d) R6B(OH)2, Na2CO3, Pd(dppf)Cl2, dioxane/water, 100 °C; e) CuBr, HBr, NaNO2,H2O, 0°C to rt; f) R3H, EtOH, 75°C; g) R6B(OH)2, Na2CO3, Pd(dppf)Cl2, dioxane/water, 100 °C or R6B(OH)2, K2CO3, Pd(PPh3)4, H2O, DME.
Structure-activity relationship studies
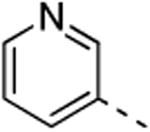
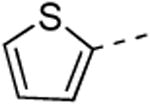
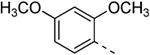
The isothiazolo[4,3-b]pyridine scaffold offers two sites for structural variation. Therefore, the structure-activity relationship (SAR) study started with the synthesis of a small library matrix of isothiazolo[4,3-b]pyridines with structural variation at positions 3 (amino, methoxy, ethanolamine, morpholine, and N-Me-piperazine) and 6 (substituted aryl and heteroaryl groups). Similarly to our primary screening strategy, all compounds were tested at a 10 μM concentration and the most promising ones were then subjected to determination of Kd values. The matrix depicted in Table 1, demonstrates that compounds 9c and 9d, resulting from the combination of a 3,4-dimethoxyphenyl or a 3-thienyl substituent at position 6 with an ethanolamino or a morpholino moiety at position 3 have a strong affinity for GAK (Kd values of 52 nM and 42 nM, respectively).
Table 1.
GAK affinity data of an isothiazolo[4,3-b]pyridine library.
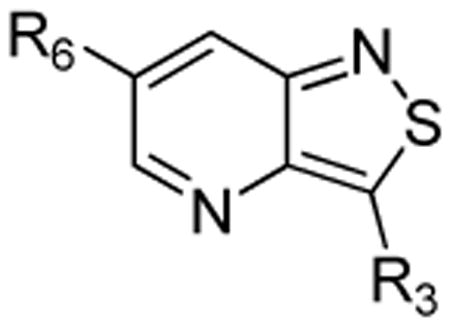
| |||||
|---|---|---|---|---|---|
| R3 | ---NH2 |
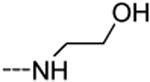
|
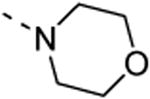
|
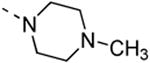
|
- -OCH3 |
| R6 | |||||
|
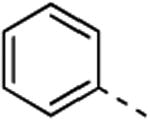
5a; %Ctrl:68 | 8a; %Ctrl:18 | 9a; %Ctrl:0,8; Kd = 0,5 μM | 10a; %Ctrl:76 | 11a; %Ctrl:18 |
|
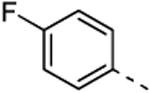
5b; %Ctrl:55 | 8b; %Ctrl:2.2 | 9b; %Ctrl:11 | 10b; %Ctrl:72 | 11b; %Ctrl:37 |
|
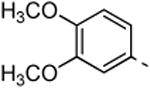
5c; %Ctrl:70 | 8c; %Ctrl:0.4; Kd = 0,12 μM | 9c; %Ctrl:0; Kd= 0,052 μM | 10c; % Ctrl:19 | 11c; %Ctrl:19 |
|
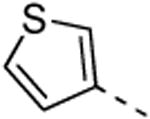
5d; %Ctrl:61 | 8d; % Ctrl 0,3; Kd = 0,3 μM | 9d; %Ctrl:0.1; Kd= 0,042 μM | 10d; %Ctrl:7.4 | 11d; %Ctrl:22 |
|
5e; %Ctrl:80 | 8e; % Ctrl:45 | 9e; %Ctrl:25 | 10e; %Ctrl:88 | 11e; %Ctrl: 93 |
Values represent the average of two independent experiments.
A dual strategy was then used for further optimization. To probe the optimal substitution pattern at position 6, the morpholino substituent at position 3 was fixed, and a wide range of substituted aryl or heteroaryl groups was evaluated. As it became clear that a 3,4-dimethoxyphenyl or a 3-thienyl substituents were optimal for GAK binding, the SAR focused on closely related derivatives of both of these aryl groups (Table 2).
Table 2.
SAR of the aryl moiety.
| Cpmd # | Structure | % Ctrl @10 μMa | Kd (μM)a |
|---|---|---|---|
| 12a |
|
0.05 | 0.047 |
| 12b |
|
0.15 | 0.2 |
| 12c |
|
6.3 | ND |
| 12d |
|
0.7 | 0.13 |
| 12e |
|
0.2 | 0.072 |
| 12f |
|
51 | ND |
| 12g |
|
0 | 0.0083 |
| 12h |
|
0 | 0.018 |
| 12i |
|
0 | 0.0089 |
| 12j |
|
0 | 0.018 |
Values represent the average of two independent experiments. ND = not determined.
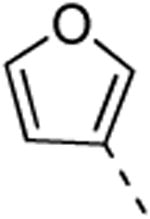
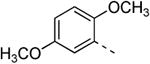
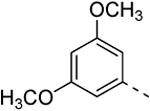
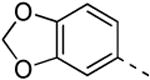
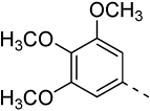
Replacing the 3-thienyl group of compound 9d by a 2-thienyl group (compound 12a) led to equipotent compounds. On the other hand, the presence of a 2-furanyl moiety led to a 5-fold decrease in GAK affinity, yielding compound 12b with a Kd value of 0.2 μM. The SAR of the dimethoxyphenyl ring was investigated by the synthesis of a number of regio-isomeric dimethoxyphenyl derivatives. Compared to the original 3,4-dimethoxyphenyl analogue 9c (Kd = 0.052 μM), the 2,4-dimethoxyphenyl analogue (compound 12c) lacks any affinity for the GAK enzyme, the 2,5-dimethoxyphenyl derivative 12d shows intermediate affinity (Kd = 0.13 μM), whereas the 3,5-dimethoxyphenyl derivative 12e demonstrates a similar GAK affinity (Kd = 0.072 μM). The dioxolane analogue 12f (a ring-closed analogue of the 3,4-dimethoxyphenyl moiety) is completely inactive. The insertion of an additional methoxy group yielded the 3,4,5-trimethoxyphenyl analogue 12g, displaying very strong binding affinity for the GAK enzyme (Kd = 0.0083 μM). In another round of SAR, the 3-methoxy group was kept intact and the 4-methoxy moiety was replaced by an ester moiety (compound 12h), an amino group (compound 12i) and a hydroxyl function (compound 12j), generating compounds with a strong affinity for the GAK enzyme, displaying Kd values of 0.018 μM, 0.0089 μM and 0.018 μM, respectively.
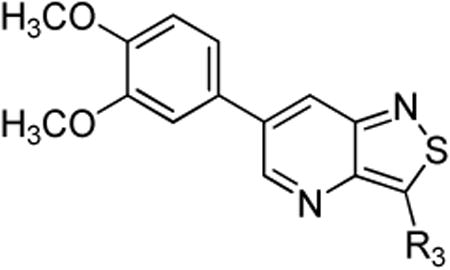
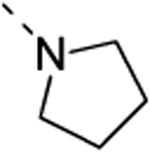
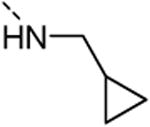
Since the first library screening demonstrated that a 3,4-dimethoxyphenyl moiety at position 6 was favorable for GAK binding, this substituent was kept intact, and a variety of amines were introduced at position 3 (Table 3). Although a free amino group at position 3 of the isothiazolo[5,4-b]pyridine scaffold is not tolerated for GAK binding (Table 1, compound 5c), the presence of small aliphatic amines (primary as well as secondary), such as dimethylamino (compound 13a), methoxyethylamino (compound 13b), diethanolamino (13c), gives rise to compounds displaying Kd values in the range of 0.14-0.3 μM. Cyclo-aliphatic amines, such as a pyrrolidino (compound 13d) and a cyclopropylmethylamino (compound 13e) yielded compounds with a higher potency, with Kd values of 100 nM or 27 nM, for compounds 13d and 13e, respectively.
Table 3.
SAR of the amine moiety.
| |||
|---|---|---|---|
|
| |||
| Cmpd# | R3 | % Ctrl (10 μM)a | Kd (μM)a |
| 13a |
|
0.1 | 0.19 |
| 13b |
|
0.1 | 0.14 |
| 13c |
|
4 | 0.32 |
| 13d |
|
0 | 0.1 |
| 13e |
|
0 | 0.027 |
| 13f |
|
2.6 | 0.27 |
| 13g |
|
0.8 | 0.23 |
| 13h |
|
0.1 | 0.11 |
| 13i |
|
0.1 | 0.063 |
| 13j |
|
0.1 | 0.088 |
| 13k |
|
81 | ND |
Values represent the average of two independent experiments. ND = not determined
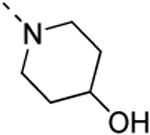
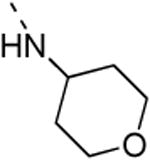
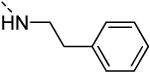
The isosteric replacement of the oxygen of the morpholine moiety by a carbon (resulting in compound 13f) or a sulfur (resulting in compound 13g) leads to a 5-fold drop in GAK affinity, when compared to the corresponding morpholine analogue 9c. To better mimic the morpholine substituent, a 4-hydroxy-piperidino and a 4-amino-pyrane moiety were introduced, giving rise to isothiazolo[4,3-b]pyridines 13i and 13j, both exhibiting a strong affinity for the GAK enzyme (Kd values of 63 nM and 88 nM, respectively). Overall, the data in Table 3 indicate quite high tolerance for structural variation at position 3, with only an aromatic phenethylamino group (compound 13k) completely lacking GAK affinity.
Although improving binding affinity is important in an optimization campaign, an overemphasis on potency has often resulted in molecules with unsuitable physicochemical properties for further development for in vivo use. We therefore complemented our lead optimization by monitoring a number of in silico parameters (Table 4). The optimized compounds had a low-molecular weight, which in combination with their potent binding affinity, yielded very ‘efficient’ LE values exceeding 0.4 kcal/mol. Moreover, the optimization campaign did not result in any gain in lipophilicity, as the most potent GAK ligands, 12g and 12i, displayed cLogP values of 2.49 and 2.28, respectively.
Table 4. In silico parameters of hit and optimized compounds.
| Cmpd # | MW | Kd (μM) | LE (kcal/mol) | cLogP |
|---|---|---|---|---|
| 6d (Hit) | 277.37 | 0.3 | 0.494 | 2.44 |
| 12g | 387.45 | 0.0083 | 0.408 | 2.49 |
| 12i | 342.42 | 0.0089 | 0.458 | 2.28 |
Selectivity profiling
Most kinase inhibitors are ATP-competitive and bind to the ATP domain, which is highly conserved across the kinome. Selectivity screens usually involve close analogues of the target kinase harboring similar ATP binding sites. However, we opted for a kinome-wide selectivity screen, using a panel of 456 kinases available at DiscoverX. As a representative example, 12g, the most potent congener, was selected for the selectivity profiling at a single concentration of 10 μM. Remarkably, no affinity of 12g was observed for any of the other members of the NAK family (AAK1: 90%; STK16: 92%; BIKE: 92% compared to the DMSO control) (Figure 2). Moreover, only 7 additional kinases appeared to interact with 12g, with a binding activity of less than 10%, when compared to DMSO control. The exact Kd values for these kinases were determined (Table 5). This large scale kinase profiling confirms that the identified lead compound is selective. Compound 12g displays the strongest affinity for GAK (Kd = 8.3 nM). A 3-fold level of selectivity is observed when compared with the affinity for KIT (Kd = 29 nM), whereas for the other kinases (CLK2, CSF1R, FLT3, MEK5, PDGFRA and PDGFRB), at least a 8-fold level of selectivity is achieved.
Figure 2. Specificity profile of compound 12g.
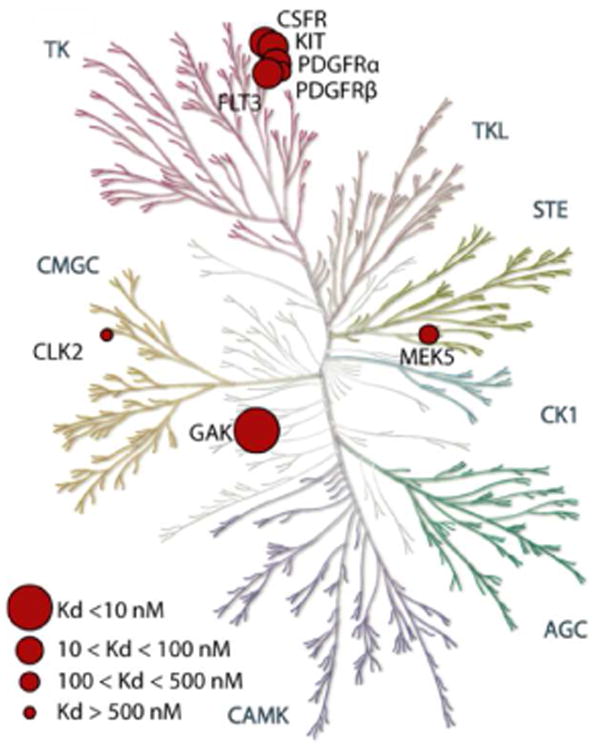
Table 5.
Kd values for off-target kinases affected by 12g.
| Kinase | Kd (nM)a |
|---|---|
| GAK | 8.3 |
| CLK2 | 710 |
| CSF1R | 320 |
| FLT3 | 110 |
| KIT | 29 |
| MEK5 | 150 |
| PDGFRA | 220 |
| PDGFRB | 70 |
Values represent the average of two independent experiments.
X-ray crystallography
To shed a light on the binding mode of the compounds to GAK, co-crystallization experiments were initiated. We have recently solved the structure of GAK in complex with a single chain antibody (nanobody), which resulted in reproducible crystallization conditions for the GAK kinase domain.22 The nanobody bound distal to the ATP binding site interacting with the lower kinase lobe (Figure 3). The lead compound 12i was co-crystallized and bound as expected to the ATP binding site of GAK. The binding mode of the inhibitor was well defined by electron density and the structure was refined to 2.1 Å resolution. Data collection and refinement statistics are provided in supplemental Table 2 (see Supporting Information). Two kinase domain:nanobody complexes were present in the asymmetric unit that interacted via the extended activation loops of GAK as described previously.22 The upper lobe of the kinase domain was flexible as indicated by high B-factors in particular in the N-terminal region (Supplemental Figure 1). The helix αC and the DFG motive were in an active conformation compatible with the type I binding mode of the inhibitor (Figure 3). The nitrogen of the isothiazolo moiety of the inhibitor formed a hydrogen bond with the backbone amide of Cys126. Interactions with small gatekeepers, which are found in only a small subset of kinases, have been explored for the development of selective kinase inhibitors.23 Therefore, our observation of this contact in the case of 12i may explain its excellent selectivity for GAK. The 4-amino-3-methoxy-phenyl substituent of 12i is oriented towards the solvent exposed area forming a number of hydrophobic contacts, in particular with Leu46 and a long range polar interaction with Arg44. Overall, 12i exhibits a good shape complementarity with the GAK ATP binding site.
Figure 3.
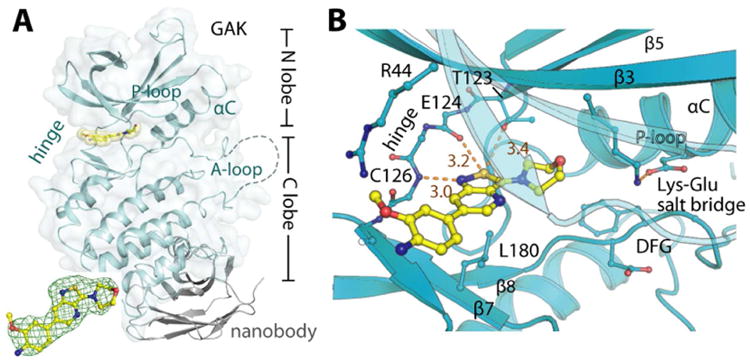
Structure of GAK in complex with 12i. A) Overview of GAK:NbGAK_4 complex with inhibitor 12i bound. The inset shows |FO| - |FC| omitted map contoured at 3σ for the bound inhibitor. B) Detailed interactions of 12i within the GAK ATP binding site.
Antiviral activity
We have recently shown that GAK is a regulator of HCV entry and assembly and therefore represents a potential target for anti-HCV treatment. Moreover, erlotinib, an approved anticancer drug known to target GAK significantly inhibited binding of HCV core to AP2M1, HCV entry, and assembly.8,11 Nevertheless, while erlotinib binds GAK with a high affinity (Kd = 3.4 nM), it binds EGFR, its primary anticancer target, with a comparable affinity (Kd = 1 nM), and several other kinases (albeit at a lower affinity).9,10 The use of erlotinib as a chemical tool to probe the role of GAK in HCV infection is therefore somewhat limited, particularly, since EGFR has also been recognized as an essential host factor for HCV infection.24 Moreover, the limited selectivity contributes to erlotinib's side effects, which may reduce its potential as an antiviral agent, particularly in long duration regimens as those required for the treatment of HCV. We therefore hypothesized that since the isothiazolo[4,3-b]pyridines 12g and 12i are structurally unrelated to erlotinib and lack anti-EGFR activity, they represent attractive chemical tools to study the role of GAK in HCV infection, and that their strong potency, promising selectivity profile and favorable drug-like properties make them a potential novel class of antiviral agents.
To further validate GAK as an antiviral target and determine the antiviral effect of these compounds on HCV infection, Huh-7.5 (human hepatoma) cells were infected with cell culture grown J6/JFH(p7-Rluc2A) HCV (HCVcc), a Renilla luciferase-containing reporter virus that replicates and produces high viral titers in Huh-7.5 cells.25 Infected cells were treated with various concentrations of 12g, 12i, or DMSO. Drug-containing media was replenished every 24 hours. Antiviral activity and cellular viability were measured by luciferase and alamarBlue-based assays, respectively, 72 hours postinfection. As shown in Figure 4A, treatment with either 12g or 12i resulted in a dose-dependent inhibition of viral replication. Half maximal effective concentration (EC50) values were 2.55±0.43μM (p=0.0002) and 2.81±0.8μM (p=0.009) for 12g and 12i, respectively. The half maximal cytotoxic concentration (CC50) values were 23.27±3.4 μM (p=0.000012) for 12g and 8.92±1.43 μM (p= 0.000044) for 12i.
Figure 4.
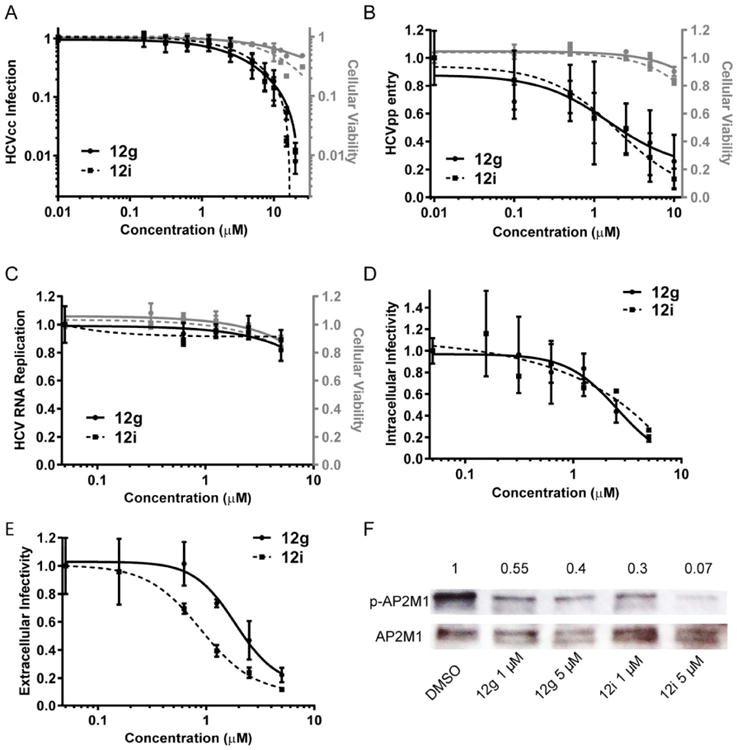
Selective GAK inhibitors inhibit HCV infection and AP2M1 phosphorylation. Dose response curves of 12g and 12i effects on infection of Huh-7.5 cells with cell culture grown HCV (HCVcc) (A), entry of pseudoparticles of HCV (HCVpp) (B), HCV RNA replication 72 hours postelectroporation with J6/JFH(p7-Rluc2A) RNA (C), and intra- (D) and extra-cellular (E) infectivity in naive cells inoculated with cell lysates or supernatants derived from the electroporated cells, respectively. Plotted in black (left y-axes) are relative luciferase values normalized to DMSO treated controls. Corresponding cellular viability, as measured by alamarBlue-based assays, are plotted in grey (right y-axes). Data reflect means and s.d. (error bars). F. The effect of the inhibitors on AP2M1 phosphorylation by Western analysis in lysates derived from Huh-7.5 cells. A representative membrane blotted with anti-phospho-AP2M1 (p-AP2M1) and anti-AP2M1 antibodies is shown. Numbers represent relative p-AP2M1/total AP2M1 protein ratio normalized to DMSO controls.
Next, we sought to pinpoint the step of the viral lifecycle that is disrupted by these GAK inhibitors. To study the effect of the GAK inhibitors on HCV entry, we measured the entry of pseudoparticles of HCV (HCVpp) (lentiviral vectors that incorporate the HCV glycoproteins on the viral envelope26) upon a 4 hour treatment with various concentrations of 12g and 12i using luciferase reporter-based assays. Huh-7.5 cells were infected with HCVpp for 1 hour on ice followed by a temperature shift to 37°C, 4 hour treatment with various concentrations of the compounds or DMSO, and medium replacement for removal of residual drugs and unbound virus. These compounds inhibited HCVpp entry in a dose-dependent manner, with half maximal effective concentrations (EC50's) of 3.6±1.2μM (p=0.03) and 2.05±0.36μM (p=0.0025), respectively, and a minimal effect on cellular viability (Figure 4B).
To determine the effect of the compounds on later steps of the HCV lifecycle and distinguish between a defect in viral RNA replication, assembly and/or release, Huh-7.5 cells were electroporated with in vitro transcribed luciferase reporter J6/JFH(p7-Rluc2A) HCV RNA24 and treated daily for 72 hours with either 12g, 12i or DMSO. HCV RNA replication was measured by luciferase assays 72 hours postelectroporation and intra- and extra-cellular infectivity were measured by luciferase assays in naïve cells inoculated with either clarified cell lysates or supernatants derived from the electroporated cells, respectively. Treatment with 12g and 12i did not affect HCV RNA replication (Figure 4C). Nevertheless, 12g and 12i demonstrated a dose-dependent effect on intracellular infectivity (Figure 4D), with EC50 values of 1.64±0.272 μM (p=0.009) and 2.43±0.72 μM (p=0.0281), respectively. Moreover, 12g and 12i significantly inhibited extracellular infectivity (Figure 4E), with EC50 values of 2.13±0.72 μM (p=0.04) and 2.47±0.9 μM (p=0.04), respectively. These data indicate that the two selective GAK inhibitors tested disrupt HCV assembly and infectious virus production without affecting HCV RNA replication.
Similar to small interfering RNAs (siRNAs) that target GAK as well as erlotinib,8,11 these compounds thus inhibit two temporally distinct steps in the HCV lifecycle: entry and assembly. The EC50 values for the antiviral effect of these compounds range from ∼1.5-3 μM, depending on the assay used. These EC50 values are slightly higher or comparable to those of erlotinib (0.5-1.5 μM)8,11 likely reflecting the absence of EGFR inhibition exhibited by erlotinib.
Taken together, these results provide a pharmacological validation of the requirement for GAK in the regulation of HCV entry and assembly. Moreover, the selective GAK inhibitors represent candidate compounds to target two steps of the HCV lifecycle: viral entry and assembly (Figure 5).
Figure 5.
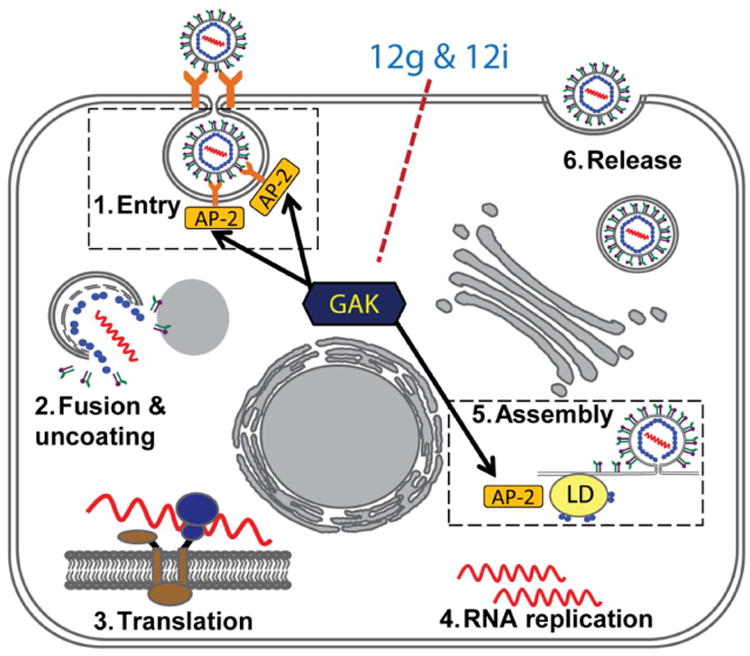
The proposed model for the mechanism by which the selective GAK inhibitors, 12g and 12i inhibit HCV infection. By directly inhibiting GAK kinase activity, 12g and 12i interfere with clathrin/AP-2-mediated endocytosis of HCV (step 1) as well as AP-2 binding to core in HCV assembly (step 5).
To validate the mechanism of action of these compounds, we studied their effect on AP2M1 phosphorylation. Huh-7.5 cells were treated with various concentrations of the compounds or DMSO. Since AP2M1 phosphorylation is transient (due to the activity of the phosphatase PP2A)27, to allow capturing of the phosphorylated AP2M1 state, these cells were incubated for 1 hour in the presence of the PP2A inhibitor, calyculin A, prior to lysis. The ratio of phosphorylated AP2M1 (p-AP2M1) to total AP2M1 was measured by quantitating Western blot band intensity. p-AP2M1 to AP2M1 ratios were reduced by either 12g or 12i in a dose-dependent manner and were significantly lower than the ratios measured in the DMSO control (Fig. 4F). These results indicate that 12g and 12i modulate AP2M1 phosphorylation, as predicted for compounds that target GAK.
Conclusion
We have previously reported that GAK is an essential host factor for HCV entry and assembly and that erlotinib, an approved anticancer drug with inhibitory activity against GAK, effectively disrupts HCV entry and assembly.8,11 Here, for the first time, we report on a specific optimization campaign towards the identification of novel GAK inhibitors. This endeavor resulted in the identification of novel and highly selective GAK inhibitors, based on an isothiazolo[4,3-b]pyridine scaffold, an unexplored chemical space. Their drug-like properties, as well as their selectivity profile, make these compounds an attractive tool to study regulatory mechanisms of intracellular traffic in cell biology and disease conditions, where GAK is known to be implicated. We used these compounds to better understand the role of GAK in HCV infection and further validate this host factor as an antiviral target. We have demonstrated that compounds 12g and 12i exhibit a potent in vitro anti-HCV activity and that their antiviral effect correlates with a decrease in AP2M1 phosphorylation. Although to the best of our knowledge, KIT has not been reported to have a role in HCV infection, it is bound by these compounds, albeit at a lower affinity (∼30nM). We therefore cannot exclude the possibility that inhibition of KIT or other cellular kinases, in addition to GAK, contributes to the antiviral effect of these compounds.
Unlike approved direct acting antivirals (DAA), which target HCV RNA replication, these compounds inhibit two distinct steps of the HCV lifecycle: entry and assembly. By displaying limited off-target binding to other kinases, these more selective GAK inhibitors have the potential to reduce drug toxicity. Moreover, inhibiting a host rather than a viral target is more likely to provide a higher genetic barrier for drug resistance than most DAAs and a comprehensive protection against all HCV genotypes. Hence, selective GAK inhibitors may potentially represent a novel class of antivirals for inclusion in future combination drug regimens for treating HCV. Last, since multiple other viruses hijack clathrin adaptor proteins-mediated pathways either for viral entry or late steps of their viral lifecycle,28,29,30 we predict that the requirement for GAK is broadly conserved among RNA viruses, and therefore that selective GAK inhibitors may display a broad-spectrum antiviral activity.
Experimental section
General
For all reactions, analytical grade solvents were used. All moisture-sensitive reactions were carried out in oven-dried glass-ware (135 °C). 1H and 13C NMR spectra: Bruker Avance 300 Mhz (1H-NMR: 300 MHz, 13C-NMR: 75 MHz), 500 MHz (1H-NMR: 500 MHz, 13C-NMR: 125 MHz) and 600 MHz (1H-NMR: 600 MHz, 13C-NMR: 150 MHz) using tetramethylsilane as internal standard for 1H-NMR spectra and (D6)-DMSO (39.5 ppm) or CDCl3 (77.2 ppm) and CD3OD (49.0 ppm) for 13C-NMR spectra. Abbreviations used are: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. Coupling constants are expressed in Hz. Mass spectra are obtained with a Finnigan LCQ Advantage Max (ion trap) mass spectrophotometer from Thermo Finnigan, San Jose, CA, USA. High resolution mass spectrometry spectra were acquired on a quadrupole orthogonal acceleration time-of-flight mass spectrometer (Synapt G2 HDMS, Waters, Milford, MA). Samples were infused at 3 μL/min and spectra were obtained in positive (or negative) ionization mode with a resolution of 15000 (FWHM) using leucine enkephalin as lock mass. Precoated aluminum sheets (Fluka Silica gel/TLC-cards, 254 nm) were used for TLC. Column chromatography (CC) was performed on ICN silica gel 63-200, 60 Å. Purity of final compounds was determined by analytical RP-HPLC analysis on a XBridge column (C-18, 5 μm, 4.6 mm × 150 mm) in combination with a Waters 600 HPLC system and a Waters 2996 photodiode array detector from Waters, Milford, Massachusetts, USA. Elution was done using a gradient mixture of H2O containing 0.2% (vol) of TFA (A) and Acetonitrile (B) (Supporting information). All compounds were at least 95% pure, with the exception of compound 11b (90.32% purity).
3-Amino-5-bromopyridine-2-carbonitrile (2)
A solution of iron powder (3.36 g, 60 mmol) in acetic acid (15 ml) was stirred at 0 °C. To this solution was added dropwise a solution of 3-nitro-5-bromopyridine-2-carbonitrile (2.51 g, 11 mmol) In acetic acid (15 ml). The reaction mixture was stirred at room temperature for two hours. Then, ethyl acetate (300 ml) was added and the mixture was filtered (paper filter). The filter cake was washed with ethylacetate. The filtrate was evaporated and partitioned between ethylacetate (500 ml) and water (250 ml). The organic phase was washed with a 1 N NaOH solution (ca. 200 ml). The combined organic phases were dried and evaporated in vacuo, yielding a mixture of two compounds, i.e. 3-amino-5-bromopyridine-2-carbonitrile (major compound) and 3-amino-5-bromopyridine-2-carboxamide (minor compound). This mixture was used as such in the next reaction.
3-Amino-5-bromo-2-pyridinecarbothioamide (3)
To a solution of a mixture of 3-amino-5-bromopyridine-2-carbonitrile and 3-amino-5-bromopyridine-2-carboxamide (crude) in ethanol (25 ml) was added phosphorus pentasulfide (2 eq; 4.84 g). The mixture was heated overnight at 75°C. The solvents were evaporated and the crude residue was purified by flash chromatography on silica, using a mixture of cyclohexane/ethyl acetate (in a ratio of 7:1) as mobile phase, yielding the title compound (3.36 gram crude). 1H-NMR (300 MHz, CDCl3): δ = 6.95 (bs, 2H, NH2), 7.27 (d, J = 1.95 Hz, 1H, arom H), 7.89 (d, J = 1.95 Hz, 1H, arom H), 9.37 (bs, 2H, NH2) ppm.
3-amino-6-bromo-isothiazolo[4,3-b]pyridine (4)
To a solution of 3-amino-5-bromo-2-pyridinecarbothioamide in methanol (50 ml) was added dropwise a 30% H2O2 solution in water (3.5 ml) at 0°C. The reaction mixture was stirred overnight at room temperature and then cooled again to 0°C. The crystals were filtered off and washed with cold methanol yielding the title compound (1.6 g, 63%). 1H-NMR (300 MHz, DMSO-d6): δ = 8.03 (d, J = 2.01 Hz, 1H, arom H), 8.08 (bs, 2H, NH2), 8.29 (d, J = 2.01 Hz, 1H, arom H) ppm. 13C-NMR (75 MHz, DMSO-d6): δ = 120.56 (Cq), 129.39 (CH), 133.23 (Cq), 143.69 (Cq), 153.58 (CH), 173.34 (Cq) ppm.
Synthesis of 3-amino-6-aryl-isothiazolo[4,3-b]pyridines (5a-e)
General procedure
To a solution of 3-amino-6-bromo-isothiazolo[4,3-b]pyridine (166 mg, 0.72 mmol) in a mixture of dioxane (10 ml) and water (1.5 ml) was added an appropriate boronic acid (2 eq), sodium carbonate (2 eq, 153 mg) and Pd(dppf)Cl2 (0.1 eq, 59 mg) The reaction mixture was stirred overnight at 100 °C. The reaction mixture was cooled down to room temperature and the reaction was partioned between ethylacetate (60 ml) and brine (30 ml). The aqueous phase was then extracted with ethyl acetate (40 ml). The combined organic phases were dried over MgSO4 and evaporated in vacuo. The residue was purified by flash chromatography on silicagel yielding the pure title compounds.
The following compounds were made according to this procedure:
3-Amino-6-phenyl-isothiazolo[4,3-b]pyridine (5a)
This compound was obtained using phenylboronic acid and the crude residue was purified by flash chromatography using a mixture of cyclohexane and ethylacetate (in a ratio of 4:1) as mobile phase, affording the title compound in 77% yield (125 mg, 0.55 mmol). 1H-NMR (300 MHz, CDCl3): δ = 5.84 (s, 2H, NH2), 7.52 (m, 3H, arom H), 7.66 (m, 2H, arom H), 7.95 (d, J = 1.98 Hz, 1H, arom H), 8.68 (d, J = 1.95 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, DMSO-d6): δ = 124.37 (CH), 127.38 (CH), 128.40 (CH), 129.18 (CH), 134.06 (Cq), 135.37 (Cq), 137.25 (Cq), 143.27 (CH), 153.67 (Cq), 172.38 (Cq) ppm. HR-MS [M+H]+ found 228.0594, calculated 228.0551.
3-Amino-6-(4-fluorophenyl)-isothiazolo[4,3-b]pyridine (5b)
This compound was obtained using 4-fluorophenylboronic acid and the crude residue was purified by flash chromatography using a mixture of cyclohexane and ethylacetate (in a ratio of 4:1) as mobile phase, affording the title compound in 76% yield (134 mg, 0.54 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 6.36 (br s, 2H, NH2), 7.34 (m, 2H, arom H), 7.38 (d, J = 3.1 Hz, 1H, arom H), 7.71 (m, 2H, arom H), 8.17 (d, J = 1.92 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, DMSO-d6): δ = 115.94 (CH), 116.23 (CH), 124.43 (CH), 129.53 (CH), 129.65 (CH), 133.76 (Cq), 134.43 (Cq), 143.19 (CH), 153.65 (Cq), 160.88 (Cq), 164.24 (Cq), 172.49 (Cq) ppm. HR-MS [M+H]+ found 246.0497, calculated 243.0457.
3-Amino-6-(3,4-dimethoxyphenyl)-isothiazolo[4,3-b]pyridine (5c)
This compound was obtained using 3,4-dimethoxyphenylboronic acid and the crude residue was purified by flash chromatography using a mixture of methanol and dichloromethane (in a ratio of 1:40) as mobile phase, affording the title compound in 57% yield (117 mg, 0.41 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.81 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 7.07 (d, J = 8.89 Hz, 1H, arom H), 7.35 (m, 2H, arom H), 7.84 (br s, 2H, NH2), 7.89 (d, J = 1.92 Hz, 1H, arom H), 8.63 (d, J = 1.95 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, DMSO-d6): δ = 55.76 (CH3), 55.82 (CH3), 111.00 (CH), 112.34 (CH), 119.76 (CH), 123.56 (CH), 129.82 (Cq), 133.77 (Cq), 135.31 (Cq), 143.53 (CH), 149.38 (2 × Cq), 153.96 (Cq), 172.20 (Cq) ppm. HR-MS [M+H]+ found 288.0800, calculated 288.0762.
3-Amino-6-(3-thienyl)-isothiazolo[4,3-b]pyridine (5d)
This compound was obtained using 3-thienylboronic acid and the crude residue was purified by flash chromatography using a mixture of cyclohexane and ethylacetate (in a ratio of 3:2) as mobile phase, affording the title compound in 69% yield (115 mg, 0.49 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 7.70 (m, 2H, arom H), 7.84 (br s, 2H, NH2), 7.98 (d, J = 1.89 Hz, 1H, arom H), 8.17 (q, J = 1.32 Hz, 1H, arom H), 8.74 (d, J = 1.95 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, DMSO-d6): δ = 122.99 (CH), 123.20 (CH), 126.60 (CH), 127.65 (CH), 130.35 (Cq), 133.71 (Cq), 138.38 (Cq), 143.17 (CH), 153.91 (Cq), 172.22 (Cq) ppm. HR-MS [M+H]+ found 243.0157, calculated 243.0115.
3-amino-6-(3-pyridyl)-isothiazolo[4,3-b]pyridine (5e)
This compound was obtained using 3-pyridylboronic acid and the crude residue was purified by flash chromatography using a mixture of methanol and dichloromethane (in a ratio of 1:20) as mobile phase, affording the title compound in 75% yield (123 mg, 0.54 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 7.52 (m, 1H, arom H), 7.95 (br s, 2H, NH2), 8.02 (d, J = 1.98 Hz, 1H, arom H), 8.24 (m, 1H, arom H), 8.66 (m, 2H, arom H), 9.03 (d, J = 1.98 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, DMSO-d6): δ = 124.06 (CH), 125.16 (CH), 132.54 (Cq), 133.01 (Cq), 134.92 (CH), 142.85 (CH), 148.26 (CH), 149.37 (CH), 153.46 (Cq), 173.63 (Cq) ppm. HR-MS [M+H]+ found 229.0537, calculated 229.0503.
3,6-di-Bromo-isothiazolo[4,3-b]pyridine (6)
A solution of 3-amino-6-bromo-isothiazolo[4,3-b]pyridine (1.1 g, 4.78 mmol) in HBr (50 ml) was stirred for 10 minutes at room temperature and then CuBr was added in one portion (1.37 g, 9.56 mmol, 2.0 eq). The resulting mixture was cooled to 0°C. A solution of sodium nitrite (0.99 g, 14.34 mmol, 3.0 eq) in H2O (15 ml) was added dropwise to the mixture over a period of 30 minutes. The reaction mixture was stirred for 2 hours at 0°C and then overnight at room temperature. Then, the mixture was cooled to 0°C and carefully neutralized with solid potassium carbonate and extracted with ethyl acetate (2 × 50 ml). The combined organic phases were dried over MgSO4 and evaporated in vacuo. The crude residue was purified by silicagel flash chromatography, using the mixture of cyclohexane and ethylacetate (in a ratio of 95:5) as mobile phase. The title compound was obtained in 78% yield (1.1 g, 3.75 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 8.70 (d, J = 2.01 Hz, 1H, arom H), 8.92 (d, J = 2.04 Hz, 1H, arom H) ppm. 13C-NMR (75 MHz, DMSO-d6): δ = 121.12 (Cq), 131.29 (CH), 137.79 (Cq), 144.54 (Cq), 153.31 (CH), 154.58 (Cq) ppm.
6-Bromo-3-substituted-isothiazolo[4,3-b]pyridines (7a-c)
General procedure
To a solution of 3,6-di-bromo-isothiazolo[4,3-b]pyridine (150 mg, 0.51 mmol) in ethanol (10 ml) was added an appropriate nitrogen nucleophile (3 eq). The reaction was stirred overnight at 75°C. The solvent was evaporated in vacuo and the crude residue was purified by silicagel flash chromatography, the mobile phase being a mixture of cyclohexane and ethylacetate (in a ratio of 4:1), yielding the pure title compounds.
The following compounds were made according to this procedure:
6-Bromo-3-morpholino-isothiazolo[4,3-b]pyridine (7a)
This compound was made using morpholine as nucleophile and the crude residue was purified using a mixture of cyclohexane and ethylacetate (in a ratio of 4:1) as mobile phase, affording the title compound in 92% yield (140 mg, 0.46 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.84 (br s, 4H, 2 × NCH2), 3.87 (br s, 4 H, 2 × OCH2), 8.15 (s, 1H, arom H), 8.37 (s, 1H, arom H) ppm.
6-Bromo-6-ethanolamino-isothiazolo[4,3-b]pyridine (7b)
This compound was made using ethanolamine as nucleophile and the crude residue was purified using a mixture of cyclohexane and ethylacetate (in a ratio gradually ranging from 1:1 to 1:4) as mobile phase, affording the title compound in 94% yield (131 mg, 0.47 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.39 (q, J = 5.75 Hz, 2H, NCH2), 3.68 (q, J = 5.68 Hz, 2H, OCH2), 4.91 (t, J = 5.57 Hz, 1H), 8.04 (d, J = 2.01 Hz, 1H, arom H), 8.29 (d, J = 1.98 Hz, 1H, arom H), 8.66 (t, J = 5.78 Hz, 1H) ppm.
6-Bromo-3-(N-methyl-piperazino)-isothiazolo[4,3-b]pyridine (7c)
This compound was made using N-methyl-piperazine as nucleophile and the crude residue was purified using a mixture of methanol and dichloromethane (in a ratio of 3:100) as mobile phase, affording the title compound in 92% yield (146 mg, 0.46 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 2.24 (s, 3H, NCH3), 2.52 (m, 4H, 2 × NCH2), 3.88 (t, J = Hz, 4 H, 2 × NCH2), 8.12 (d, J = 2.01 Hz, 1H, arom H), 8.36 (d, J = 1.23 Hz, 1H, arom H) ppm.
Synthesis of 3-ethanolamino-6-aryl-isothiazolo[4,3-b]pyridines (8a-e)
General procedure
To a solution of 3-ethanolamino-6-bromo-isothiazolo[4,3-b]pyridine (0.37 mmol, 101 mg) in dioxane (5 ml) and water (1 ml) was added an appropriate boronic acid (2 eq), sodium carbonate (2 eq, 78 mg) and Pd(dppf)Cl2 (0.01 eq, 30 mg). The reaction was overnight at 75°C. The reaction mixture was diluted with ethyl acetate (50 ml) and brine (30 ml). The aqueous phase was extracted with ethyl acetate (50 ml). The organic phases were dried and evaporated. The crude residue was purified by silicagel flash chromatography yielding the pure title compounds.
The following compounds were made according to this procedure:
3-Ethanolamino-6-phenyl-isothiazolo[4,3-b]pyridine (8a)
This compound was prepared using phenylboronic acid and was purified using a mixture of dichloromethane and methanol (in a ratio 20:1), affording the title compound in 99% yield (99 mg, 0.36 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.41 (q, J = 5.80 Hz, 2H, NCH2), 3.71 (q, J = 5.61 Hz, 2H, OCH2), 4.95 (t, J = 5.52 Hz, 1H), 7.49 (m, 5H, arom H), 7.80 (br d, J = 7.23 Hz, 1H, arom H), 7.91 (s, 1H, arom H), 8.50 (br t, 1H), 8.63 (d, J = 1.59 Hz, 1H, arom H) ppm. 13C-NMR (150 Mhz, CDCl3): δ = 50.09 (CH2), 59.84 (CH2), 125.97 (CH), 127.42 (CH), 128.47 (CH), 129.14 (CH), 132.86 (Cq), 136.81 (Cq), 137.46 (Cq), 144.10 (CH), 154.71 (Cq), 172.35 (Cq) ppm. HR-MS [M+H]+ found 272.0852, calculated 272.0552.
3-Ethanolamino-6-(3-(4-fluorophenyl))-isothiazolo[4,3-b]pyridine (8b)
This compound was prepared using 4-fluorophenylboronic acid and was purified using a mixture of dichloromethane and methanol (in a ratio 20:1), affording the title compound in 81% yield (86 mg, 0.29 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.30 (2H, NCH2, hidden under H2O peak), 3.71 (q, J = 5.61 Hz, 2H, OCH2), 4.96 (t, J = 5.45 Hz, 1H), 7.35 (t, J = 8.64 Hz, 1H, arom H), 7.88 (m, 3H, arom H), 8.47 (br t, 1H), 8.62 (br s, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 50.06 (CH2), 60.28 (CH2), 116.10 (CH), 116.25 (CH), 125.57 (CH), 129.14 (CH), 129.19 (CH), 133.43 (Cq), 133.74 (Cq), 135.94 (Cq), 144.19 (CH), 153.53 (Cq), 162.26 (Cq), 163.90 (Cq), 172.30 (Cq) ppm. HR-MS [M+H]+ found 290.0765, calculated 290.0719.
3-Ethanolamino-6-(3,4-dimethoxyphenyl)-isothiazolo[4,3-b]pyridine (8c)
This compound was prepared using 3,4-dimethoxyphenylboronic acid and was purified using a mixture of dichloromethane and methanol (in a ratio 30:1), affording the title compound in 95% yield (116 mg, 0.35 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.42 (q, J = 5.62 Hz, 2H, NCH2), 3.70 (q, J = 5.42 Hz, 2H, OCH2), 3.81 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 4.93 (t, J = 5.67 Hz, 1H), 7.07 (d, J = 8.34 Hz, 1H, arom H), 7.36 (m, 2H, arom H), 7.92 (s, 1H, arom H), 8.40 (t, J = 5.55 Hz, 1 arom H), 8.64 (br s, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 50.06 (CH2), 56.01 (CH3), 56.05 (CH3), 59.75 (CH2), 110.40 (CH), 111.63 (CH), 119.93 (CH), 125.18 (CH), 130.13 (Cq), 132.53 (Cq), 136.55 (Cq), 144.01 (CH), 149.50 (Cq), 149.49 (Cq), 154.82 (Cq), 172.29 (Cq) ppm. HR-MS [M+H]+ found 332.1067, calculated 332.1063.
3-Ethanolamino-6-(3-thienyl)-isothiazolo[4,3-b]pyridine (8d)
This compound was prepared using 3-thienylboronic acid and was purified using a mixture of dichloromethane and methanol (in a ratio 30:1), affording the title compound in 90% yield (92 mg, 0.33 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.40 (q, J = 5.58 Hz, 2H, NCH2), 3.70 (q, J = 5.64 Hz, 2H, OCH2), 4.93 (t, J = 5.49 Hz, 1H), 7.74 (m, 2H, arom H), 8.01 (br s, 1H, arom H), 8.19 (br s, 1 arom H), 8.42 (br t, 1H), 8.75 (br s, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 50.08 (CH2), 60.18 (CH2), 122.30 (CH), 124.47 (CH), 126.15 (CH), 127.13 (CH), 131.51 (Cq), 134.20 (Cq), 138.54 (Cq), 143.86 (CH), 154.73 (Cq), 172.20 (Cq) ppm. HR-MS [M+H]+ found 278.1415, calculated 278.0416.
3-Ethanolamino-6-(3-pyridyl)-isothiazolo[4,3-b]pyridine (8e)
This compound was prepared using 3-pyridyl boronic acid and was purified using a mixture of dichloromethane and methanol (in a ratio gradually ranging from 20:1 to 10:1), affording the title compound in 90% yield (90 mg, 0.33 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.42 (q, J = 5.67 Hz, 2H, NCH2), 3.71 (t, J = 5.64 Hz, 2H, OCH2), 4.95 (br s, 1H), 7.54 (m, 1H, arom H), 8.05 (br s, 1H, arom H), 8.25 (br d, J = 7.95 Hz, 1H, arom H), 8.52 (br t, 1H), 8.61 (m, 2H, arom H), 9.04 (d, J = 2.13 Hz 1H, arom H) ppm. 13C-NMR (75 MHz, DMSO-d6): δ = 50.70 (CH2), 59.16 (CH2), 124.08 (CH), 125.05 (CH), 132.76 (Cq), 132.97 (Cq), 133.90 (Cq), 134.98 (CH), 142.52 (CH), 148.22 (CH), 149.34 (CH), 153.81 (Cq), 173.35 (Cq) ppm. HR-MS [M+H]+ found 273.0807, calculated 273.0804.
Synthesis of 3-morpholino-6-aryl-isothiazolo[4,3-b]pyridines (9a-e)
General procedure
To a solution of 3-morpholino-6-bromo-isothiazolo[4,3-b]pyridine (0.37 mmol, 111 mg) in DME (5 ml) and water (1 ml) was added an appropriate boronic acid (2 eq), sodium carbonate (2 eq, 78 mg) and Pd(dppf)Cl2 (0.01 eq, 30 mg). The reaction was heated overnight at 75°C. The reaction mixture was diluted with ethyl acetate (50 ml) and brine (30 ml). The aqueous phase was extracted with ethyl acetate (50 ml). The organic phases were dried and evaporated. The crude residue was purified by silicagel flash chromatography, yielding the pure title compounds.
The following compounds were made according to this procedure:
3-Morpholino-6-phenyl-isothiazolo[4,3-b]pyridine (9a)
This compound was prepared using phenylboronic acid and was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 2:1, affording the title compound in 78% yield (85 mg. 0.28 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.86 (br s, 4 H, 2 × NCH2), 3.90 (br s, 4H, 2 × OCH2), 7.50 (m, 3H, arom H), 7.82 (m, 2H, arom H), 8.01 (d, J = 2.1 Hz, 1H, arom H), 8.72 (d, J = 2.1 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 50.40 (CH2), 66.20 (CH2), 125.69 (CH), 127.38 (CH), 128.39 (CH), 129.13 (CH), 134.35 (Cq), 135.81 (Cq), 137.56 (Cq), 144.54 (CH), 156.23 (Cq), 173.32 (Cq) ppm. HR-MS [M+H]+ found 298.1010, calculated 298.1008.
3-Morpholino-6-(4-fluoro-phenyl)-isothiazolo[4,3-b]pyridine (9b)
This compound was prepared using 4-fluorophenylboronic acid and was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 3:1, affording the title compound in 95% yield (110 mg, 0.35 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.85 (br s, 4H, 2 × NCH2), 3.89 (br s, 4H, 2 × OCH2), 7.34 (t, J = 8.82 Hz, 2H, arom H), 7.87 (m, 2H, arom H), 8.00 (d, J = 2.04 Hz, 1H, arom H), 8.69 (d, J = 2.04 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 50.38 (CH2), 66.18 (CH2), 116.08 (CH), 116.23 (CH), 125.52 (CH), 129.03 (CH), 129.09 (CH), 133.65 (Cq), 134.31 (Cq), 144.17 (CH), 156.05 (Cq), 162.22 (Cq), 163.86 (Cq), 173.38 (Cq) ppm. HR-MS [M+H]+ found 316.0912, calculated 316.0914.
3-Morpholino-6-(3,4-dimethoxyphenyl)-isothiazolo[4,3-b]pyridine (9c)
This compound was prepared using 3,4-dimethoxyphenylboronic acid and was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 2:1, affording the title compound in 72% yield (95 mg, 0.26 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.82 (s, 3H, OCH3), 3.89 (br s, 11H, 2 × NCH2, 2 × OCH2 and OCH3), 7.09 (d, J = 8.04 Hz, 1H, arom H), 7.39 (m, 2H, arom H), 8.02 (d, J = 2.01 Hz, 1 H, arom H), 8.75 (d, J = 1.95 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 50.37 (CH2), 55.97 (CH3), 56.00 (CH3), 66.18 (CH2), 110.33 (CH), 111.64 (CH), 119.84 (CH), 124.80 (CH), 130.23 (Cq), 134.06 (Cq), 135.55 (Cq), 144.51 (CH), 149.47 (Cq), 149.53 (Cq), 156.31 (Cq), 173.21 (Cq) ppm. HR-MS [M+H]+ found 358.1223, calculated 358.1219.
3-Morpholino-6-(3-thienyl)-isothiazolo[4,3-b]pyridine (9d)
This compound was prepared using 3-thienylboronic acid and was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 2:1, affording the title compound in 71% yield (79 mg, 0.26 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.86 (br s, 4 H, 2 × NCH2), 3.88 (br s, 4H, 2 × OCH2), 7.73 (m, 1H, arom H), 7.77 (m, 1H, arom H), 8.09 (d, J = 2.01 Hz, 1H, arom H), 8.21 (m, 1H, arom H), 8.84 (d, J = 2.04 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 50.36 (CH2), 66.19 (CH2), 122.22 (CH), 124.29 (CH), 126.08 (CH), 127.07 (CH), 130.46 (Cq), 134.06 (Cq), 138.54 (Cq), 143.99 (CH), 156.24 (Cq), 173.23 (Cq) ppm. HR-MS [M+H]+ found 304.0575, calculated 304.0572.
3-Morpholino-6-(3-pyridyl))-isothiazolo[4,3-b]pyridine (9e)
This compound was prepared using 3-pyridinyl boronic acid and was purified using a mixture of methanol/dichloromethane in a ratio of 1:25, affording the title compound in 80% yield (88 mg, 0.29 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.86 (br s, 4H, 2 × NCH2), 3.91 (br s, 4H, 2 × OCH2), 7.55 (m, 1H, arom H), 8.15 (d, J = 1.95 Hz, 2H, arom H), 8.26 (br d, 1 H, arom H), 8.65 (br d, 1H, arom H), 8.76 (d, J = 1.92 Hz, 1H, arom H), 9.05 (br s, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 50.37 (CH2), 66.15 (CH2), 123.82 (CH), 126.22 (CH), 132.59 (Cq), 133.33 (Cq), 134.60 (CH), 134.71 (Cq), 143.39 (CH), 148.36 (CH), 149.50 (CH), 155.70 (Cq), 173.56 (Cq) ppm. HR-MS [M+H]+ found 299.0959, calculated 299.0961.
Synthesis of 3-(N-Me-piperazinyl)-6-aryl-isothiazolo[4,3-b]pyridines 10a-10e
General procedure
To a solution of 3-(N-methylpiperazino)-6-bromo-isothiazolo[4,3-b]pyridine (100 mg, 0.32 mmol) in dioxane (6 ml) and water (2.5 ml) was added an appropriate boronic acid (2 eq), sodium carbonate (2 eq, 68 mg) and Pd(dppf)Cl2 (0.01 eq, 26 mg). The reaction mixture was heated at 95°C overnight. The reaction mixture was diluted with ethyl acetate (50 ml) and brine (30 ml). The aqueous phase was extracted with ethyl acetate (50 ml). The organic phases were dried and evaporated. The crude residue was purified by silicagel flash chromatography yielding the pure title compounds
The following compounds were made according to this procedure:
3-(N-Me-piperazinyl)-6-phenyl-isothiazolo[4,3-b]pyridine (10a)
This compound was prepared using phenylboronic acid and the crude residue was purified using a mixture of methanol and dichloromethane (in a ratio of 3:100), affording the title compound in 91% yield (90 mg, 0.29 mmol). 1H-NMR (300 MHz, CDCl3): δ = 2.39 (s, 3H, NCH3), 2.67 (t, J = 5.01 Hz, 4H, 2 × NCH2), 4.00 (t, J = 4.99 Hz, 4H, 2 × NCH2), 7.47 (m, 3H, arom H), 7.65 (m, 2H, arom H), 7.89 (d, J = 1.83 Hz, 1H, arom H), 8.63 (d, J = 1.83 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 46.19 (CH3), 50.25 (CH2), 54.21 (CH2), 125.61 (CH), 127.35 (CH), 128.32 (CH), 129.09 (CH), 134.19 (Cq), 135.64 (Cq), 137.64 (Cq), 144.13 (CH), 156.20 (Cq), 173.21 (Cq) ppm. HR-MS [M+H]+ found 311.1330, calculated 311.1324.
3-(N-Me-piperazinyl)-6-(4-fluorophenyl)-isothiazolo[4,3-b]pyridine (10b)
This compound was prepared using 4-fluorophenylboronic acid and the crude residue was purified using a mixture of methanol and dichloromethane (in a ratio of 3:100), affording the title compound in 96% yield (100 mg, 0.30 mmol). 1H-NMR (300 MHz, CDCl3): δ = 2.40 (s, 3H, NCH3), 2.68 (br s, 4H, 2 × NCH2), 4.00 (br s, 4H, 2 × NCH2), 7.19 (t, J = 8.61 Hz, 1H, arom H), 7.59 (m, 2H, arom H), 7.84 (d, J = 2.01 Hz, 1H, arom H), 8.57 (d, J = 2.01 Hz, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 46.18 (CH3), 50.24 (CH2), 54.20 (CH2), 116.05 (CH), 116.19 (CH), 125.46 (CH), 129.01 (CH), 129.06 (CH), 133.76 (Cq), 134.68 (Cq), 143.78 (CH), 156.03 (Cq), 162.18 (Cq), 163.83 (Cq), 173.26 (Cq) ppm. HR-MS [M+H]+ found 329.1231, calculated 329.1230.
3-(N-Me-piperazinyl)-6-(3,4-dimethoxyphenyl)-isothiazolo[4,3-b]pyridine (10c)
This compound was prepared using 3,4-dimethoxyphenyl boronic acid and the crude residue was purified using a mixture of methanol and dichloromethane (in a ratio of 1:30), affording the title compound in 66% yield (78 mg, 0.21 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 2.27 (s, 3H, NCH3), 2.56 (t, J = 4.91 Hz, 4H, 2 × NCH2), 3.82 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 3.92 (t, J = 4.88 Hz, 4H, 2 × NCH2), 7.09 (d, J = 8.43 Hz, 1H, arom H), 7.38 (m, 2H, arom H), 7.99 (d, J = 1.98 Hz, 1H, arom H), 8.75 (d, J = 2.01 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 46.20 (CH3), 50.24 (CH2), 54.22 (CH2), 55.96 (CH3), 55.99 (CH3), 110.33 (CH), 111.64 (CH), 119.81 (CH), 124.76 (CH), 130.34 (Cq), 133.93 (Cq), 135.39 (Cq), 144.11 (CH), 149.45 (Cq), 149.47 (Cq), 156.30 (Cq), 173.12 (Cq) ppm. HR-MS [M+H]+ found 371.1532, calculated 371.1536.
3-(N-Me-piperazinyl)-6-(3-thienyl)-isothiazolo[4,3-b]pyridine (10d)
This compound was prepared using 3-thienylboronic acid and the crude residue was purified using a mixture of methanol and dichloromethane (in a ratio of 3:100), affording the title compound in 84% yield (84 mg, 0.26 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 2.26 (s, 3H, NCH3), 2.55 (t, J = 4.91 Hz, 4H, 2 × NCH2), 3.91 (t, J = 4.88 Hz, 4H, 2 × NCH2), 7.72 (m, 2H, arom H), 8.07 (br s, 1H, arom H), 8.07 (br s, 1H, arom H), 8.19 (br s, 1H, arom H), 8.83 (d, J = 1.98 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 46.18 (CH3), 50.20 (CH2), 54.20 (CH2), 122.12 (CH), 124.24 (CH), 126.09 (CH), 127.00 (CH), 130.31 (Cq), 133.91 (Cq), 138.62 (Cq), 143.59 (CH), 156.21 (Cq), 173.10 (Cq) ppm. HR-MS [M+H]+ found 317.0893, calculated 317.0850.
3-(N-Me-piperazinyl)-6-(3-pyridyl)-isothiazolo[4,3-b]pyridine (10e)
This compound was prepared using 3-pyridylboronic acid and the crude residue was purified using a mixture of methanol and dichloromethane (in a ratio of 1:20), affording the title compound in 83% yield (82 mg, 0.26 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 2.26 (s, 3H, NCH3), 2.55 (t, J = 4.85 Hz, 4H, 2 × NCH2), 3.92 (t, J = 4.80 Hz, 4H, 2 × NCH2), 7.54 (m, 1H, arom H), 8.12 (d, J = 1.98 Hz, 1H, arom H), 8.24 (d, J = 7.86 Hz, 1H, arom H), 8.65 (d, J = 4.74 Hz, 1H, arom H), 8.75 (d, J = 1.92 Hz, 1H, arom H), 9.04 (d, J = 1.95 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 46.17 (CH3), 50.24 (CH2), 54.17 (CH2), 123.79 (CH), 126.15 (CH), 132.44 (Cq), 133.41 (Cq), 134.58 (CH, Cq), 142.99 (CH), 148.36 (CH), 149.45 (CH), 155.68 (Cq), 173.43 (Cq) ppm. HR-MS [M+H]+ found 312.1277, calculated 312.1277.
3-Methoxy-6-bromo-isothiazolo[4,3-b]pyridine (7d)
To a solution of 3,6-dibromo-isothiazolo[4,3-b]pyridine (700 mg, 2.38 mmol) in absolute methanol (50 ml) was added carefully at 0°C sodium methoxide (2.5 eq, 322 mg) in small portions. The resulting reaction mixture was stirred overnight at room temperature and then heated at 55°C for 8 hours. The reaction was cooled down to room temperature, neutralized with a 5% HCl solution and evaporated in vacuo. The residue was divided between ethyl acetate (250 ml) and water (150 ml). The organic phase was dried and evaporated. The crude residue was purified by silica gel flash chromatography, the mobile phase being a mixture of cyclohexane and ethylacetate (in a ratio gradually ranging from 5:1 to 4:1), yielding the pure title compound in 96% yield (558 mg, 2.27 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 4.35 (s, 3H, OCH3), 8.37 (d, J = 2.01 Hz, 1H, arom H), 8.60 (d, J = 1.98 Hz, 1H, arom H) ppm.
3-Methoxy-6-aryl-isothiazolo[4,3-b]pyridines (11a-e)
General procedure
To a solution of 3-methoxy-6-bromo-isothiazolo[4,3-b]pyridine (100 mg, 0.41 mmol) in dioxane (5 ml) and water (1 ml) was added an appropriate boronic acid (2 eq), sodium carbonate (2 eq, 87 mg) and Pd(dppf)Cl2 (0.01 eq, 33 mg). The reaction mixture was heated at 95°C overnight. The reaction mixture was diluted with ethyl acetate (50 ml) and brine (30 ml). The aqueous phase was extracted with ethyl acetate (50 ml). The organic phases were dried and evaporated. The crude residue was purified by silicagel flash chromatography yielding the pure title compounds.
The following compounds were made according to this procedure:
3-Methoxy-6-phenyl-isothiazolo[4,3-b]pyridine (11a)
This compound was prepared using phenylboronic acid and the crude residue was purified using a mixture of cyclohexane and ethylacetate (in a ratio of 2:1), affording the title compound in 87% yield (86 mg, 0.35 mmol). 1H-NMR (300 MHz, CDCl3): δ = 4.37 (s, 3H, OCH3), 7.50 (m, 3H, arom H), 7.68 (m, 2H, arom H), 8.00 (d, J = 2.01 Hz, 1H, arom H), 8.86 (d, J = 1.98 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 62.37 (CH3), 125.55 (CH), 127.15 (CH), 128.28 (CH), 128.84 (CH), 133.08 (Cq), 136.47 (Cq), 136.85 (Cq), 148.37 (CH), 154.54 (Cq), 182.58 (Cq) ppm. HR-MS [M+H]+ found 243.0584, calculated 243.0547.
3-Methoxy-6-(4-fluoro-phenyl)-isothiazolo[4,3-b]pyridine (11b)
This compound was prepared using 4-fluoro-phenylboronic acid and the crude residue was purified using a mixture of cyclohexane and ethylacetate (in a ratio of 2:1), affording the title compound in 76% yield (81 mg, 0.31 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 4.36 (s, 3H, OCH3), 7.37 (t, J = 8.79, 3H, arom H), 7.93 (m, 2H, arom H), 8.18 (d, J = 1.98 Hz, 1H, arom H), 8.93 (d, J = 1.92 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 62.75 (CH3), 116.18 (CH), 116.33 (CH), 125.77 (CH), 129.21 (CH), 129.27 (CH), 133.31 (Cq), 135.83 (Cq), 148.39 (CH), 154.72 (Cq), 162.35 (Cq), 164.00 (Cq), 183.04 (Cq) ppm. HR-MS [M+H]+ found 261.0503, calculated 261.0453.
3-Methoxy-6-(3,4-dimethoxyphenyl)-isothiazolo[4,3-b]pyridine (11c)
This compound was prepared using 3,4-dimethoxyphenylboronic acid and the crude residue was purified using a mixture of cyclohexane and ethylacetae (in a ratio of 1:1), affording the title compound in 74% yield (91 mg, 0.30 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.82 (s, 3H, OCH3), 3.89 (s, 3H, OCH3), 4.35 (s, 3H, OCH3), 7.09 (d, J = 9 Hz, 1H, arom H), 7.42 (m, 2H, arom H), 8.16 (d, J = 1.92 Hz, 1H, arom H), 8.97 (d, J = 1.92 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 56.02 (CH3), 56.04 (CH3), 62.69 (CH2), 110.42 (CH), 111.67 (CH), 120.11 (CH), 125.02 (CH), 129.90 (Cq), 133.18 (Cq), 136.58 (Cq), 148.71 (CH), 149.55 (Cq), 149.74 (Cq), 155.01 (Cq), 182.83 (Cq) ppm. HR-MS [M+H]+ found 303.0796, calculated 303.0797.
3-Methoxy-6-(3-thienyl)-isothiazolo[4,3-b]pyridine (11d)
This compound was prepared using 3-thienyllboronic acid and the crude residue was purified using a mixture of cyclohexane and ethylacetae (in a ratio of 2:1), affording the title compound in 75% yield (74 mg, 0.30 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 4.35 (s, 3H, OCH3), 7.74 (m, 1H, arom H), 7.82 (m, 1H, arom H), 8.25 (d, J = 1.98 Hz, 1H, arom H), 8.29 (m, 1H, arom H), 9.06 (d, J = 1.95 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 62.70 (CH3), 122.73 (CH), 124.44 (CH), 126.08 (CH), 127.26 (CH), 131.43 (Cq), 133.17 (Cq), 138.19 (Cq), 148.18 (CH), 154.92 (Cq), 182.84 (Cq) ppm. HR-MS [M+H]+ found 249.0156, calculated 249.0150.
3-Methoxy-6-(3-pyridyl)-isothiazolo[4,3-b]pyridine (11e)
This compound was prepared using 3-pyridylboronic acid and the crude residue was purified using a mixture of methanol and dichloromethane (in a ratio of 1:30), affording the title compound in 55% yield (54 mg, 0.22 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 4.40 (s, 3H, OCH3), 7.57 (m, 1H, arom H), 8.31 (m, 2H, arom H), 8.67 (d, J = 4.23 Hz, 1H, arom H), 8.99 (d, J = 1.71 Hz, 1H, arom H), 9.09 (d, J = 1.89 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 62.87 (CH2), 123.86 (CH), 126.60 (CH), 133.05 (Cq), 133.64 (Cq), 133.87 (Cq), 134.77 (CH), 147.75 (CH), 148.49 (CH), 149.78 (CH), 154.39 (Cq), 183.38 (Cq) ppm. HR-MS [M+H]+ found 244.0542, calculated 244.0500.
Synthesis of 3-morpholino-6-aryl-isothiazolo[4,3-b]pyridines (12a-j)
General procedure
To a solution of 3-morpholino-6-bromo-isothiazolo[4,3-b]pyridine in DME (2 ml) was added an appropriate boronic acid (2 eq or 1.5 eq) and potassium carbonate (2 eq, 1M solution in H2O). Mixture was degassed and Pd(PPh3)4 (10 mol %) was added. The reaction was heated at 80°C. After the completion of reaction, solvents were evaporated. The crude residue was purified by silicagel flash chromatography, yielding the pure title compounds.
The following compounds were made according to this procedure:
4-(6-(Thiophen-2-yl)isothiazolo[4,3-b]pyridin-3-yl)morpholine (12a)
This compound was prepared from 3-morpholino-6-bromo-isothiazolo[4,3-b]pyridine (90 mg, 0.3 mmol) using thiophene-2-boronic acid (0.6 mmol, 76 mg), 1M K2CO3 (0.6 ml) and Pd(PPh3)4 (0.03 mmol, 34 mg). The crude product was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 1:1, affording the title compound in 78 % yield (71 mg, 0.234 mmol). 1H-NMR (300 MHz, CDCl3): δ = 3.96 (m, 8H, 4×CH2), 7.16 (dd, J = 3.66 Hz, J = 5.10 Hz, 1H, arom H), 7.42 (dd, J = 1.08 Hz, J = 5.10 Hz, 1H, arom H), 7.49 (dd, J = 3.66 Hz, J = 1.08 Hz, 1H, arom H), 7.93 (d, J = 2.07 Hz, 1H, arom H), 8.69 (d, J = 2.10 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 50.35 (CH2), 66.17 (CH2), 123.49 (CH), 124.99 (CH), 126.56 (CH), 128.39 (CH), 129.36 (Cq), 134.08 (Cq), 140.42 (Cq), 143.12 (CH), 155.91 (Cq), 173.24 (Cq) ppm. HR-MS [M+H]+ found 304.0577, calculated 304.0572.
4-(6-(Furan-3-yl)isothiazolo[4,3-b]pyridin-3-yl)morpholine (12b)
This compound was prepared from 3-morpholino-6-bromo-isothiazolo[4,3-b]pyridine (40 mg, 0.133 mmol) using furan-3-boronic acid (0.2 mmol, 23 mg), 1M K2CO3 (0.26 ml) and Pd(PPh3)4 (0.013 mmol, 36 mg). The product was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 2:1, affording the title compound in 51 % yield (19 mg, 0.067 mmol). 1H-NMR (300 MHz, CDCl3): δ = 3.96 (m, 8H, 4×CH2), 6.79 (s, 1H, arom H), 7.56 (m, 1H, arom H), 7.81 (d, J = 2.01 Hz, 1H, arom H), 7.90 (s, 1H, arom H), 8.56 (d, J = 2.01 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 50.37 (CH2), 66.19 (CH2), 108.55 (CH), 123.25 (Cq), 123.59 (CH), 127.58 (Cq), 133.94 (Cq), 139.58 (CH), 143.40 (CH), 144.26 (CH), 156.19 (Cq), 173.23 (Cq) ppm. HR-MS [M+H]+ found 288.0803, calculated 288.0801.
4-(6-(2,4-Dimethoxyphenyl)isothiazolo[4,3-b]pyridin-3-yl)morpholine (12c)
This compound was prepared from 3-morpholino-6-bromo-isothiazolo[4,3-b]pyridine (90 mg, 0.3 mmol) using 2,4-dimethoxyphenylboronic acid (0.6 mmol, 109 mg), 1M K2CO3 (0.6 ml) and Pd(PPh3)4 (0.03 mmol, 34 mg). The crude product was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 2:1, affording the title compound in 73 % yield (79 mg, 0.221 mmol). 1H-NMR (300 MHz, CDCl3): δ = 3.84 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 3.96 (m, 8H, 4×CH2), 6.60-6.64 (m, 2H, arom H), 7.33 (d, J = 8.19 Hz, 1H, arom H), 7.84 (d, J = 1.95 Hz, 1H, arom H), 8.57 (d, J = 1.92 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 50.14 (CH2), 55.17 (CH3), 55.22 (CH3), 65.92 (CH2), 98.76 (CH), 104.77 (CH), 119.29 (Cq), 127.22 (CH), 131.03 (CH), 133.16 (Cq), 133.52 (Cq), 146.66 (CH), 156.22 (Cq), 157.56 (Cq), 161.00 (Cq), 172.70 (Cq) ppm. HR-MS [M+H]+ found 358.1224, calculated 358.1219.
4-(6-(2,5-Dimethoxyphenyl)isothiazolo[4,3-b]pyridin-3-yl)morpholine (12d)
This compound was prepared from 3-morpholino-6-bromo-isothiazolo[4,3-b]pyridine (90 mg, 0.3 mmol) using 2,5-dimethoxyphenylboronic acid (0.6 mmol, 109 mg), 1M K2CO3 (0.6 ml) and Pd(PPh3)4 (0.03 mmol, 34 mg). The product was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 2:1, affording the title compound in 69 % yield (74 mg, 0.207 mmol). 1H-NMR (300 MHz, CDCl3): δ = 3.80 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 3.97 (m, 8H, 4×CH2), 6.95-6.97 (m, 3H, arom H), 7.89 (d, J = 1.95 Hz, 1H, arom H), 8.58 (d, J = 1.92 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 50.13 (CH2), 55.51 (CH3), 55.89 (CH3), 65.92 (CH2), 112.35 (CH), 114.15 (CH), 116.19 (CH), 123.33 (Cq), 127.36 (Cq), 127.77 (CH), 133.55 (Cq), 146.23 (CH), 150.71 (Cq), 153.60 (Cq), 155.98 (Cq), 172.84 (Cq) ppm. HR-MS [M+H]+ found 358.1221, calculated 358.1219.
4-(6-(3,5-Dimethoxyphenyl)isothiazolo[4,3-b]pyridin-3-yl)morpholine (12e)
This compound was prepared from 3-morpholino-6-bromo-isothiazolo[4,3-b]pyridine (60 mg, 0.2 mmol) using 3,5-dimethoxyphenylboronic acid (0.4 mmol, 72 mg), 1M K2CO3 (0.4 ml) and Pd(PPh3)4 (0.02 mmol, 23 mg). The crude product was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 2:1, affording the title compound in 70 % yield (50 mg, 0.140 mmol). 1H-NMR (300 MHz, CDCl3): δ = 3.88 (s, 6H, 2 × OCH3), 3.98 (m, 8H, 4×CH2), 6.55 (m, 1H, arom H), 6.80 (d, J = 2.19 Hz, 2H, arom H), 7.92 (d, J = 1.98 Hz, 1H, arom H), 8.63 (d, J = 2.01 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 50.09 (CH2), 55.16 (CH3), 65.88 (CH2), 99.97 (CH), 105.32 (CH), 125.46 (CH), 134.18 (Cq), 135.48 (Cq), 139.32 (Cq), 144.14 (CH), 155.83 (Cq), 160.99 (Cq), 173.01 (Cq) ppm. HR-MS [M+H]+ found 358.1218, calculated 358.1219.
4-(6-(Benzo[d][1,3]dioxol-5-yl)isothiazolo[4,3-b]pyridin-3-yl)morpholine (12f)
This compound was prepared from 3-morpholino-6-bromo-isothiazolo[4,3-b]pyridine (60 mg, 0.2 mmol) using 3,4-methylenedioxyphenylboronic acid (0.4 mmol, 66 mg), 1M K2CO3 (0.4 ml) and Pd(PPh3)4 (0.02 mmol, 23 mg). Product was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 2:1, affording the title compound in 64 % yield (43 mg, 0.128 mmol). 1H-NMR (300 MHz, CDCl3): δ = 3.96 (m, 8H, 4×CH2), 6.05 (s, 2H, CH2), 6.94 (d, J = 7.86 Hz, 1H, arom H), 7.15 (m, 2H, arom H), 7.83 (d, J = 2.01 Hz, 1H, arom H), 8.58 (d, J = 2.04 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 50.38 (CH2), 66.19 (CH2), 101.41 (CH2), 107.66 (CH), 108.94 (CH), 121.23 (CH), 124.98 (CH), 131.65 (Cq), 134.11 (Cq), 135.55 (Cq), 144.47 (CH), 148.05 (Cq), 148.45 (Cq), 156.22 (Cq), 173.24 (Cq). HR-MS [M+H]+ found 342.0905, calculated 342.0906.
4-(6-(3,4,5-Trimethoxyphenyl)isothiazolo[4,3-b]pyridin-3-yl)morpholine (12g)
This compound was prepared from 3-morpholino-6-bromo-isothiazolo[4,3-b]pyridine (76 mg, 0.25 mmol) using 3,4,5-trimethoxyphenylboronic acid (0.5 mmol, 107 mg), 1M K2CO3 (0.5 ml) and Pd(PPh3)4 (0.025 mmol, 29 mg). The crude product was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 2:1, affording the title compound in 56 % yield (54 mg, 0.141 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.72 (s, 3H, OCH3), 3.86 (s, 6H, 2 × OCH3), 3.90 (m, 8H, 4×CH2), 7.10 (s, 2H, arom H), 8.08 (d, J = 2.01 Hz, 1H, arom H), 8.78 (d, J = 1.98 Hz, 1H, arom H) ppm. 13C-NMR (75 MHz, DMSO-d6): δ = 50.15 (CH2), 56.24 (OCH3), 60.21 (OCH3), 65.52 (CH2), 104.97 (CH), 124.83 (CH), 132.39 (C), 133.59 (C), 135.00 (C), 138.11 (C), 144.37 (CH), 153.53 (C), 156.68 (C), 172.18 (C) ppm. HR-MS [M+H]+ found 388.1323, calculated 388.1325.
2-Methoxy-4-(3-morpholinoisothiazolo[4,3-b]pyridin-6-yl)phenyl acetate (12h)
This compound was prepared from 3-morpholino-6-bromo-isothiazolo[4,3-b]pyridine (60 mg, 0.2 mmol) using 4-acetoxy-3-methoxyphenylboronic acid pinacol ester (0.4 mmol, 116 mg), 1M K2CO3 (0.4 ml) and Pd(PPh3)4 (0.02 mmol, 23 mg). Product was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 2:1, affording the title compound in 58 % yield (45 mg, 0.116 mmol). 1H-NMR (300 MHz, CDCl3): δ = 2.37 (s, 3H, CH3(acetyl)), 3.93 (s, 3H, OCH3), 3.98 (m, 8H, 4×CH2), 7.16-7.28 (m, 3H, arom H), 7.91 (d, J = 1.98 Hz, 1H, arom H), 8.53 (d, J = 2.01 Hz, 1H, arom H) ppm. 13C-NMR (150 MHz, CDCl3): δ = 20.67 (CH3), 50.38 (CH2), 55.97 (CH3), 66.18 (CH2), 111.51 (CH), 119.80 (CH), 123.44 (CH), 125.71 (CH), 134.40 (Cq), 135.32 (Cq), 136.58 (Cq), 140.06 (Cq), 144.27 (CH), 151.53 (Cq), 156.06 (Cq), 169.00 (CO), 173.36 (Cq) ppm. HR-MS [M+H]+ found 386.1165, calculated 386.1168.
2-Methoxy-4-(3-morpholinoisothiazolo[4,3-b]pyridin-6-yl)aniline (12i)
This compound was prepared from 3-morpholino-6-bromo-isothiazolo[4,3-b]pyridine (60 mg, 0.2 mmol) using 4-amino-3-methoxyphenylboronic acid pinacol ester (0.4 mmol, 99 mg), 1M K2CO3 (0.4 ml) and Pd(PPh3)4 (0.02 mmol, 23 mg). The crude product was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 2:1, affording the title compound in 58 % yield (40 mg, 0.116 mmol). 1H-NMR (300 MHz, DMSO-d6): δ = 3.89 (m, 11H, 4×CH2 and OCH3), 5.09 (br s, 2H, NH2), 6.75 (d, J = 8.1 Hz, 1H, arom H), 7.22 (m, 2H, arom H), 7.90 (d, J = 1.86 Hz, 1H, arom H), 8.74 (d, J = 1.86 Hz, 1H, arom H) ppm. 13C-NMR (75 MHz, DMSO-d6): δ = 50.12 (CH2), 55.63 (CH3), 65.54 (CH2), 109.43 (CH), 113.91 (CH), 120.21 (CH), 122.21 (CH), 124.09 (CH), 132.86 (Cq), 135.49 (Cq), 138.89 (Cq), 144.44 (CH), 146.85 (Cq), 156.18 (Cq), 172.53 (Cq) ppm. HR-MS [M+H]+ found 343.1222, calculated 343.1223.
2-Methoxy-4-(3-morpholinoisothiazolo[4,3-b]pyridin-6-yl)phenol (12j)
This compound was prepared from 3-morpholino-6-bromo-isothiazolo[4,3-b]pyridine (60 mg, 0.2 mmol) using methoxy-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol (0.3 mmol, 75 mg), 1M K2CO3 (0.4 ml) and Pd(PPh3)4 (0.02 mmol, 23 mg). The crude product was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 3:2, affording the title compound in 44 % yield (30 mg, 0.087 mmol). 1H-NMR (300 MHz, CDCl3): δ = 3.97 (m, 11H, 4×CH2, CH3), 5.93 (s, 1H, OH), 7.04 (d, J = 8.16 Hz, 1H, arom H), 7.14 (d, J = 1.95 Hz, 1H, arom H), 7.19 (dd, J = 2.04 Hz, J = 8.16 Hz, 1H, arom H), 7.87 (d, J = 2.07 Hz, 1H, arom H), 8.64 (d, J = 2.07 Hz, 1H, arom H) ppm. 13C-NMR (75 MHz, DMSO-d6): δ = 50.43 (CH2), 56.05 (OCH3), 66.23 (CH2), 109.80 (CH), 115.14 (CH), 120.61 (CH), 124.74 (CH), 129.75 (C), 132.12 (C), 135.78 (C), 144.62 (CH), 146.37 (C), 147.15 (C), 156.37 (C), 173.25 (C) ppm. HR-MS [M+H]+ found 344.1067, calculated 344.1063.
Synthesis of 3-substituted-6-(3,4-dimethoxyphenyl)-isothiazolo[4,3-b]pyridine (13a-k)
General procedure A
To a solution of 3,6-di-bromo-isothiazolo[4,3-b]pyridine in ethanol was added an appropriate nitrogen nucleophile (3 eq). The reaction was stirred at 75°C. After reaction finished, solvent was evaporated in vacuo and the crude residue was purified by silicagel flash chromatography yielding the 3-substituted-6-bromo-isothiazolo[4,3-b]pyridine derivatives. To a solution of 3-substituted-6-bromo-isothiazolo[4,3-b]pyridine in DME (2 ml) was added 3,4-dimethoxyphenyl boronic acid (1.5 eq) and potassium carbonate (2 eq, 1M solution in H2O). The mixture was degassed and Pd(PPh3)4 (10 mol %) was added. The reaction was heated at 80°C. After the completion of reaction, solvents were evaporated. The crude residue was purified by silicagel flash chromatography, yielding the pure title compounds.
The following compounds were made according to this procedure: 13b, 13c, 13d, 13e, 13i, 13j, 13k.
General procedure B
To a solution of 3,6-di-bromo-isothiazolo[4,3-b]pyridine in ethanol was added an appropriate nitrogen nucleophile (3 eq). The reaction was stirred at 70°C. After reaction finished, solvent was evaporated in vacuo and the crude residue was purified by silicagel flash chromatography yielding the 3-substituted-6-bromo-isothiazolo[4,3-b]pyridine derivatives. To a solution of 3- substituted-6-bromo-isothiazolo[4,3-b]pyridine in dioxane (2 ml) was added 3,4- dimethoxyphenyl boronic acid (1.5 eq) and potassium carbonate (3 eq). The mixture was degassed and Pd(dppf)Cl2 (5 mol %) was added. The reaction was heated at 100°C. After the completion of reaction, solvents were evaporated. The crude residue was purified by silicagel flash chromatography, yielding the pure title compounds.
The following compounds were made according to this procedure: 13a, 13f, 13g and 13h.
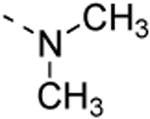
6-(3,4-dimethoxyphenyl)-N,N-dimethylisothiazolo[4,3-b]pyridin-3-amine (13a)
6-Bromo-N,N-dimethylisothiazolo[4,3-b]pyridin-3-amine was prepared from 3,6-di-bromo-isothiazolo[4,3-b]pyridine (0.5 mmol, 146 mg) and dimethylamine (44μl, 1.5mmol) in EtOH (10 ml). The product was purified using a mixture of cyclohexane/ethylacetate in a ratio of 4:1, affording the 6-bromo-N,N-dimethylisothiazolo[4,3-b]pyridin-3-amine in 98 % yield (379 mg, 0.49 mmol). The title compound was prepared from 6-bromo-N,N-dimethylisothiazolo[4,3-b]pyridin-3-amine (52 mg, 0.2 mmol) using 3,4-dimethoxyphenylboronic acid (0.3 mmol, 54.5 mg), K2CO3 (83mg, 0.6 mmol) and Pd(dppf)Cl2 (0.01 mmol, 7.4 mg). The crude product was purified using a mixture of cyclohexane/ethylacetate in a ratio of 1:1, affording the title compound in 72 % yield (46.6 mg, 1.5 mmol). 1H-NMR (300 MHz, CDCl3) δ = 3.49 (s, 6H, 2 × CH3), 3.92 (s, 3H, OCH3), 3.94 (s, 3H, OCH3), 6.97 (d, J = 8.3 Hz, 1H, arom H), 7.15 (s, 1H, arom H), 7.21 (dd, J = 2.1 Hz, J = 8.3 Hz, 1H arom H), 7.80 (d, J = 2.0 Hz, 1H, arom H), 8.56 (d, J = 2.0 Hz, 1H, arom H) ppm. 13C-NMR (300 MHz, CDCl3), δ = 43.0 (CH3), 56.1 (OCH3), 56.1 (OCH3), 110.5 (CH), 111.8 (CH), 119.9 (CH), 124.6 (CH), 130.6 (C), 133.5 (C), 135.3 (C), 143.4 (CH), 149.5 (C), 156.2 (C), 173.2 (C) ppm. HR-MS: [M + H]+ found 316.1113, calculated 316.1114.
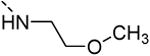
6-(3,4-dimethoxyphenyl)-N-(2-methoxyethyl)isothiazolo[4,3-b]pyridin-3-amine (13b)
6-Bromo-N-(2-methoxyethyl)isothiazolo[4,3-b]pyridin-3-amine was prepared from 3,6-di-bromo-isothiazolo[4,3-b]pyridine (0.5 mmol, 146 mg) and 2-methoxyethylamine (0.13 ml, 1.5 mmol) in EtOH (10 ml). The crude product was purified using a mixture of cyclohexane/ethylacetate in a ratio of 4:1, affording the 6-bromo-N-(2-methoxyethyl)isothiazolo[4,3-b]pyridin-3-amine in 83 % yield (120 mg, 0.416 mmol). The title compound was prepared from 6-bromo-N-(2-methoxyethyl)isothiazolo[4,3-b]pyridin-3-amine (57 mg, 0.2 mmol) using 3,4-dimethoxyphenylboronic acid (0.3 mmol, 54.5 mg), 1 M K2CO3 (0.40 ml) and Pd(PPh3)4 (0.02 mmol, 23 mg). The product was purified using a mixture of cyclohexane/ethylacetate in a ratio of 1:1, affording the title compound in 22 % yield (15 mg, 0.043 mmol). H-NMR (300 MHz, CDCl3): 5 = 3.40 (s, 3H, OCH3), 3.55 (m, 2H, CH2), 3.58 (m, 2H, CH2), 3.95 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 6.53 (m, 1H, NH), 6.99 (d, J = 8.31 Hz, 1H, arom H), 7.15 (d, 1H, J = 1.98 Hz, arom H), 7.21 (dd, J = 1.98 Hz, J = 8.22 Hz, 1H, arom H), 7.87 (d, J = 1.80 Hz, 1H, arom H), 8.60 (d, J = 1.80 Hz, 1H, arom H) ppm. 13C-NMR (125 MHz, DMSO-d6): 5 = 47.33 (CH2), 55.70 (OCH3), 58.67 (OCH3), 69.76 (CH2), 110.24 (CH), 111.38 (CH), 119.64 (CH), 124.25 (CH), 130.24 (C), 133.24 (C), 136.37 (C), 144.33 (CH), 149.20 (C), 149.23 (C), 154.40 (C), 171.72 (C) ppm. HR-MS [M+H]+ found 346.1223, calculated 346.1219.
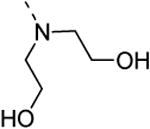
2,2′-(6-(3,4-dimethoxyphenyl)isothiazolo[4,3-b]pyridin-3-ylazanediyl)diethanol (13c)
2,2′-(6-Bromoisothiazolo[4,3-b]pyridin-3-ylazanediyl)diethanol was prepared from 3,6-di-bromo-isothiazolo[4,3-b]pyridine (0.5 mmol, 146 mg) and diethanolamine (0.145 ml, 1.5 mmol) in EtOH (10 ml). The crude product was purified using a mixture of DCM/MeOH in a ratio of 95:5, affording 2,2′-(6-bromoisothiazolo[4,3-b]pyridin-3-ylazanediyl)diethanol in 81 % yield (129 mg, 0.405 mmol). The title compound was prepared from 2,2′-(6-bromoisothiazolo[4,3-b]pyridin-3-ylazanediyl)diethanol (63.6 mg, 0.2 mmol) using 3,4-dimethoxyphenylboronic acid (0.3 mmol, 54.5 mg), 1M K2CO3 (0.4 ml) and Pd(PPh3)4 (0.02 mmol, 23 mg). The crude product was purified using a mixture of DCM/MeOH in a ratio of 100:1, affording the title compound in 60 % yield (45 mg, 0.12 mmol). 1H-NMR (300 MHz, CDCl3): δ = 3.96 (s, 3H, OCH3), 3.98 (s, 3H, OCH3), 4.05 (m, 8H, 4×CH2), 4.20 (m, 2H, 2×OH), 7.02 (d, J = 8.31 Hz, 1H, arom H), 7.15 (d, J = 2.04 Hz, 1H, arom H), 7.24 (dd, J = 8.28, J = 2.13 Hz, 1H, arom H), 7.86 (d, J = 2.04 Hz, 1H, arom H), 8.56 (d, J = 2.04 Hz, 1H, arom H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 55.70 (OCH3), 57.80 (CH2), 59.36 (CH2), 109.98 (CH), 111.38 (CH), 119.52 (CH), 124.54 (CH), 129.60 (C), 132.80 (C), 135.39 (C), 143.58 (CH), 149.21 (C), 149.33 (C), 155.62 (C), 172.05 (C) ppm. HR-MS [M+H]+ found 376.1322, calculated 376.1252.
6-(3,4-dimethoxyphenyl)-3-(pyrrolidin-1-yl)isothiazolo[4,3-b]pyridine (13d)
6-Bromo-3-(pyrrolidin-1-yl)isothiazolo[4,3-b]pyridine was prepared from 3,6-di-bromo-isothiazolo[4,3-b]pyridine (0.5 mmol, 146 mg) and pyrrolidine (0.04 ml, 1.5 mmol) in EtOH (10ml). The crude product was purified using a mixture of cyclohexane/ethylacetate in a ratio of 4:1, affording the 6-bromo-3-(pyrrolidin-1-yl)isothiazolo[4,3-b]pyridine in 90 % yield (128 mg, 0.450 mmol). The title compound was prepared from 6-bromo-3-(pyrrolidin-1-yl)isothiazolo[4,3-b]pyridine (71 mg, 0.25 mmol) using 3,4-dimethoxyphenylboronic acid (0.375 mmol, 68 mg), 1M K2CO3 (0.50 ml) and Pd(PPh3)4 (0.025 mmol, 29 mg). The crude product was purified using a mixture of cyclohexane/ethylacetate in a ratio of 3:2, affording the title compound in 63 % yield (54 mg, 0.158 mmol). 1H-NMR (300 MHz, CDCl3): δ = 2.17 (m, 4H, 2×CH2), 3.87 (m, 4H, 2×CH2), 3.96 (s, 3H, OCH3), 3.98 (s, 3H, OCH3), 7.00 (d, J = 8.28 Hz, 1H, arom H), 7.18 (d, 1H, J = 2.01 Hz, arom H), 7.26 (dd, J = 8.52, J = 2.28 Hz, 1H, arom H), 7.82 (d, J = 1.95 Hz, 1H, arom H), 8.58 (d, J = 1.95 Hz, 1H, arom H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 25.37 (CH2), 51.68 (CH2), 55.63 (OCH3), 55.67 (OCH3), 110.12 (CH), 111.36 (CH), 119.45 (CH), 123.73 (CH), 130.89 (C), 133.44 (C), 135.20 (C), 142.98 (CH), 149.06 (C), 149.11 (C), 155.50 (C), 169.25 (C) ppm. HR-MS [M+H]+ found 342.1273, calculated 342.1270.
N-(cyclopropylmethyl)-6-(3,4-dimethoxyphenyl)isothiazolo[4,3-b]pyridin-3-amine (13e)
6-Bromo-N-(cyclopropylmethyl)isothiazolo[4,3-b]pyridin-3-amine was prepared from 3,6-di-bromo-isothiazolo[4,3-b]pyridine (0.5 mmol, 146 mg) and aminomethylcyclopropane (0.086 ml, 3.0 mmol) in EtOH (10 ml). The crude product was purified using a mixture of cyclohexane/ethylacetate in a ratio of 9:1, affording the 6-bromo-N-(cyclopropylmethyl)isothiazolo[4,3-b]pyridin-3-amine in 84 % yield (120 mg, 0.422 mmol). The title compound was prepared from 6-bromo-N-(cyclopropylmethyl)isothiazolo[4,3-b]pyridin-3-amine (85 mg, 0.3 mmol) using 3,4-dimethoxyphenylboronic acid (0.45 mmol, 82 mg), 1M K2CO3 (0.60 ml) and Pd(PPh3)4 (0.03 mmol, 34 mg). The crude residue was purified using a mixture of cyclohexane/ethylacetate in a ratio of 4:1, affording the title compound in 73 % yield (75 mg, 0.210 mmol). 1H-NMR (300 MHz, CDCl3): δ = 0.67 (q, 2H, CH2), 0.69 (q, 2H, CH2), 1.25 (m, 1H, CH), 3.24 (q, 2H, NCH2), 3.96 (s, 3H, OCH3), 3.98 (s, 3H, OCH3), 6.36 (t, 1H, NH), 7.02 (d, J = 8.34 Hz, 1H, arom H), 7.17 (d, J = 2.07 Hz, 1H, arom H), 7.25 (dd, J = 8.28, J = 2.1 Hz, 1H, arom H), 7.88 (d, J = 1.92 Hz, 1H, arom H), 8.59 (d, J = 1.95 Hz, 1H, arom H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 3.93 (CH2), 10.42 (CH), 53.11 (CH2), 56.07 (OCH3), 110.58 (CH), 111.43 (CH), 120.00 (CH), 124.71 (CH), 130.58 (C), 132.14 (C), 136.74 (C), 144.39 (CH), 149.59 (C), 149.65 (C), 154.71 (C), 171.77 (C) ppm. HR-MS [M+H]+ found 342.1269, calculated 342.1270.
6-(3,4-dimethoxyphenyl)-3-(piperidin-1-yl)isothiazolo[4,3-b]pyridine (13f)
6-Bromo-3-(piperidin-1-yl)isothiazolo[4,3-b]pyridine was prepared from 3,6-di-bromo-isothiazolo[4,3-b]pyridine (0.5 mmol, 146 mg) and piperidine (148 μl, 1.5 mmol) in EtOH (10 ml). The crude product was purified using a mixture of cyclohexane/ethylacetate in a ratio of 4:1, affording 6-bromo-N,N-dimethylisothiazolo[4,3-b]pyridin-3-amine in 84 % yield (125 mg, 0.42 mmol). The title compound was prepared from 6-bromo-3-(piperidin-1-yl)isothiazolo[4,3-b]pyridine (60 mg, 0.2 mmol) using 3,4-dimethoxyphenylboronic acid (0.3 mmol, 54.5 mg), K2CO3 (83 mg, 0.6 mmol) and Pd(dppf)Cl2 (0.01 mmol, 7.4 mg). The crude product was purified using a mixture of cyclohexane/ethylacetate in a ratio of 1:1, affording the title compound in 62 % yield (43 mg, 0.12 mmol). 1H-NMR (300 MHz, CDCl3) δ = 1.74 (m, 6H, 3 × CH2), 3.88 (br s, 4H, 2 × NCH2), 3.91 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 6.95 (d, J = 8.31 Hz, 1H, arom H), 7.13 (s, 1H, arom H), 7.19 (dd, J = 1.93 Hz, J = 8.26 Hz, 1H arom H), 7.80 (d, J = 1.98 Hz, 1H, arom H), 8.57 (d, J = 1.98 Hz, 1H, arom H) ppm. 13C-NMR (300 MHz, CDCl3), δ = 24.0 (CH2), 25.3 (CH2), 51.8 (CH2), 56.0 (CH3), 56.0 (CH3), 110.4 (CH), 111.7 (CH), 119.8 (CH), 124.6 (CH), 130.5 (C), 133.7 (C), 135.3 (C), 143.5 (CH), 149.5 (C), 156.3 (C), 173.5 (C) ppm. HR-MS [M + H]+ found 356.1426, calculated 356.1427.
6-(3,4-dimethoxyphenyl)-3-thiomorpholinoisothiazolo[4,3-b]pyridine (13g)
6-Bromo-3-thiomorpholinoisothiazolo[4,3-b]pyridine was prepared from 3,6-di-bromo-isothiazolo[4,3-b]pyridine (0.5 mmol, 146 mg) and thiomorpholine (151μl, 1.5mmol) in EtOH (10 ml). The product was purified using a mixture of cyclohexane/ethylacetate in a ratio of 4:1, affording the 6-bromo-3-thiomorpholinoisothiazolo[4,3-b]pyridine in 85 % yield (134.5 mg, 0.425 mmol). The title compound was prepared from 6-bromo-3-thiomorpholinoisothiazolo[4,3-b]pyridine (63 mg, 0.2 mmol) using 3,4-dimethoxyphenylboronic acid (0.3 mmol, 54.5 mg), K2CO3 (83 mg, 0.6 mmol) and Pd(dppf)Cl2 (0.01 mmol, 7.4 mg). The crude residue was purified using a mixture of cyclohexane/ethylacetate in a ratio of 1:1, affording the title compound in 51 % yield (38 mg, 0.102 mmol). 1H-NMR (300 MHz, CDCl3) δ = 2.85 (t, J = 4.97 Hz 4H, 2 × SCH2), 3.92 (s, 3H, OCH3), 3.94 (s, 3H, OCH3), 4.29 (t, J = 4.99 Hz 4H, 2 × NCH2), 6.96 (d, J = 8.25 Hz, 1H, arom H), 7.13 (s, 1H, arom H), 7.20 (dd, J = 1.45, J = 8.26 Hz, 1H, arom H), 7.81 (d, J = 1.95 Hz, 1H, arom H), 8.58 (d, J = 1.41 Hz, 1H, arom H) ppm. 13C-NMR (300 MHz, CDCl3), δ = 26.5 (CH2), 53.2 (CH2), 56.0 (CH3), 56.0 (CH3), 110.4 (CH), 111.7 (CH), 119.9 (CH), 124.8 (CH), 130.3 (C), 133.7 (C), 135.6 (C), 144.2 (CH), 149.5 (C), 149.6 (C), 156.6 (C), 172.3 (C) ppm. HR-MS [M + H]+ found 374.0987, calculated 374.0991.
(2R,6S)-4-(6-(3,4-dimethoxyphenyl)isothiazolo[4,3-b]pyridin-3-yl)-2,6-dimethylmorpholine (13h)
(2R,6S)-4-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)-2,6-dimethylmorpholine was prepared from 3,6-di-bromo-isothiazolo[4,3-b]pyridine (0.5 mmol, 146 mg) and cis-2,6-dimethylmopholine (185 μl, 1.5 mmol) in EtOH (10ml). The crude product was purified using a mixture of cyclohexane/ethylacetate in a ratio of 4:1, affording the (2R,6S)-4-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)-2,6-dimethylmorpholine in 74 % yield (121.4 mg, 0.37 mmol). The title compound was prepared from (2R,6S)-4-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)-2,6-dimethylmorpholine (65 mg, 0.2 mmol) using 3,4-dimethoxyphenylboronic acid (0.3 mmol, 54.5 mg), K2CO3 (83 mg, 0.6 mmol) and Pd(dppf)Cl2 (0.01 mmol, 7.4 mg). The crude product was purified using a mixture of cyclohexane/ethylacetate in a ratio of 1:1, affording the title compound in 40 % yield (31 mg, 0.08 mmol). 1H-NMR (300 MHz, CDCl3) δ = 1.31 (d, J = 6.3 Hz, 6H, 2 × CH3), 2.90 (t, J = 11.7 Hz, 2H, 2 × NCH), 3.94 (m, 8H, 2 × OCH3, 2 × OCH), 4.50, (d, J = 12.6 Hz, 2 × NCH), 6.98 (d, J = 8.4 Hz, 1H, arom H), 7.15 (d, J = 2.1 Hz, 1H, arom H), 7.23 (dd, J = 2.2 Hz, J = 8.3 Hz, 1H arom H), 7.84 (d, J = 2.1 Hz, 1H, arom H), 8.62 (d, J = 2.1 Hz, 1H, arom H) ppm. 13C-NMR (300 MHz, CDCl3), δ = 18.9 (CH3), 55.5 (CH2), 56.1 (CH3), 56.1 (CH3), 71.3 (CH), 110.5 (CH), 111.8 (CH), 119.9 (CH), 124.8 (CH), 130.4 (C), 134.0 (C), 135.6 (C), 144.4 (CH), 149.6 (C), 156.5 (C), 172.9 (C) ppm. HR-MS [M + H]+ found 386.1523, calculated 386.1533.
1-(6-(3,4-dimethoxyphenyl)isothiazolo[4,3-b]pyridin-3-yl)piperidin-4-ol (13i)
1-(6-Bromoisothiazolo[4,3-b]pyridin-3-yl)piperidin-4-ol was prepared from 3,6-di-bromo-isothiazolo[4,3-b]pyridine (0.5 mmol, 146 mg) and 4-hydroxypiperidine (1.5 mmol, 151 mg) in EtOH (10 ml). The product was purified using a mixture of DCM/MeOH in a ratio of 95:5, affording 1-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)piperidin-4-ol in 82 % yield (130 mg, 0.414 mmol). The title compound was prepared from 1-(6-bromoisothiazolo[4,3-b]pyridin-3-yl)piperidin-4-ol (63 mg, 0.2 mmol) using 3,4-dimethoxyphenylboronic acid (0.3 mmol, 54.5 mg), 1M K2CO3 (0.4 ml) and Pd(PPh3)4 (0.02 mmol, 23 mg). The product was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 1:1, affording the title compound in 57 % yield (42.8 mg, 0.115 mmol). 1H-NMR (300 MHz, CDCl3): δ = 1.83 (m, 2H, CH2), 2.09 (m, 2H, CH2), 3.70 (m, 2H, CH2), 3.94 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 4.06 (m, 1H, CH), 4.35 (m, 2H, CH2), 6.97 (d, J = 8.34 Hz, 1H, arom H), 7.17 (d, J = 2.07 Hz, 1H, arom H), 7.24 (dd, J = 8.28 Hz, J = 2.07 Hz, 1H, arom H), 7.86 (d, J = 2.01 Hz, 1H, arom H), 8.61 (d, J = 2.04 Hz, 1H, arom H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 33.06 (CH2), 47.76 (CH2), 55.69 (OCH3), 66.07 (CH) 110.11 (CH), 111.40 (CH), 119.56 (CH), 124.22 (CH), 129.97 (C), 133.34 (C), 135.22 (C), 143.58 (CH), 149.18 (C), 149.24 (C), 155.84 (C), 172.69 (C) ppm. HR-MS [M+H]+ found 372.1378, calculated 372.1376.
6-(3,4-dimethoxyphenyl)-N-(tetrahydro-2H-pyran-4-yl)isothiazolo[4,3-b]pyridin-3-amine (13j)
6-Bromo-N-(tetrahydro-2H-pyran-4-yl)isothiazolo[4,3-b]pyridin-3-amine was prepared from 3,6-di-bromo-isothiazolo[4,3-b]pyridine (0.5 mmol, 146 mg) and 4-aminopyrane (0.151 ml, 1.5 mmol) in EtOH (10 ml). The crude product was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 3:1, affording 6-bromo-N-(tetrahydro-2H-pyran-4-yl)isothiazolo[4,3-b]pyridin-3-amine in 40 % yield (64 mg, 0.203 mmol). The title compound was prepared from 6-bromo-N-(tetrahydro-2H-pyran-4-yl)isothiazolo[4,3-b]pyridin-3-amine (64 mg, 0.203 mmol) using 3,4-dimethoxyphenylboronic acid (0.304 mmol, 55.3 mg), 1M K2CO3 (0.406 ml) and Pd(PPh3)4 (0.02 mmol, 23 mg). The crude product was purified using a mixture of cyclohexane/ethyl acetate in a ratio of 1:1, affording the title compound in 16 % yield (5.5 mg, 0.015 mmol). 1H-NMR (300 MHz, CDCl3): δ = 1.72 (m, 2H, 2×CH2), 2.25 (m, 2H, CH2), 3.54 (m, 3H, CH, CH2) 3.95 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 4.12 (m, 2H, CH2), 6.26 (d, J = 7.59, 1H, NH) 6.99 (d, J = 8.34 Hz, 1H, arom H), 7.16 (d, J = 2.04 Hz, 1H, arom H), 7.24 (dd, J = 8.28 Hz, J = 2.04 Hz, 1H, arom H), 7.89 (d, J = 1.89 Hz, 1H, arom H), 8.59 (d, J = 1.89 Hz, 1H, arom H) ppm. 13C-NMR (75 MHz, CDCl3): δ = 31.90 (CH2), 54.18 (CH), 55.73 (CH3), 66.06 (CH2), 110.20 (CH), 111.38 (CH), 119.66 (CH), 124.33 (CH), 130.02 (Cq), 133.15 (Cq), 136.63 (Cq), 144.33 (CH), 149.23 (Cq), 149.32 (Cq), 154.19 (Cq), 169.57 (Cq) ppm. HR-MS [M+H]+ found 372.1377, calculated 372.1376.
6-(3,4-dimethoxyphenyl)-N-phenethylisothiazolo[4,3-b]pyridin-3-amine (13k)
6-Bromo-N-phenethylisothiazolo[4,3-b]pyridin-3-amine was prepared from 3,6-di-bromo-isothiazolo[4,3-b]pyridine (0.5 mmol, 146 mg) and phenethylamine (0.188 ml, 1.5 mmol) in EtOH (10ml). The crude product was purified using a mixture of cyclohexane/ethylacetate in a ratio of 4:1, affording the 6-bromo-N-phenethylisothiazolo[4,3-b]pyridin-3-amine in 77 % yield (130 mg, 0.389 mmol). The title compound was prepared from 6-bromo-N-phenethylisothiazolo[4,3-b]pyridin-3-amine (130 mg, 0.389 mmol) using 3,4-dimethoxyphenylboronic acid (0.583 mmol, 106 mg), 1M K2CO3 (0.78 ml) and Pd(PPh3)4 (0.038 mmol, 45 mg). The crude residue was purified using a mixture of cyclohexane/ethylacetate in a ratio of 9:1, affording the title compound in 25 % yield (39 mg, 0.099 mmol). 1H-NMR (300 MHz, CDCl3): δ = 3.11 (t, J = 7.05 Hz, J = 6.99 Hz, 1H, CH2), 3.66 (m, 2H, CH2), 3.95 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 6.36 (m, 1H, NH) 7.01 (d, J = 8.31 Hz, 1H, arom H), 7.15 (d, 1H, J = 1.89 Hz, arom H), 7.21-7.37 (m, 6H, arom H), 7.87 (d, J = 1.86 Hz, 1H, arom H), 8.66 (d, J = 1.83 Hz, 1H, arom H) ppm. 13C-NMR (125 MHz, CDCl3): δ = 34.60 (CH2), 49.04 (CH2), 55.70 (OCH3), 110.23 (CH), 111.38 (CH), 119.64 (CH), 124.22 (CH), 126.64 (CH), 128.38 (CH), 128.56 (CH), 130.21 (C), 133.15 (C), 136.42 (C), 137.43 (C), 144.30 (CH), 149.20 (C), 149.24 (C), 154.33 (C), 171.39 (C) ppm. HR-MS [M+H]+ found 392.1422, calculated 392.1427.
Structure determination
Recombinant GAK (aa 14-351) was mixed with the GAK-specific NbGAK_4 nanobody (a gift from Serge Muyldermans, Vrije Universiteit Brussel), and the complex was purified as previously described.21 The GAK:NbGAK_4 complex was pre-incubated with 1 mM of compound 12i, and crystallized using the sitting drop vapour diffusion method at 20°C and the reservoir solution containing 9% broad-molecular-weight PEG smears (mixture of PEGs with their molecular weight ranging from 400-10000 Da), 0.1 M MES pH 6.2 and 0.15 M CaCl2. Viable crystals were cryo-protected with mother liquor supplemented with 22% ethylene glycol and flash-frozen in liquid nitrogen. Diffraction data collected at Diamond Light Source, beamline I04-1, were processed and scaled with XDS31 and SCALA from the CCP4 suite32, respectively. Initial structure solution was solved by molecular replacement using PHASER33 and the coordinate of the GAK:NbGAK_4 complex.21 Manual model building in COOT34 alternated with refinement in REFMAC35 was performed, and the geometric correctness of the complete model was verified with MOLPROBITY.36 Data collection and refinement statistics are summarized in Table 2 of the Supporting Information.
Plasmids
pFL-J6/JFH(p7-Rluc2A) was a gift from Dr. C.M. Rice.24 Plasmids used in the HCVpp entry assays (pNL4-3.Luc.R-E, pcDM8 and pcDM8-E1E2) were a gift from Dr. Shoshana Levy.
Cells
Huh-7.5 cells and 293T cells were grown in Dulbecco's modified Eagle medium (DMEM; Mediatech) supplemented with 10% fetal bovine serum (Omega Scientific), nonessential amino acids (Gibco), 1% L-glutamine (Gibco), and 1% penicillin-streptomycin (Gibco), and maintained in 5% CO2 at 37°C.
In vitro transcription of viral RNA, transfection, and HCVcc generation
HCV RNA was generated and delivered into Huh-7.5 cells, as previously described.37 Briefly, RNA was reverse transcribed from XbaI-linearized J6/JFH(p7-Rluc2A) template using the T7 MEGAscript kit according to the manufacturer's instructions (Ambion). Viral RNA was purified using the RNeasy kit (Qiagen). 6×106 Huh7.5 cells were washed three times with ice-cold RNase-free PBS (BioWhittaker) and electroporated (0.82 kV, five 99 μs pulses) with 2 μg of viral RNA in a 2 mm-gap cuvette (BTX) using a BTX-830 electroporator. After a 15 min recovery at room temperature, cells were resuspended in pre-warmed growth medium and plated into 96 or 6-well culture plates. For HCVcc generation, cells were diluted in 30ml of prewarmed growth medium and plated. Viral supernatants were collected daily from up to 5 passages of the electroporated cells, filtered through a 0.22 micron cellulose nitrate filter, and kept at -80°C. Viral titers were determined by limiting dilution and immunohistochemical staining using an antibody directed to core. 50% tissue culture infectious dose (TCID50) was calculated, as described.36 Results are expressed as TCID50/ml.
HCVcc infection
6×103 Huh-7.5 cells seeded in 96-well plates were infected in triplicates with HCVcc J6/JFH(p7-Rluc2A) at MOI (multiplicity of infection) of 0.1 in the presence of serial dilutions of the compounds. Culture medium was replaced daily with medium containing serial dilutions of the inhibitors. HCVcc infection was measured by standard luciferase assays at 72 hours postinfection, using a Renilla luciferase substrate and a Tecan luminometer (Tecan) according to the manufacturers' protocols. Alternatively, 4 hours postinfection, cells were washed and medium was replaced, followed by standard luciferase assays at 24 hours postinfection.
HCV RNA replication by luciferase assays
HCV RNA replication was measured at 72 hours postelectroporation, as described.8 Electroporated cells plated in quadruplicates in 96-well plates were washed twice with PBS and lysed with 30μl of Renilla lysis buffer (Promega). Following 15 minute shaking at room temperature, luciferase activity was quantified by standard luciferase assays.
Extracellular and Intracellular infectivity
Huh-7.5 cells electroporated with J6/JFH(p7-Rluc2A) RNA and plated in 6-well dishes were treated every 24 hours with GAK inhibitors for a total of 72 hours. For measurements of extracellular infectivity, supernatants were harvested, filtered using a 0.22-μm-pore size filter and used to infect naïve Huh-7.5 cells in triplicates. For intracellular infectivity measurements, electroporated cells were trypsinized, collected by centrifugation, resuspended in 500 μl medium, lysed by 4 freeze-thaw cycles, and pelleted at 3,650×g. Clarified supernatants were diluted in complete medium and used to inoculate naive Huh-7.5 cells in triplicates. At 72 hours postinfection cells were lysed and luciferase activity quantified as above.
HCVpp production and entry assays
HCVpp (H77c strain, genotype 1a) were generated as described previously.26 Briefly, 293T cells were transfected with a 1:1 ratio of plasmids encoding HIV provirus expressing luciferase and HCV E1E2 envelope glycoproteins. Supernatants were harvested 48 hours posttransfection and filtered. Huh-7.5 cells were infected with HCVpp and 8 mg/ml polybrene (Sigma) for 4 hours. Cell lysates were collected at 48 hours after HCVpp infection, and firefly luciferase (Promega) activity was measured using a Tecan luminometer (Tecan).
Viability assay
Following treatment with GAK inhibitors, Huh-7.5 cells either infected with HCVcc and HCVpp or electroporated with HCV RNA were incubated for 2–4 hours with media supplemented with 10% AlamarBlue reagent (TREK Diagnostic Systems) at 37°C. Fluorescence at 560nm was measured via FLEXstation II 384 (Molecular Devices, Inc.) as readout of cellular metabolic activity.
The effect of the compounds on AP2M1 phosphorylation
Huh-7.5 cells were treated with various concentrations of the compounds or DMSO in serum free medium. To allow capturing of the phosphorylated AP2M1 state, 100nM of the PP2A inhibitor calyculin A was added to all wells 30 min after drug administration. 1 hour later cells were lysed and samples subjected to SDS-PAGE and blotting with antibodies targeting AP2M1 (Santa Cruz Biotechnology) and p-AP2M1 (Cell Signaling).
Supplementary Material
Acknowledgments
This research was supported by a grant from the IWT (Agentschap voor Innovatie door Wetenschap en Technologie-Vlaanderen; Grant IWT-SBO 100014; to P.H). Mass spectrometry was made possible by the support of the Hercules Foundation of the Flemish Government (grant 20100225–7).
This work was supported in part by grants from Stanford Clinical and Translational Science Award (CTSA) to Spectrum (UL1 TR001085), Stanford SPARK program, Stanford Bio-X, Stanford Translational Research and Applied Medicine Program, grant 2013100 from the Doris Duke Charitable Foundation, and grant RSG-14-11 0-0 1-MPC from the American Cancer Society to S.E.
G.N. is supported by the Child Health Research Institute, Luclie Packard Foundation for Children's Health, as well as the Stanford CSTA (grant number UL1 TR000093). We thank Dr. Charles M. Rice for kindly providing us the pFL-J6/JFH, pFL-J6/JFH(p7-Rluc2A) plasmid, Apath LLC (St. Louis, MO) for providing Huh-7.5 cells, and Dr. Shoshana Levy for providing the plasmids used in the HCVpp entry assays (pNL4-3.Luc.R-E, pcDM8 and pcDM8-E1E2).
SK is supported by the SGC, a registered charity (number 1097737) that receives funds from AbbVie, Bayer, Boehringer Ingelheim, the Canada Foundation for Innovation, the Canadian Institutes for Health Research, Genome Canada, GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust [092809/Z/10/Z]. AC is supported by the European Union FP7 Grant No. 278568 “PRIMES” (Protein interaction machines in oncogenic EGF receptor signalling).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- AAK1
adaptor-associated kinase
- AcOH
acetic acid
- AP-2
Adaptor protein complex 2
- ATP
adenosine triphosphate
- BIKE
BMP-2 inducible kinase
- CCVs
clathrin-coated vesicles
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- EGFR
Epidermal Growth Factor Receptor
- GAK
Cyclin-G associated kinase
- HCV
Hepatitis C virus
- Kd
binding affinity constant
- LE
ligand efficiency
- NAK
numb-associated kinase
- ND
not determined
- rt
room temperature
- SAR
structure-activity relationship
- THF
tetrahydrofuran
- STK16/MPSK1
Serine/threonine kinase 16/myristoylated and palmitoylated serine/threonine kinase 1
Footnotes
Supporting Information: NMR spectra of representative compounds, HPLC purity data of final compounds, as well as data collection and refinement statistics for the X-ray crystallography. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kanaoka Y, Kimura SH, Okazaki I, Ikeda M, Nojima H. GAK: a cyclin G associated kinase contains a tensin/auxilin-like domain. FEBS Lett. 1997;402:73–80. doi: 10.1016/s0014-5793(96)01484-6. [DOI] [PubMed] [Google Scholar]
- 2.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 3.Lee DW, Zhao X, Zhang F, Eisenberg E, Greene LE. Depletion of GAK/auxilin 2 inhibits receptor-mediated endocytosis and recruitment of both clathrin and clathrin adaptors. J Cell Sci. 2005;118:4311–4321. doi: 10.1242/jcs.02548. [DOI] [PubMed] [Google Scholar]
- 4.Korolchuk VI, Banting G. CK2 and GAK/auxilin2 are major protein kinases in clathrin-coated vesicles. Traffic. 2002;3:428–439. doi: 10.1034/j.1600-0854.2002.30606.x. [DOI] [PubMed] [Google Scholar]
- 5.Zhang CX, Engqvist-Goldstein ÅEY, Carreno S, Owen DJ, Smythe E, Drubin DG. Multiple roles for cyclin G-associated kinase in clathrin-mediated sorting events. Traffic. 2005;6:1103–1113. doi: 10.1111/j.1600-0854.2005.00346.x. [DOI] [PubMed] [Google Scholar]
- 6.Zhao X, Greener T, Al-Hasani H, Cushman SW, Eisenberg E, Greene LE. Expression of auxilin or AP180 inhibits endocytosis by mislocalizing clathrin: evidence for formation of nascent pits containing AP1 or AP2 but not clathrin. J Cell Sci. 2001;114:353–365. doi: 10.1242/jcs.114.2.353. [DOI] [PubMed] [Google Scholar]
- 7.Zhang L, Gjoerup O, Roberts TM. The serine threonine kinase cyclin G-associated kinase regulates epidermal growth factor receptor signaling. Proc Natl Acad Sci. 2004;101:10296–10301. doi: 10.1073/pnas.0403175101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neveu G, Barouch-Bentov R, Ziv-Av A, Gerber D, Jacob Y, Einav S. Identification and targeting of an interaction between a tyrosine motif within hepatitis C virus core protein and AP2M1 essential for viral assembly. PLoS Pathog. 2012;8(8):e1002845. doi: 10.1371/journal.ppat.1002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 10.(2008) SuperNova Life Science, Human Kinome Heat Map, http://www.supernovalifescience.com/HM/HM%2041.pdf
- 11.Neveu G, Ziv-Av A, Barouch-Bentov R, Bekerman E, Mulholland J, Einav S. AAK1 and GAK regulate hepatitis C virus entry and are potential drug targets. J Virology. doi: 10.1128/JVI.02705-14. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fabian MA, Biggs WH, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lélias J, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HP, Zarrinkar PP, Lockhart DJ. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 13.Bellei B, Pitisci A, Migliano E, Cardinali G, Picardo M. Pyridinyl imidazole compounds interfere with melanosomes sorting through the inhibition of cyclin G-associated kinase, a regulator of cathepsins maturation. Cellular Signalling. 2014;26:716–723. doi: 10.1016/j.cellsig.2013.12.023. [DOI] [PubMed] [Google Scholar]
- 14.Meiby E, Knapp S, Elkins JM, Ohlson S. Fragment screening of cyclin G-associated kinase by weak affinity chromatography. Anal Bioanal Chem. 2012;404:2417–2425. doi: 10.1007/s00216-012-6335-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Sakurai MA, Ozaki Y, Okuzaki D, Naito Y, Sasakura T, Okamoto A, Tabara H, Inoue T, Hagiyama M, Ito A, Yabuta N, Nojima H. Gefitinib and luteolin cause growth arrest of human prostate cancer PC-3 cells via inhibition of cyclin G-associated kinase and induction of miR-630. PLoS One. 2014;9(6):e100124. doi: 10.1371/journal.pone.0100124. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Susa M, Choy E, Liu X, Schwab J, Hornicek FJ, Mankin H, Duan Z. Cyclin G-associated kinase is necessary for osteosarcoma cell proliferation and receptor trafficking. Mol Cancer Ther. 2010;9:3342–3350. doi: 10.1158/1535-7163.MCT-10-0637. [DOI] [PubMed] [Google Scholar]
- 16.Dzamko N, Zhou J, Huang Y, Halliday GM. Parkinson's disease-implicated kinases in the brain; insights into disease pathogenesis. Front Mol Neurosci. 2014;24:7–57. doi: 10.3389/fnmol.2014.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuduk SD, Di Marco CN, Chang RK, Ray WJ, Mab L, Wittmann M, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MT. Heterocyclic fused pyridone carboxylic acid M1 positive allosteric modulators. Bioorg Med Chem Lett. 2010;20:2533–2537. doi: 10.1016/j.bmcl.2010.02.096. [DOI] [PubMed] [Google Scholar]
- 18.Wohlfahrt G, Tormakangas O, Salo H, Hoglund L, Karjalainen A, Knuutila P, Holm P, Rasku S, Vesalainen A. Androgen receptor modulating compounds. WO2011/051540
- 19.Zhang N, Ayral-Kaloustian S, Mansour T, Nguyen T, Niu C, Rosfjord E, Suayan R, Tsou H. 2-aryl and 2-heteroarylthiazolyl compounds, methods for their preparation and use there of. WO2009/120826
- 20.Taurins A, Tan Khouw V. Isothiazolopyridines. I. synthesis and spectra of isothiazolo[3,4-b]-, 3-amino-isothiazolo[4,3-b]-, isothiazolo[5,4-b]-, and 3-methylisothiazolo[5,4-c]pyridines. Preparation and spectra of some 2,3- and 3,4-disubstitutedpyridines. Can J Chem. 1973;51:1741–1748. [Google Scholar]
- 21.Miyaura N, Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem Rev. 1995;95:2457–2483. [Google Scholar]
- 22.Chaikuad A, Keates T, Vincke C, Kaufholz M, Zenn M, Zimmermann B, Gutiérrez C, Zhang RG, Hatzos-Skintges C, Joachimiak A, Muyldermans S, Herberg FW, Knapp S, Müller S. Structure of cyclin G-associated kinase (GAK) trapped in different conformations using nanobodies. Biochem J. 2014;459:59–69. doi: 10.1042/BJ20131399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Z, Wu H, Wang L, Liu Y, Knapp S, Liu Q, Gray NS. Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery? ACS Chem Biol. 2014;9:1230–1241. doi: 10.1021/cb500129t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset F, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoël M, Raffelsberger W, Poch O, Mckeating JA, Brino LA, Baumert TF. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murray CL, Jones CT, Tassello J, Rice CM. Alanine scanning of the hepatitis C virus core protein reveals numerous residues essential for production of infectious virus. J Virol. 2007;81:10220–10231. doi: 10.1128/JVI.00793-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med. 2003;197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ricotta D, Hansen J, Preiss C, Teichert D, Höning S. Characterization of a Protein Phosphatase 2A Holoenzyme That Dephosphorylates the Clathrin Adaptors AP-1 and AP-2. J Biol Chem. 2008;283:5510–5517. doi: 10.1074/jbc.M707166200. [DOI] [PubMed] [Google Scholar]
- 28.Bhattacharyya S, Warfield KL, Ruthel G, Bavari S, Aman MJ, Hope TJ. Ebola virus uses clathrin-mediated endocytosis as an entry pathway. Virology. 2010;401:18–28. doi: 10.1016/j.virol.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin M, Park J, Lee S, Park B, Shin J, Song KJ, Ahn TI, Hwang SY, Ahn BY, Ahn K. Hantaan virus enters cells by clathrin-dependent receptor-mediated endocytosis. Virology. 2002;294:60–69. doi: 10.1006/viro.2001.1303. [DOI] [PubMed] [Google Scholar]
- 30.Boge M, Wyss SP, Bonifacino JS, Thali M. A membrane-proximal tyrosine-based signal mediates internalization of the HIV-1 envelope glycoprotein via interaction with the AP-2 clathrin adaptor. J Biol Chem. 1998;273:15773–15778. doi: 10.1074/jbc.273.25.15773. [DOI] [PubMed] [Google Scholar]
- 31.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 33.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 36.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.