

Abstract
Background
Infection with HIV can result in a debilitating CNS disorder known as HIV dementia (HIV-D). Since the advent of highly active antiretroviral therapy (HAART), the incidence of HIV-D has declined, but the prevalence continues to increase. In this new era of HIV-D, traditional biomarkers such as CSF viral load and monocyte chemotactic protein 1 levels are less likely to be associated with dementia in patients on HAART and biomarkers that can predict HIV-D have not yet been identified.
Objective
To identify biomarkers that are associated with and can predict HIV-D.
Methods
We grouped patients with HIV based on changes in cognitive status over a 1-year period and analyzed sphingolipid, sterol, triglyceride, antioxidant, and lipid peroxidation levels in CSF.
Results
We found that increased levels of the vitamin E and triglyceride C52 predicted the onset or worsening of dementia. Elevated levels of sphingomyelin were associated with inactive dementia. Elevated levels of ceramide and the accumulation of 4-hydroxynonenals were associated with active dementia.
Conclusions
We interpret these findings to indicate that early in the pathogenesis of HIV dementia, there is an up-regulation of endogenous antioxidant defenses in brain. The failure of this attempted neuroprotective mechanism leads to the accumulation of sphingomyelin and moderate cognitive dysfunction. The breakdown of this enlarged pool of sphingomyelin to ceramide and the accumulation of highly reactive aldehydes are associated with declining cognitive function. Thus, elevations in endogenous protective mechanisms may identify patients who are at increased risk of the development of HIV dementia.
In the post–highly active antiretroviral therapy (HAART) era, the severity of cognitive dysfunction in patients infected with HIV has declined and the course of dementia is more variable. A milder form of cognitive impairment, HIV-associated minor cognitive motor disorder (MC/MD), is now the most frequent form of cognitive impairment in patients infected with HIV.1 These clinical observations suggest that the biopathology of HIV dementia (HIV-D) may be different in the pre- and the post-HAART era and demonstrates the importance of defining new predictive and associative biomarkers that may predict the onset, progression, or relative risk of HIV-D. Based on previous findings that sphingomyelin, ceramide, sterol, and lipid peroxidation products accumulate in the brains of patients with HIV-D,2,3 we hypothesized that these lipids may also be useful biomarkers to predict dementia in this patient population.
Using quantitative electrospray ionization tandem mass spectrometry, we sought to determine whether changes in sphingolipid, sterol, and oxidant balance are associated with, and can predict, changes in the cognitive status of patients with HIV-D. Our findings suggest that elevations of sphingomyelin and ceramide and the accumulation of lipid peroxidation products are associated with declining cognitive status. A decrease in the cholesterol ester C16 and increases in tocopherol and triglyceride C52 predict the worsening of cognitive status. These biomarkers may identify patients who are at greater risk of HIV-associated cognitive dysfunction who would benefit from neuroprotective therapy.
METHODS
Human CSF samples
A total of 48 CSF samples were obtained from the Johns Hopkins HIV neurology repository from patients included in the North Eastern AIDS Dementia (NEAD) cohort. All CSF samples were centrifuged, and cell-free CSF was aliquoted and immediately frozen at −80 °C. The patient population consisted of 14 women and 34 men; six were white, 41 were African American, and one was Hispanic. Risk factors for HIV infection were distributed among IV drug users (n = 22), homosexual transmission (n = 11), heterosexual transmission (n = 14), dual risk factors of IV drug users/heterosexual transmission (n = 6), IV drug use/homosexual transmission (n = 2), and unknown (n = 8). Exclusion criteria for subjects in the NEAD cohort included current or past opportunistic CNS infection at study entry, with the exception of treated neurosyphilis, a history of or current severe affective disorder believed to explain the subject’s cognitive impairment, history of chronic neurologic disorder such as MS, head injury with loss of consciousness for longer than 1 hour, stroke, or uncontrolled epilepsy. The majority of patients were on HAART; a complete list of combinational drug use by each participant is provided in tables E-1 and E-2 (available on the Neurology Web site at www.neurology.org). Based on a complete neurologic examination, neuropsychological testing, and functional assessments performed in the NEAD cohort,4 HIV patients were categorized at each of three visits (interspersed by 6 months) using the Memorial Sloan-Kettering Scale (MSK). The MSK ratings were assigned by a consensus conference including a neurologist and neuropsychologist. Raters were blinded to all the CSF biomarker data. Patients were classified as either normal or HIV+ impaired but not severe enough to meet criteria for HIV-D (MSK score, 0 to 0.5), HIV+ mild dementia (MSK score, 1), and HIV+ moderate dementia (MSK score, 2). Based on changes in MSK score from visit 1 to visit 2, patients were further classified into no dementia (HIV-ND; n = 21); active dementia (HIV-AD; n = 10), defined as a transition from a nondemented status to dementia within the past 6 months; and inactive dementia (HIV-ID; n = 17), defined as a dementia status unchanged over the preceding 6-month period. This categorization was used to identify biochemical changes that are associated with dementia. Based on changes in MSK scores from visit 2 to visit 3, patients were classified as stable (MSK-stb; n = 22), defined as a dementia status unchanged over the 6-month period; worse (MSK-wor; n = 11), defined by an MSK score that increased from visit 2 to visit 3; and improved (MSK-imp; n = 7), defined by an MSK score that decreased from visit 2 to visit 3 (neuropsychological data for visit 3 were not available for eight patients due to death or study discontinuation). This categorization was used to identify biochemical changes that were predictive of the onset or worsening of dementia. CSF drawn at visit 2 was used for analysis (figure 1). Data on plasma viral RNA was available for 37 patients and CSF viral RNA for 23 patients. All patients were similar in age: HIV-ND = 45 ± 7 years, HIV-ID = 44 ± 8 years, HIV-AD = 50 ± 6 years; education, HIV-ND = 11.9 ± 2 years, HIV-ID = 11.8 ± 2 years, and HIV-AD = 12.5 ± 1.5 years. Demographic information of patients by group is shown in table 1.
Figure 1. Schematic representation of study design.
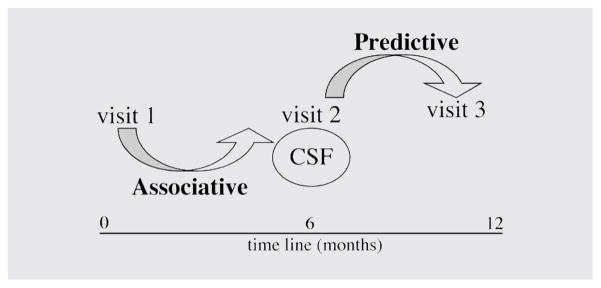
Patients were classified using the Memorial Sloan-Kettering Scale (MSK). Associative biomarkers were identified by grouping patients based on changes in MSK score from visit 1 to visit 2 into no dementia (HIV-ND); active dementia (HIV-AD; n = 10), defined as a transition from a nondemented status to dementia within the last 6 months; and inactive dementia (HIV-ID; n = 17), defined as a dementia status unchanged over the preceding 6-month period. Predictive biomarkers were identified by grouping patients based on changes in MSK scores from visit 2 to visit 3 as stable (HIV-stb), defined as a dementia status unchanged over the 6-month period; worsening cognitive function (MSK-wor), defined by an MSK score that increased from visit 2 to visit 3; and improved cognitive function (MSK-imp), defined by an MSK score that decreased from visit 2 to visit 3. CSF drawn at visit 2 was used for analysis.
Table.
Demographic statistics of patients by group
| Associative grouping | Age, y | Educ, y | Gender (M/F) | CD4 (cells/mm3) | Plasma viral load, copies/mL (range) | CSF viral load, copies/mL (range) | MSK score |
|---|---|---|---|---|---|---|---|
| HIV-ND | 45 ± 7 | 12 ± 2 | 18 M/3 F | 159 ± 141 | 10,000 (130–280,000) | 3,300 (150–80,000) | 0.1 ± 0.2 |
|
| |||||||
| HIV-ID | 44 ± 8 | 12 ± 2 | 11 M/6 F | 156 ± 146 | 83,000 (300–150,000) | 4,800 (590–80,000) | 1.4 ± 0.7 |
| HIV-AD | 50 ± 6 | 13 ± 2 | 5 M/5 F | 137 ± 115 | 110,000 (25,000–1,000,000) | 7,600 (530–210,000) | 1.4 ± 0.6 |
|
| |||||||
| Predictive grouping | |||||||
|
| |||||||
| MSK-stb | 46 ± 8 | 12 ± 2 | 15 M/3 F | 176 ± 151 | 14,700 (130–1,100,000) | 3,850 (150–210,000) | 0.9 ± 0.8 |
|
| |||||||
| MSK-imp | 49 ± 6 | 11 ± 2 | 5 M/2 F | 76 ± 61 | 9,800 (400–190,000) | 1,650 (1000–17,000) | 0.4 ± 0.4 |
|
| |||||||
| MSK-wor | 45 ± 5 | 13 ± 2 | 4 M/6 F | 115 ± 111 | 120,000 (300–730,000) | 5,75093 (300–80,000) | 1.0 ± 1.0 |
Age, education (Educ), CD4 count, and MSK (Memorial Sloan-Kettering Scale) score are represented as mean ± SD. Plasma and CSF viral load are expressed as median score and range.
ND = no dementia; ID = inactive dementia; AD = active dementia; MSK = Memorial Sloan-Kettering Scale; stb = stable; imp = improved; wor = worse.
Lipid extraction and measurements of sphingolipids, phospholipids, sterols, and lipid peroxides
Total lipids from CSF samples were prepared as previously described3 by diluting 500 μL of CSF in three volumes of 100% methanol containing 30 mM ammonium acetate and then vortexing. Four volumes of chloroform were then added, and the mixture was vortexed and then centrifuged at 1,000g for 10 minutes. The bottom (chloroform) layer was removed and analyzed by direct injection into a tandem mass spectrometer. Electrospray ionization tandem mass-spectrometry (ESI/MS/MS) analyses were performed using methods similar to those used in our previous studies.2,3 Samples were injected using a Harvard Apparatus pump at 15 μL/min into an electrospray ionization (i.e., Turbo Ion Spray module) Sciex API 3000 triple-stage quadruple tandem mass spectrometer from Sciex Inc. (Thornhill, Ontario, Canada) operated in the positive mode. The ion spray voltage (V) was 5,500 at a temperature of 80 °C with a nebulizer gas of 8 psi, curtain gas of 8 psi, and the collision gas set at 4 psi. The declustering potential was 80 V, the focusing potential 400 V, the entrance potential −10 V, the collision energy 30 V, and the collision cell exit potential was 18 V. The MS/MS scanned from 300 to 2,000 atomic mass units (amu) per second at a step of 0.1 amu. Each species of sphingolipids, phospholipids, cholesterol esters, and lipid peroxides initially was identified by a Q1 mass scan and then by precursor ion scanning or neutral loss scanning of a purified standard. Samples were injected into the ESI/MS/MS for 3 minutes, where the mass counts accumulated and the sum of the total counts under each peak was used to quantify each species. Cholesterol; cholesterol ester standards C16:0, C18:0, and C18:1; and cholesteryl-arachidonate (C20:0) were purchased from Sigma. Sphingomyelins; ceramides C16:0, C18:0, C20:0, C22:0, C24:0, and C24:1; phosphatidylcholine C16:0 to C18:1 and C18:0 to C18:1; phosphatidylethanolamine C16:0 to C18:1; phosphatidylglycerol C16:0 to C18:1; phosphatidylserine C16:0 to C18:1; phosphatidylinositol C16:0 to C18:1; and phosphatidic acid C16:0 to C18:1; and triglyceride C52 (palmitic, oleic, stearic) were purchased from Avanti Polar Lipids (Alabaster, AL). Palmitoyl-lactosyl ceramide C16:0 to C16:0, stearoyl-lactosyl ceramide C16:0 to C18:0, lignoceryl-glucosyl ceramide C16:0 to C24:0, lignoceryl-galactosyl ceramide C16:0 to C24:0, and stearoyl-galactosyl ceramide sulfate C18: to C24:0 were purchased from Matreya Inc. (Pleasant Gap, PA). 4-Hydroxynonenol and adducts, 8-isoprostane, stearidonic acid, 8-iso-15-keto prostaglandin (PG)E2, 8-iso-15-keto PGF2α, 8-iso-15(R) PGF2α, 8-iso PGA1, 8-iso PGE1, 8-iso PGE2, and 8-iso PGE2 isopropyl ester were purchased from Cayman Chemicals.
Monocyte chemotactic protein 1 (MCP-1) ELISA
MCP-1 levels in CSF were determined using a Quantikine human MCP-1/CCL2 immunoassay (R&D Systems Inc., Minneapolis, MN) according to the manufacturer’s instructions for cell culture supernatant samples. Briefly, each CSF sample was thawed on ice, and 250 μL of each sample was mixed with an equal volume of RD5L calibrator diluent. Two hundred micro-liters per well of each diluted sample was plated in duplicate wells. Standards were prepared in RD5L calibrator diluent. Readings at 450 nm were acquired on a VersaMax tunable microplate reader (Molecular Devices, Sunnyvale, CA) and quantified by densitometry (SOFTmax Pro, version 3.13).
RESULTS
Viral load and chemokine measures associated with HIV-D
Based on previous reports that have suggested an association between viral load, cytokine balance, and cognitive function in HIV-D, we sought to determine whether viral load and MCP-1 imbalance were associated with dementia progression in patients from the NEAD cohort. We found no significant association between plasma or CSF viral load, MCP-1 levels, and cognitive status (figure 2A through C). Specifically, there was no difference in plasma or CSF viral load in patients with HIV-ND compared with patients with HIV-ID and a trend toward an increase in plasma viral load in patients with HIV-AD (figure 2A and B). MCP-1 levels in CSF were comparable among patients with HIV-ND, HIV-ID, and HIV-AD (figure 2C).
Figure 2. Viral load and monocyte chemotactic protein 1 (MCP-1) levels are not associated with an active dementia and cannot predict changes in cognitive status.

(A to C) Plasma (A) or CSF (B) viral load or CSF MCP-1 levels (C) were not associated with changes in cognitive status when we compared HIV-infected patients with no dementia (HIV-ND), inactive dementia (HIV-ID), or active dementia (HIV-AD). (D through F) Plasma (D) or CSF (E) viral load, or MCP-1 levels (F) were not predictive of changes in cognitive status when we compared HIV-infected patients with HIV-ID, worsening cognitive function (MSK-wor), or improved cognitive function (MSK-imp) MSK = Memorial Sloan-Kettering Scale.
CSF and plasma viral load or MCP-1 levels were also not predictive of changes in cognitive status (figure 2D through F). There was a trend toward increased plasma viral load in patients with MSK-wor compared with patients with MSK-stb and MSK-imp (figure 2D) and comparable CSF viral loads in all three groups of patients (figure 2E). MCP-1 levels were not different among patients with MSK-stb, MSK-wor, and MSK-imp (figure 2F).
Dysfunctions of sphingolipid and sterol metabolism are associated with active dementia in patients infected with HIV
We identified biomarkers that are associated with the progression of dementia by categorizing patients into HIV-ND, HIV-ID, and HIV-AD based on changes in cognitive status that occurred during the 6-month period preceding lumbar tap. In patients with HIV-ID, we found significant increases in sphingomyelin C16:0, C18:0, C20:0, C22:0, and C24:0 (figure 3A) vs patients with HIV-ND or HIV-AD. Although sphingomyelin concentrations were not different in patients with HIV-AD compared with patients with HIV-ND, there were significant increases in ceramide C18:0, C22:0, C24:0, C24:1, and sulfatide in the HIV-AD group (figure 3B), suggesting that a conversion of sphingomyelin to ceramide is associated with the progression of dementia. Consistent with this hypothesis, we found an increase of sphingomyelinase activity in the CSF of patients with HIV-AD (figure 3C). There were no differences in free cholesterol or the cholesterol esters C16:0 and C18:1 or triglyceride C52 when we compared patients with HIV-ND with those with HIV-ID to HIV-AD ( figure 3D and E). There was, however, a reduction in the concentration of triglyceride C52 in the CSF of patients with HIV-AD compared with patients with HIV-ND or HIV-ID (figure 3F). CSF levels of the lysine and histidine adducts of 4-hydroynonenal were increased and the endogenous antioxidant vitamin E was decreased in patients with HIV-AD (figure 3G). Together these results suggest that a stable dementia is associated with the accumulation of sphingomyelin. An active dementia is associated with dysfunctional sphingolipid metabolism that results in the accumulation of sphingomyelin, ceramide, and 4-hydroxynonenal concurrent with a reduced antioxidant potential.
Figure 3. Increased CSF levels of ceramide and 4-hydroxynoneal (4-HNE) are associated with active dementia.

CSF levels of sphingolipids, sterols, triglyceride C52, reactive aldehyde, and tocopherol were determined by electrospray ionization tandem mass spectrometry (ESI/MS/MS). (A) Levels of sphingomyelin C16:0, C18:0, C20:0, C22:0, and C24:0 were increased in patients with inactive dementia (HIV-ID) compared with patients with no dementia (HIV-ND) or patients with active dementia (HIV-AD). (B) The ceramides C22:0, C24:0, and C24:1 and sulfatide were increased in HIV-AD patients compared with HIV-ND and HIV-ID patients. (C) Sphingomyelinase activity was increased in HIV-AD compared with HIV-ND or HIV-ID patients. (D) Levels of free cholesterol and the cholesterol esters C16:0 and C18:1 were similar in all HIV patient groups. (E) Levels of 25-hydroxycholesterol in CSF were decreased in HIV-AD patients compared with HIV-ND and HIV-ID groups. 25-OH = 25-hydroxy. (F) Triglyceride C52 levels were similar in all patient groups. (G) CSF levels of the reactive aldehydes 2-pentilpyrrole histidine-hydroxynoneal and 2-pentilpyrrole lysine-hydroxynoneal were increased in HIV-AD patients compared with HIV-ND and HIV-ID patients. (H) Vitamin E levels were decreased in patients with HIV-AD compared with HIV-ND and HIV-ID patient groups. **p < 0.01; ***p < 0.001. Analysis of variance with Tukey post hoc comparisons.
Accumulations of tocopherol and triglyceride C52 predict the worsening of dementia in HIV-1–infected patients
We identified biomarkers that predict the progression of dementia by categorizing patients into MSK-stb, MSK-imp, and MSK-wor based on changes in cognitive status that occurred during the 6-month period following lumbar tap. There were no differences in the concentrations of sphingomyelin or ceramide when we compared patients with MSK-stb to patients with MSK-imp and MSK-wor (figure 4A and B). Levels of free cholesterol and the cholesterol ester C:18:1 were similar in all groups; however, there was a decline in the amount of the cholesterol ester C16:0 in patients with MSK-wor compared with patients with MSK-stb or MSK-imp (figure 4C). There were no differences among groups in levels of 25-hydroxycholesterol (figure 4D). The concentration of triglyceride C52 was increased in patients with MSK-wor compared with the MSK-stb group (figure 4E). Although levels of the reactive aldehyde 4-hydroxynonenal were comparable between the three groups, levels of the endogenous antioxidant vitamin E were elevated in patients with MSK-wor compared with patients with MSK-stb or MSK-imp (figure 4F and G). These findings suggest that decreased cholesterol storage and the accumulations of triglyceride C52 and vitamin E may predict the onset or worsening of dementia in HIV-infected patients.
Figure 4. Increased CSF levels of triglyceride C52 and vitamin E predict the onset or worsening of dementia in HIV-infected patients.

(A and B) There were no differences in the levels of any sphingomyelins or ceramides tested when we compared MSK-stb, MSK-imp and MSK-wor patient groups. (C) Although levels of free cholesterol were similar in all groups, the cholesterol esters C16:0 and C18:1 were decreased in MSK-wor patients compared with MSK-stb and MSK-imp patient groups. (D) Levels of 25-hydroxycholesterol were similar in all patient groups. (E) The long-chain triglyceride C52 was increased in MSK-wor patients compared with MSK-stb or MSK-imp. (F) 4-Hydroxynonenal (4-HNE) levels were similar in all patient groups. (G) The antioxidant vitamin E was increased in the CSF of patients with MSK-wor compared with the MSK-stb and MSK-imp patient groups. 25-OH = 25-hydroxy; ND = no dementia; MSK = Memorial Sloan-Kettering Scale; stb = stable; imp = improved; wor = worse.
DISCUSSION
Since the advent of HAART, the cognitive manifestations of HIV-D are less severe and the course of HIV-D appears to be more variable. The onset of dementia in patients infected with HIV is no longer predictive of mortality, and there are frequently bidirectional transitions in cognitive status. In this new, more complex era of HIV-D, traditional biomarkers such as CSF viral load and MCP-1 levels are less likely to be associated with dementia in patients on HAART. Furthermore, there are no other biomarkers that can predict the onset or worsening of HIV-D. The biomarkers that we identified that can predict the onset or worsening of dementia or are associated with a worsening dementia in HIV-infected patients are depicted in figure 5. The ability to identify patients who are at high risk of developing dementia is critical for the development of early and effective treatments designed to maintain normal cognition and good quality of life.
Figure 5. Biomarkers that are predictive of or are associated with dementia.
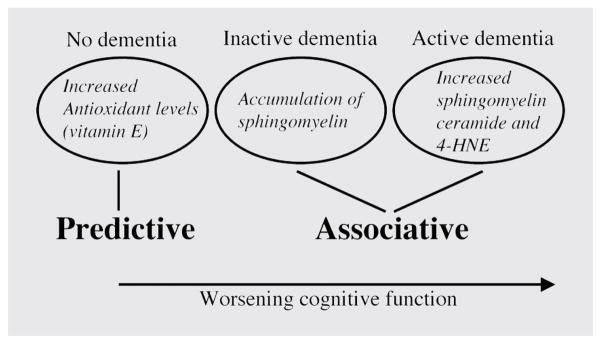
Early in the course of HIV dementia (HIV-D), when no dementia is apparent, increased levels of endogenous antioxidants (vitamin E), may predict the worsening of cognitive function. Mild cognitive impairment is associated with the accumulation of sphingomyelin and decreased levels of vitamin E (inactive dementia). Worsening cognitive function is associated with increased levels of sphingomyelin, ceramide, and the reactive aldehyde 4-hydroxynonenal (4-HNE).
Abnormalities in sphingolipid metabolism have been reported in the brains and CSF of patients with HIV-D.3 Tissue culture models of HIV-D have shown that the HIV-1 coat protein gp120 and the trans acting protein Tat can increase levels of sphingomyelin in 6 hours followed by increases of ceramide within 12 hours followed by accumulation of reactive aldehydes in 24 hours.3,5 Using pharmacologic and biochemical methods, it was found that neutral sphingomyelinase was mechanistically involved in gp120 and Tat-induced accumulations of ceramide.3,5 Neutral sphingomyelinase is thought to reside on the inner leaflet of the plasma membrane and can be activated by inflammatory cytokines such as tumor necrosis factor α, interleukin-1, and FasL to promote the hydrolysis of the cell membrane phospholipid sphingomyelin to ceramide. CNS inflammation is thought to play a prominent role in the etiology of HIV-D and may be an early event that leads to neural dysfunction.6 Consistent with this hypothesis, we found accumulations of sphingomyelin in the CSF of patients with HIV-ID, suggesting that there may be an early neuropathogenic event that interferes with normal sphingolipid metabolism. These findings are reminiscent of accumulations of sphingomyelin that occur in Niemann-Pick disease. In Niemann-Pick sphingomyelinosis (types A and B), a deficiency of the lysosomal acidic sphingomyelinase leads to the widespread lysosomal deposition of sphingomyelin.7 Recently, it was reported that the lysosomal apparatus in the subcortical white matter in brains from persons with AIDS is expanded.8 Using a representative lysosomal glycosidase, they found a correlation between neuro-cognitive impairment score and brain frontal lobe white matter lysosomal enzyme activity. Combined with our results, these findings suggest that there may be an inhibition of sphingomyelinase or an overproduction of sphingolipids that results in the accumulation of sphingomyelin in lysosymes early in the progression of HIV-D. A subsequent inflammatory/oxidative event in the CNS would convert this enlarged pool of sphingomyelin to ceramide, leading to neuronal dysfunction and death. A second consequence of sphingomyelinase catabolizing sphingomyelin is a disruption of the association between cholesterol and sphingomyelin. The release of free cholesterol in this reaction can result in the accumulation of cholesterol esters in vacuoles with triglycerides (i.e., lipid droplets). Cholesterol and triglycerides are normally removed from cells by lipid carriers such as apoproteins, of which ApoE is the most abundant in the brain. Because the movement of sterols is rate limited by the availability of lipid carriers, large increases in cholesterol and triglycerides can overwhelm the system and accumulate in cells as storage forms of sterols and triglycerides. Thus, changes in sterols levels may reflect neurodegenerative changes. Indeed, disruption of cholesterol and triglyceride balance can modify synapse integrity,9 increase neuronal vulnerability to insults,10 and may play roles in the pathogenesis of Alzheimer disease, ALS, and HIV-D.2,11,12
The nervous system is particularly susceptible to the damaging effects of free radicals for several reasons. The brain contains high concentrations of polyunsaturated fatty acids that are susceptible to lipid peroxidation. This lipid composition, coupled with large amounts of oxygen and a relatively deficient antioxidant system (compared with other tissues), renders neural cells vulnerable to oxidative damage. Thus, the antioxidant defenses present in brain are critically important to protect neural tissues from oxidative damage. The critical role of vitamin E in neurologic health and disease was recognized several decades ago.13,14 Since then, vitamin E research has been highly focused on the antioxidant properties of α-tocopherol (or its analog Trolox), although there are at least eight substances with vitamin E activity: α-, β-, γ-, and δ-tocopherol and α-, β-, γ-, and δ-tocotrienol. The neuroprotective properties of vitamin E have been documented in a number of neurodegenerative disease models including HIV-1 protein-induced neuronal toxicity.15–18 In our analysis of CSF from patients infected with HIV patients, we found that increases of vitamin E predicted the onset or decline in cognitive function of HIV-D patients. Based on these observations, we believe that increases in vitamin E in the CSF of patients with declining cognitive function reflects an endogenous antioxidant defense that is increasing in response to an ongoing neuropathology. If our hypothesis is correct, then it would be reasonable to expect that increases of other endogenous antioxidants would likewise predict the onset or worsening of dementia in HIV-1–infected patients. These studies are under way in a larger group of patients infected with HIV.
A second biomarker that predicted the worsening of dementia in HIV-AD patients was triglyceride C52. In a recent aging study, it was found that plasma triglyceride C52 increased linearly with age, whereas the levels of other long-chain triglycerides (C50, C54, and C56) were unchanged.19 One interpretation of these findings is that the accumulation of triglyceride C52 with age protected neural cells in the aging brain. Similar neuroprotective results have been reported in rodent seizure models, where a ketogenic diet is neuroprotective presumably by changes in fatty acid metabolism that result in increased concentrations of polyunsaturated fatty acids in the brain.20 The effects of a ketogenic diet in transgenic models of HIV-D may provide insight into the role of lipid metabolism in aging patients with HIV-D.
Progressive multifocal leukoencephalopathy (PML) is a demyelinating disease that can occur in immunocompromised patients such as those afflicted with AIDS. Although the exact biochemical changes in white matter that are responsible for PML are not known, the accumulation of long-chain fatty acids in other demyelinating diseases including adrenoleukodystrophy21,22 and solvent vapor abuse leukoencephalopathy23 have been described. The accumulations of long-chain sphingomyelins and ceramides that we observed in the CSF of HIV patients with inactive and active dementia may thus reflect ongoing damage to white matter in the CNS.
Supplementary Material
Acknowledgments
Supported in part by the National Institute on Aging Intramural Research Program and by the NIH grants AG023471 and MH068388 to N.J.H., MH071150 to J.CM., and NS49465 to N.S.
The authors thank Patrick Cottingham for technical assistance.
Footnotes
Disclosure: The authors report no conflicts of interest.
References
- 1.McArthur JC, McDermott MP, McClernon D, et al. Attenuated central nervous system infection in advanced HIV/AIDS with combination antiretroviral therapy. Arch Neurol. 2004;61:1687–1696. doi: 10.1001/archneur.61.11.1687. [DOI] [PubMed] [Google Scholar]
- 2.Cutler RG, Haughey NJ, Tammara A, et al. Dysregulation of sphingolipid and sterol metabolism by ApoE4 in HIV dementia. Neurology. 2004;63:626–630. doi: 10.1212/01.wnl.0000134662.19883.06. [DOI] [PubMed] [Google Scholar]
- 3.Haughey NJ, Cutler RG, Tamara A, et al. Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann Neurol. 2004;55:257–267. doi: 10.1002/ana.10828. [DOI] [PubMed] [Google Scholar]
- 4.Sacktor N. The epidemiology of human immunodeficiency virus-associated neurological disease in the era of highly active antiretroviral therapy. J Neurovirol. 2002;8(suppl 2):115–121. doi: 10.1080/13550280290101094. [DOI] [PubMed] [Google Scholar]
- 5.Jana A, Pahan K. Human immunodeficiency virus type 1 gp120 induces apoptosis in human primary neurons through redox-regulated activation of neutral sphingomyelinase. J Neurosci. 2004;24:9531–9540. doi: 10.1523/JNEUROSCI.3085-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalez-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 7.da Silva V, Vassella F, Bischoff A, Spycher M, Wiesmann UN, Herschkowitz N. Niemann-Pick’s disease. Clinical, biochemical and ultrastructural findings in a case of the infantile form. J Neurol. 1975;211:61–68. doi: 10.1007/BF00312464. [DOI] [PubMed] [Google Scholar]
- 8.Gelman BB, Soukup VM, Holzer CE, 3rd, et al. Potential role for white matter lysosome expansion in HIV-associated dementia. J Acquir Immune Defic Syndr. 2005;39:422–425. doi: 10.1097/01.qai.0000164250.41475.f2. [DOI] [PubMed] [Google Scholar]
- 9.Mauch DH, Nagler K, Schumacher S, et al. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- 10.Chou YC, Lin SB, Tsai LH, Tsai HI, Lin CM. Cholesterol deficiency increases the vulnerability of hippocampal glia in primary culture to glutamate-induced excitotoxicity. Neurochem Int. 2003;43:197–209. doi: 10.1016/s0197-0186(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 11.Marx J. Alzheimer’s disease. Bad for the heart, bad for the mind? Science. 2001;294:508–509. doi: 10.1126/science.294.5542.508. [DOI] [PubMed] [Google Scholar]
- 12.Cutler RG, Pedersen WA, Camandola S, Rothstein JD, Mattson MP. Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Ann Neurol. 2002;52:448–457. doi: 10.1002/ana.10312. [DOI] [PubMed] [Google Scholar]
- 13.Muller DP, Goss-Sampson MA. Role of vitamin E in neural tissue. Ann NY Acad Sci. 1989;570:146–155. doi: 10.1111/j.1749-6632.1989.tb14915.x. [DOI] [PubMed] [Google Scholar]
- 14.Muller DP, Goss-Sampson MA. Neurochemical, neurophysiological, and neuropathological studies in vitamin E deficiency. Crit Rev Neurobiol. 1990;5:239–263. [PubMed] [Google Scholar]
- 15.Behl C. Vitamin E and other antioxidants in neuroprotection. Int J Vitam Nutr Res. 1999;69:213–219. doi: 10.1024/0300-9831.69.3.213. [DOI] [PubMed] [Google Scholar]
- 16.Viviani B, Corsini E, Binaglia M, Galli CL, Marinovich M. Reactive oxygen species generated by glia are responsible for neuron death induced by human immunodeficiency virus-glycoprotein 120 in vitro. Neuroscience. 2001;107:51–58. doi: 10.1016/s0306-4522(01)00332-3. [DOI] [PubMed] [Google Scholar]
- 17.Sen CK, Khanna S, Roy S. Tocotrienol: the natural vitamin E to defend the nervous system? Ann NY Acad Sci. 2004;1031:127–142. doi: 10.1196/annals.1331.013. [DOI] [PubMed] [Google Scholar]
- 18.Turchan J, Pocernich CB, Gairola C, et al. Oxidative stress in HIV demented patients and protection ex vivo with novel antioxidants. Neurology. 2003;60:307–314. doi: 10.1212/01.wnl.0000042048.85204.3d. [DOI] [PubMed] [Google Scholar]
- 19.Carlile SI, Kudchodkar BJ, Wang CS, Lacko AG. Age-related changes in the rate of esterification of plasma cholesterol in Fischer-344 rats. Mech Ageing Dev. 1986;33:211–220. doi: 10.1016/0047-6374(86)90028-x. [DOI] [PubMed] [Google Scholar]
- 20.Dell CA, Likhodii SS, Musa K, Ryan MA, Burnham WM, Cunnane SC. Lipid and fatty acid profiles in rats consuming different high-fat ketogenic diets. Lipids. 2001;36:373–378. doi: 10.1007/s11745-001-0730-8. [DOI] [PubMed] [Google Scholar]
- 21.Schaumburg HH, Powers JM, Raine CS, et al. Adrenoleu-kodystrophy: a clinical, pathological and biochemical study. Adv Exp Med Biol. 1976;68:379–387. doi: 10.1007/978-1-4684-7735-1_25. [DOI] [PubMed] [Google Scholar]
- 22.Moser HW, Moser AB, Kawamura N, et al. Adrenoleu-kodystrophy: studies of the phenotype, genetics and biochemistry. Johns Hopkins Med J. 1980;147:217–224. [PubMed] [Google Scholar]
- 23.Kornfeld M, Moser AB, Moser HW, Kleinschmidt-DeMasters B, Nolte K, Phelps A. Solvent vapor abuse leukoencephalopathy. Comparison to adrenoleukodystrophy. J Neuropathol Exp Neurol. 1994;53:389–398. doi: 10.1097/00005072-199407000-00011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.