

Abstract
Airway inflammation, lung remodeling, and Airway Hyperresponsiveness (AHR) are major features of asthma and Chronic Obstructive Pulmonary Disease (COPD). The inflammatory response to allergens, air pollutants, and other insults is likely to play a key role in promoting structural changes in the lung including the overabundance of Airway Smooth Muscle (ASM) seen in asthmatics. These alterations or remodeling could, in turn, impact the immunmodulatory actions of the ASM, the ASM's contractile properties, and the development of AHR. New evidences suggest that airway inflammation and AHR are not tightly related to each other and that the structural component of the airway, mainly the ASM, is a chief driver of AHR. Members of the S100/calgranulins family have been implicated in the regulation of inflammation and cell apoptosis in various systems. S100A12 is highly expressed in neutrophils and is one of the most abundant proteins in the lungs of patients with asthma or COPD. Studies with genetic engineered mice with smooth muscle cell-targeted expression of human S100A12 revealed that S100A12 reduces airway smooth muscle amounts and dampens airway inflammation and airway hyperreactivity in a model of allergic lung inflammation. Thus, targeting airway smooth muscle for instance through delivery of pro-apoptotic S100A12 could represent an attractive means to promote ASM apoptosis and to reduce ASM abundance in asthmatics.
Introduction
The incidence of respiratory disorders including asthma and Chronic Obstructive Pulmonary Disease (COPD) has dramatically increased worldwide over the last decades [1]. Asthma is an inflammatory lung disease associated with reversible airway obstruction, structural changes or remodeling, and Airway Hyperresponsiveness (AHR) to a variety of stimuli [2]. The relationship between airway inflammation and AHR has been the focus of numerous studies [3] in humans and also in various animal models. AHR is an abnormal, exaggerated constrictor response that can be attributed to changes in the structure of the airway [4] and the inflammatory processes [5]. The augmented sensitivity and greater ability of the airway of asthmatics to narrow in response to contractile agents is likely the result of multiple synergistic events that take place within the airway milieu and that are influenced by intrapulmonary and extrapulmonary events. Although in general asthmatics with more severe airway disease often have higher intensity of AHR, the degree of AHR largely varies among patients or even within the same individual.
The ability of the smooth muscle to generate forceful contractions is a chief component in causing narrowing during regulation of airway diameter and vascular tone. We previously found that exogenous expression of human S100A12 in aortic smooth muscle of mice causes loss of contractile SM fibers, which contributes to weakening of the aortic wall with development of vascular aneurysms [6]. Indeed, members of the S100/calgranulin family such as S100A8 (Calgranulin A, myeloid-related protein-8), S100A9 (Calgranulin B, myeloid related protein 14), and S100A12 (Calgranulin C, EN-RAGE) have been implicated in the regulation of inflammation and apoptosis [7,8]. These Ca2+-binding proteins are commonly considered pro-inflammatory at least in part because they can bind to pattern recognition Receptor for Advanced Glycation End products (RAGE) [7] and to Toll-Like receptor 4 (TLR4) [9]. Importantly, S100/calgranulins are highly expressed endogenously in neutrophils wherein S100A8/9 and S100A12 compose up to 40% and up to 5% of the cytosolic proteins, respectively, and are induced in other cells including monocytes, macrophages, epithelial cells, endothelial cells, and smooth muscle cells in response to cytokine stimulation. Moreover, S100/calgranulins are one of the most abundant proteins found in the Bronchoalveloar Lavage Fluid (BALF) or sputum of patients with inflamed lungs like those with asthma, COPD, and Acute Respiratory Distress Syndrome (ARDS) [10-12]. Because of their abundance in inflamed lungs and their pro-apoptotic function, it is tempting to speculate that S100/calgranulins might not only function as markers of inflammatory cell infiltration but they could also negatively impact the amount of the smooth muscle in the airways. This review summarizes the interrelationship between the smooth muscle function with lung inflammation as well as some of unexpected findings observed in allergen sensitized and challenged transgenic mice that express human S100A12 in their smooth muscle.
Airway Inflammation can Promote AHR
A wide array of factors and interactions including host-related, developmental, environmental, and microbial (virus and bacteria) may influence the processes of inflammation and AHR. It has been presumed that if inflammation is perpetuated, the cellular components of the underlying injury become important agents in the development of structural changes in the airway and can lead to worsening AHR in susceptible individuals. During inflammation, release of mediators such as histamine and leukotrienes could modulate ASM contractility [13] as well as airway remodeling through the action of chemokines and cytokines [14]. In asthmatics, inhalation of allergens or occupational sensitizers causes an inflammatory response in the airways, mainly eosinophilic, which is associated with an increase in AHR [15]. A temporal relationship of airway inflammatory events and increase in airway responsiveness after exposure to allergens has been well documented [16] and we will not discuss it here.
During infancy, a high serum level of IgE is a strong predisposing factor for the development of AHR [17]. Onset or worsening of AHR occurs after viral respiratory infections, which is associated with the presence of increased inflammatory cells in the sputum [18]. The potential mechanisms of these exaggerated responses may involve epithelial cell death and epithelial barrier damage, which could result in reduction of epithelial-derived relaxing factors such as PGE2 and in exposure of sensory terminals to the inflamed environment [19]. Additionally, it was shown in animal models that parainfluenza virus infection has detrimental effects through both the blockade in the ability of β2-adrenoceptor agonists to inhibit contraction [20] and an increase in the number of secreting mast cells [21]. Indeed, impaired adrenergic responses and enhanced mast cell activity are tightly associated with sustained AHR. Interestingly, studies have shown that healthy and asthmatic subjects experience epithelial damage after vaccination with influenza virus but AHR develops only in the asthmatics [22]. This indicates that additional factors including genetic, neural, or humoral could participate in the initiation and persistence of AHR.
S100 Proteins, RAGE, and Lung Inflammation
S100A12 is a 12 kDa protein that belongs to the large family of calcium binding EF-hand proteins. It is primarily expressed in the cytosol of myeloid cells and can be secreted from cells upon activation of protein kinase C. Expression and release of S100A12 often occurs in response to cytokine stimulation, cell injury or cell death thus serving as an “alarmin” signal to activate the innate and adaptive immune systems and to promote either tissue repair or removal of injured cells. When S100A12 binds to its receptor RAGE, which is present in a wide variety of cell types, it stimulates those cells to secrete multiple pro-inflammatory cytokines such as TNFα, IL-1β, and IL-6 [7]. Stimulation with recombinant S100A12 activates human and murine mast cells, leading to release of histamine and cytokines, even in cells lacking RAGE [11]. Together, and because S100A12 is highly abundant in inflamed lungs, it was suggested that S100A12 could be a candidate pathologic mediator of the inflammatory process in asthma.
Despite the role of S100A12 in activating inflammatory pathways in multiple cell types, definite evidence that S100A12 directly mediates allergic inflammation in the lung is lacking. Because S100A12 is very abundant in neutrophil granulocytes and is secreted upon activation, it is difficult to establish whether augmented levels of S100A12 in subjects with asthma are simply reflecting an increased cellular inflammatory milieu, or whether S100A12 is directly and actively involved in the exacerbation process, and therefore, could represent a candidate for anti-inflammatory intervention as suggested previously [23,24]. To directly test the role of S100A12 in mediating lung inflammation and / or affecting airway responsiveness, we exploited the fact that mice do not contain the S100A12 gene [25] and generated transgenic mice that express the human S100A12 in smooth muscle tissues, directed by the SM22α promoter (SM22α-S100A12 mice) [26]. Surprisingly, we found that mice expressing S100A12 in their smooth muscles actually have less allergen-induced lung inflammation, decreased mucus production, and diminished eosinophilia after ovalbumin (OVA) sensitization and challenge, a commonly used experimental asthma model. Remarkably, SM22α-S100A12 mice exhibited dilated airways and thinner airway wall with loss of smooth muscle (Figure 1, modified from Ref [26]); these features share a reassembly with the phenotype observed in the vasculature [6]. Airway smooth muscle myocytes are relatively resistant to programmed cell death, and to our knowledge, there is no quantitative information on human ASM myocytes apoptosis in vivo. It is known however, that the Extracellular Matrix (ECM) plays both a protecting and a promoting role on apoptosis of ASM cells, at least in vitro. The ECM of human airways contains fibronectin, elastin, and collagen I and IV, all of which are deposited by ASM myocytes in cell culture. These proteins are important anti-apoptotic components that provide survival signals to ASM cells [27]. On the other hand, it was also shown that human ASM myocytes seeded on decorin exhibit increased apoptosis [28]. We demonstrated that the reduced amounts of ASM in SM22α-S100A12 mice likely stemmed from cell apoptosis because isolated cultured human ASM cells treated with S100A12 showed augmented expression of the death receptor Fas [26]. We reported earlier that increased expression of Fas in cultured airway smooth muscle myocytes resulted in enhanced cell death when stimulated with TNFα [29]. Importantly, thinning of the airway smooth muscle layer as observed in the SM22α-S100A12 mice was associated with impaired airway contractile response to the bronchoconstrictor agent methacholine. Because the ASM is a rich source of pro-inflammatory molecules, it is conceivable that reduced amounts of ASM in SM22α-S100A12 mice might account directly or indirectly for the improved inflammation score in the lungs and for the reduction in eosinophilia in the BALF of mice subjected to OVA sensitization and challenge. However, transgenic expression of S100A12 in smooth muscle did enhance some features of lung inflammation like increased elastolysis of the airway elastic fibers and enhanced perivascular fibrosis, which underscores the pleiotropic role of S100A12 [26]. While our results raise the possibility that S100A12 may not enhance all aspects of allergic lung inflammation, it should be noticed that our experimental model has some limitations because of the forced, non-physiological over-expression of S100A12 in the smooth muscle. Thus, to further understand the role of S100A12 in asthma, we generated transgenic mice that produce and release S100A12 from the cells that normally express this protein in humans rather than from forced expression in smooth muscle. Our BAC-S100 transgenic mice express human S100A12 in its own genetic context within the S100 gene cluster contained in a bacterial artificial chromosome and is driven by the endogenous promoters. In this model, human S100A8/9 and S100A12 are expressed in myeloid cells and are present in the serum at concentrations similar to those found in human conditions. Analysis of the extent of lung inflammation and airway responsiveness in allergen sensitized and challenged BAC-S100 mice will be crucial to uncover the function of human S100A12 in lung inflammation without the limitations associated with protein over-expression using the smooth muscle specific promoter SM22α, and should therefore dissect the separate roles of airway smooth muscle loss and of S100A12 abundance in inflamed lungs. Preliminary results utilizing the experimental model of OVA-induced allergic lung inflammation revealed no difference in lung inflammation or AHR between BAC-S100 mice and wild type littermate mice, suggesting that myeloid derived S100A8/9 and S100A12 proteins do not exacerbate allergic lung inflammation (unpublished data).
Figure 1.
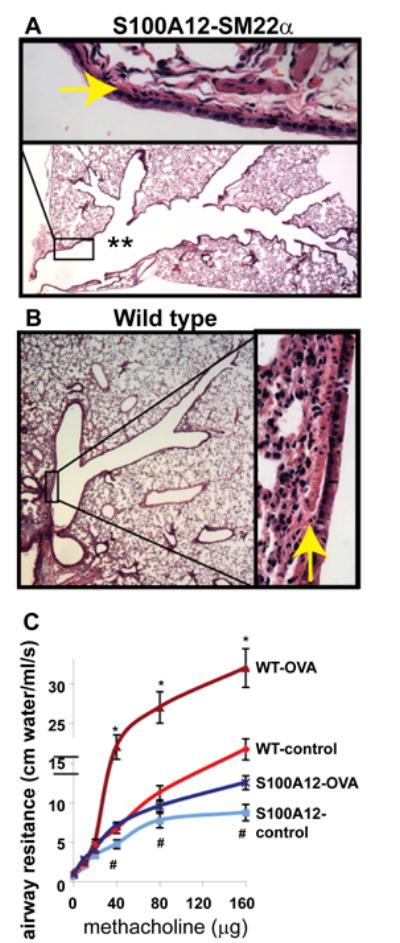
SM22α-S100A12 transgenic mice have dilated airways (** in A), reduced smooth muscle abundance (arrows in A and B), and impaired response to bronchoconstrictor methacholine at baseline (control) or after allergen sensitization and challenge (OVA) compared to wild type littermate mice (modified from reference 26).
Several rodent models of inflammatory respiratory diseases have shown large abundance of S100A8/9 and other members of the S100 family in inflamed lungs [30,31] consistent with the findings in human lung diseases. However, administration of neutralizing antibodies to S100A8 or S100A9 as potential therapeutic strategy had no effect on airway hyperresponiveness, had minimal effect on lung tissue inflammation, and reduced only slightly eosinophil or neutrophil cell counts in BALF [31,32]. Moreover, Yin et al. found that injection of recombinant S100A9-thioredoxin fusion protein at doses from 0.1-10 μg/kg reduced airway resistance in OVA sensitized and challenged rats, although neither the route of administration of recombinant S100A9 nor analysis of surrogate markers of lung inflammation were reported [33]. The authors hypothesized that a direct effect of S100A9 on airway smooth muscle cell contractility occurred in vivo because isolated rat trachea preparations showed impaired constriction in response to S100A9 added to the incubation medium [33]. Although there is mounting evidence that S100/calgranulins are induced in inflammatory lung disease, their complex biological functions are partially understood. S100 proteins' actions would likely depend on several factors including their structure/function properties, access to cell surface receptors, and composition of the cellular milieu. For instance, it was shown that S100A8, S100A9 and to a lesser extend S100A12, are susceptible to reversible protein modification in response to generation of Reactive Oxygen Species and Reactive Nitrogen Species (ROS/RNS) that involve cysteine, methionine, lysine, histidine, arginine, and proline residues. These oxidative modifications alter, at least in vitro, the biological function of S100 proteins resulting in limited inflammation [34,35]. In a recent review about oxidative modifications of S100 proteins, Geczy and colleagues hypothesized on the elevated serum levels of S100/calgranulins in chronic inflammatory diseases and suggested that S100A8/9 might represent a component of the anti-oxidative defense system, due to their ability to quench ROS/RNS [36].
Mechanistically, S100 proteins exert their actions by binding to RAGE, a multi-ligand receptor that also associates with other structurally different ligands including Advanced Glycation End products (AGE), High Mobility Group Box 1 protein (HMGB1), amyloid β fibrils, and Mac-1 (reviewed in [37]). RAGE was first isolated from lung tissue [38] and is endogenously expressed at high levels in lung alveolar type I cells [39]. Moreover, bovine whole lung protein extract was used to search for “natural” ligands to RAGE, an approach that led to the discovery of amphoterin (HMGB1) and S100A12 as binding proteins to RAGE more than a decade ago [7,40]. However, neither the precise role of RAGE nor its natural ligands within the lung have been well defined. Only recently an association between reduced pulmonary RAGE signaling and the development of pulmonary fibrosis were reported in humans and in mice [41]. While it remains unclear why RAGE deficiency has detrimental effects on lung function, these findings strongly suggest that the high abundance of RAGE in the lung might exert a homeostatic function. The role of RAGE has not been well studied in the context of lung biology, and to our knowledge, RAGE function has not been formerly tested in mouse models of allergic lung inflammation. Interestingly, the high expression of RAGE in normal lung is in marked contrast to the vasculature, in which RAGE is minimally expressed in healthy blood vessels but is up-regulated in response to injury, hyperglycemia, hypoxia, cytokine exposure, in chronic inflammatory diseases, and with aging. Studies from our laboratory and from other groups have focused on RAGE signaling in the vasculature, and consistently found activation of RAGE in endothelial cells, vascular smooth muscle cells, and inflammatory cells mediating an inflammatory phenotype. Consistent with a pro-inflammatory and disease-mediating role of RAGE and its ligands in the vasculature, several studies reported beneficial effects of RAGE deficiency in atherosclerosis, sepsis, wound healing, and organ transplantation in mice [37,42,43]. Whereas experimental evidence implicates RAGE as a prime player in orchestrating vascular inflammation associated with atherosclerosis, it raises the questions of whether, why, and how RAGE is operating differently in the lung. Future investigations using mice with conditional deficiency of RAGE in specific cell types would bring insights into the selective roles of RAGE in the vasculature and in the lung.
The plasma and body fluids of humans contain considerable amounts of soluble RAGE, isoforms of RAGE that harbor the ligand binding V-domain but lacks the transmembrane and intracellular domains of RAGE [44]. The access of S100 proteins and other RAGE ligands to cell surface receptors is affected by the presence and concentration of this decoy receptor soluble RAGE. There are at least two forms of soluble RAGE, each generated by distinct mechanisms. Total soluble RAGE is mostly derived from proteolytic cleavage of cell surface RAGE, and endogenous secretory RAGE (esRAGE) derives from spliced variants of RAGE mRNA. esRAGE has amino acids specific to splice variants [45] and a recent report describes the full range of human RAGE gene splice variants [46]. The study of circulating forms of soluble RAGE in cardiovascular and other inflammatory diseases has become an area of high interest. However, mixed results have been published suggesting either low or high levels of soluble RAGE as putative biomarkers for the presence or extent of inflammatory lung disease. For example, the study by Sukkar in patients with neutrophilic asthma or neutrophilic COPD found a marked deficiency of sRAGE in the BALF and reduced serum levels of sRAGE [47]. One possible explanation for this finding is enhanced proteolytic degradation of sRAGE in patients with neutrophilc lung disease, since recombinant sRAGE instilled into the lungs of patients with neutrophilic asthma was only partially recovered in this study. Because sRAGE is a decoy for ligands that normally would activate (pro-inflammatory) cell surface receptors such as RAGE or TLR, it is attractive to speculate that correcting sRAGE deficiency could represent a therapeutic strategy for neutrophilic asthma and neutrophilic COPD. However, and in contrast to the findings of reduced sRAGE in patients with neutrophilic asthma/COPD found by Sukkar, a study by Watanabe et al. showed elevated levels of esRAGE in the sputum of 44 asthmatic patients [48]. Intriguing differences between the two studies is the lower amount of neutrophils in the sputum in the study by Watanabe, which could have contributed to lesser proteolytic degradation of sRAGE/esRAGE, and the use of different ELISA's for the measurement of sRAGE and esRAGE. Similar conflicting results of either elevated or reduced sRAGE have been reported in studies of cardiovascular disease, particularly in diabetes. Therefore, more investigation is needed to better understand the role of sRAGE in disease, as it becomes clear that the plasma levels and turnover of soluble RAGE depend on many factors, including (i) renal function, as this contributes of sRAGE/esRAGE clearance; (ii) the amount of cell surface RAGE and the activity of proteases (ADAMTS10 and other enzymes), as RAGE is the substrate for proteolytic shedding that produces circulating forms of sRAGE; (iii) the degradation rate of sRAGE/esRAGE; (iv) the presence of medications or other drugs; (v) the contribution of different spliced variants of esRAGE; and (vi) the sensitivity and specificity of different assays to analyze total sRAGE or esRAGE. Moreover, our laboratory extensively studied the G82S coding polymorphism of RAGE that maps to the major site of the ligand-binding V-domain. RAGE harboring the G82S substitution exhibits enhanced binding to S100A12 [49] suggesting that not only the concentration of RAGE ligands and soluble RAGE but also the presence of certain polymorphisms within the receptor could modulate lung inflammation. Taken together, the high abundance of RAGE and its ligands in the lung deserve further studies to better understand their function in lung biology and disease.
Airway Smooth Muscle as Contractile and Immuno-modulatory Unit
The findings about the relationship between AHR and chronic airway inflammation have been inconsistent [50]. In some instances, weak positive correlations were observed between the degree of AHR and the presence of one or more of the inflammatory markers, mainly eosinophils, T lymphocytes, and epithelial shedding. Other studies showed that AHR could be elicited in the absence of demonstrable inflammatory cells in the airway lumen or mucosa [51], or after interventions that reduced inflammation [52], or even in an antigen-free setting [53]. Moreover, a pre-clinical study reported a negative association of AHR with inflammation [54]. Therefore, because lung inflammation is most likely a single contributor to airflow limitation, a renewed interest has emerged in understanding the influence of the smooth muscle and/or the characteristics of airway smooth muscle to the presence of AHR [3].
As mentioned above, several studies point out that it is the structural status of the airway rather than the presence of inflammation that is important to AHR. Sont et al. showed that inhaled corticosteroids directed toward reducing AHR improved lung function and correlated with reduced airway wall remodeling [55]. On the other hand, administration of anti-IL5 antibody that effectively decreased blood and sputum eosinophil counts resulted in no change in pulmonary functions or AHR [56]. Interestingly, a study of asthmatics after allergen challenge demonstrated that alterations in AHR seemed to be associated more with structural lung modifications, as measured by procollagen-expressing cells, than with early recruitment of luminal eosinophils and ensuing inflammation [57]. Although Oliver's group [58] and others have presented strong evidence that the increased muscle mass in asthmatics is a dominant factor contributing to AHR, abnormal intrinsic properties of airway smooth muscle myocytes [4] as well as alternative mechanisms involving neurohumoral control of ASM tone [59] should not be excluded. Moreover, it is conceivable that AHR independent of inflammation could develop due to influences beyond ASM contractility or autonomic regulation. For example, methacholine hyperresponsiveness after lung transplantation is commonly observed, and it is not clearly related to airway inflammation [60]. Interestingly, a relationship of AHR with different inflammatory cell types (metachromatic cells, eosinophils) and their distinct locations within the airway wall (submucosa, lumen) has also been reported [61].
Numerous studies have showed that human ASM cells can contribute to the inflammatory process by secreting an array of inflammatory cytokines and chemokines [62], extracellular matrix components [63] and cell adhesion molecules [64]. These mediators in turn, modulate lung inflammation, smooth muscle myocyte gene expression, and AHR [65]. Several immunoactive substances, some of which have been detected at elevated levels in asthmatic subjects, are secreted by airway smooth muscle cells [66]. Molecules that promote recruitment, activation, and survival of inflammatory cells like eotaxin, RANTES, IL-5, and IL-8 [67], or that modify various aspects of airway physiology like antigen processing Th1-Th2 switch including IL-4, TGF-β, GM-CSF, and IL-1β, or that exert anti-inflammatory actions like IL-10, have all been reported to be produced, at least in culture, by ASM myocytes. In vivo, these molecules modulate submucosal airway remodeling and inflammation and suggest an autocrine function for ASM that may induce airway remodeling through a mechanism of hyperplasia [68,69]. Thus, these mediators could influence transient or sustained hyperresponsiveness upon exposure to triggers.
In addition to its participation in the acquired immunity through the ability to bind IgE and elicit the secretion of IL-1β, IL-6, IL-8, eotoxin, TNFα, TSLP, and other inflammatory mediators, the ASM myocyte also contributes to the innate immunity. Indeed, ASM cells express mRNA of Toll-Like Receptor- (TLR) 1 to 10 [70]. Engagement of TLR2 and TLR4 with peptidoglycan and lipopolysaccaride (LPS), and of TLR3 and TLR8/9 with RNA induces IL-8 and eotaxin release from airway myocytes [70]. Epidemiological studies indicate that exposure to LPS, a main component of gram-negative bacteria, appears to be protective in children under particular living settings [71], or to promote asthma in adults in some permissive occupational environments, underlying the complexity of pathways and genes involved in adaptive and innate immune responses [72].
Therefore, the smooth muscle myocyte is emerging as a key structural cell that is well situated within the airway to quickly and robustly modulate the airway microenvironment and potentially affect the initiation, perpetuation, amplification and resolution of airway inflammation.
Therapeutic Interventions to Modulate Smooth Muscle Abundance
It has been demonstrated that bronchial thermoplasty, a novel intervention that ablates ASM with heat, produced sustained relief from airway bronchoconstriction and symptomatic improvement in patients suffering from moderate and severe asthma [73,74]. As described above, we recently reported that S100A12 induces apoptosis of human airway smooth muscle myocytes, and that transgenic expression of human S100A12 in pulmonary smooth muscle of mice results in attenuated native and allergen-induced airway responsiveness and exhibit enlarged airways with less airway smooth muscle compared to wild type mice. Therefore, it is tempting to speculate that the diminution of the abundance of ASM in SM22α-S100A12 mice explains, at least partially, both the protective effect on bronchoconstriction and the partial inflammation developed within the lung. Thus, therapeutic administration of S100A12 protein to the airway smooth muscle could be envisioned as a means to reduce muscle mass and consequently, improve chronic lung inflammation and constrictor responsiveness. Indeed, S100A12 and related S100A8/9 are known to promote apoptosis in a variety of cells [8]. This effect stems at least in part from enhanced oxidative stress because S100A8/9 activates p67phox in phagocytes [75] and our work suggests a similar function for S100A12 in smooth muscle cells through activation of NADPH oxidase subunit Nox-1 [76]. We speculate that in human asthma, the extensive remodeling of the airway wall and/or the presence (or absence) of unknown factors prevent S100A12 protein expressed in eosinophilis and neutrophils to reach and/or interact effectively with the airway smooth muscle cells to induce apoptosis. On the other hand, we hypothesize that in SM22α-S100A12 mice, over-expressed S10012 from the smooth muscle is brought in close contact with airway myocytes enabling the promotion of myocyte cell death. Hence, we would like to propose that nanotechnology-based delivery of S100A12 protein to the airway smooth muscle of patients with poorly controlled asthma could result in ablation of excessive airway smooth muscle, potentially antagonizing exaggerated contractility and inflammation associated with abnormal ASM abundance.
Nanoparticle-based delivery systems offer cell-specific targeting, reduced toxicity, and increased half-life of encapsulated agents. Nanoparticles can carry bioactive molecules (drugs, proteins, antibodies, nucleic acids) and are able to enhance specificity and efficacy of cargo delivery [77]. For respiratory disorders, major barriers to pulmonary delivery are the degree of branching of the airways and the presence of mucus. Cargo delivery, optimally within the diseased area of the airway, could be achieved by functionalizing the nanoparticles with ligands that allow the particles to recognize, dock, and release the cargo to specific cells. Unfortunately, the identification of ASM-specific markers has been elusive to date. We generated a series of freshly formed complexes of liposomes and nanoparticles, called naposomes, as means to deliver DNA into cultured airway smooth muscle myocytes. Using PEGylated-PLGA nanoparticles that were loaded with the fluorescent marker FITC and a plasmid containing the CMV promoter driving the lacZ gene, which encodes β-galactosidase, we demonstrated β-galactosidase activity in human airway epithelial cells and in human ASM myocytes treated with these naposomes. In mice, we and others showed that nanoparticles enter the airway wall (Figure 2) and the lung parenchyma after intratracheal administration [78]. Moreover, using a co-culture system of confluent epithelial cells physically separated from ASM cells, we demonstrated that ASM myocytes were able to uptake fluorescein-labeled naposomes that were delivered through the epithelial layer (unpublished).
Figure 2.
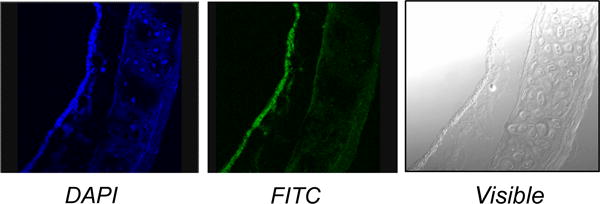
Localization of nanoparticles in mouse airway. After 48 hours delivery, fluorescence microscopy examination revealed FITC-labeled nanoparticles within the airway epithelium and subepithelial structures. Nuclei are in blue. (Magnification, 20×)
In conclusion, because S100 proteins can activate RAGE- and TLR-dependent pathways, they are capable of promoting pro-inflammatory effects. Paradoxically, S100 proteins are also able of initiating pro-apoptotic events, which ultimately could counteract S100 proteins' pro-inflammatory action (Figure 3). Therefore, a more comprehensive understanding of the complex interactions and functions of S100 proteins with the smooth muscle myocyte during allergic asthma is clearly needed. Systems biology and bioinformatics approaches as well as investigations using human specimens and animal models of lung inflammation and remodeling will be useful in achieving this goal. An important lesson learned from our studies with S100A12 is that higher abundance of certain pro-inflammatory proteins in the inflamed lung does not automatically make them targets for anti-inflammatory therapies. Rather, we proposed that therapies aimed at reducing the overabundant airway smooth muscle may yield beneficial effects in asthmatics [79,80].
Figure 3.
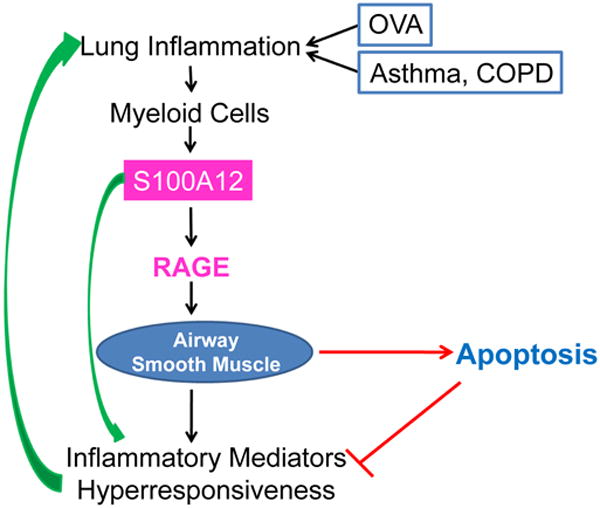
Lung inflammation and airway hyperresponsiveness are cardinal features of asthma and COPD; they are also elicited in mouse model of OVA-sensitization and challenge. Enhanced expression of S100A12 in the lung could mediate the inflammatory responses or serve as a marker of the inflammatory process. Additionally, stimulation of ASM with S100A12 induced myocyte cell death, which in turn would limit the abundance of smooth muscle mass with the concomitant reduction in contractile capacity and production of immunomodulators. COPD, chronic obstructive pulmonary disease; OVA, ovalbumin; RAGE, Receptor for Advanced Glycation End products.
Acknowledgments
Funding: NIH K01HL092588 (BCM), K08-HL090917 (MAH), NIH-CTSA UL1 RR024999 (BCM), GlaxoSmithKline Research & Education Foundation for Cardiovascular Diseases (MAH).
References
- 1.Eder W, Ege MJ, von Mutius E. The asthma epidemic. N Eng J Med. 2006;355:2226–2235. doi: 10.1056/NEJMra054308. [DOI] [PubMed] [Google Scholar]
- 2.Shifren A, Witt C, Christie C, Castro M. Mechanisms of Remodeling in Asthmatic Airways. J Allergy (Cairo) 2012;2012:316049. doi: 10.1155/2012/316049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Busse WW. The relationship of airway hyperresponsiveness and airway inflammation. Airway hyperresponsiveness in asthma: its measurement and clinical significance. Chest. 2010;138:4S–10S. doi: 10.1378/chest.10-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seow CY, Schellenberg RR, Paré PD. Structural and functional changes in the airway smooth muscle of asthmatic subjects. Am J Respir Crit Care Med. 1998;158:S179–S186. doi: 10.1164/ajrccm.158.supplement_2.13tac160. [DOI] [PubMed] [Google Scholar]
- 5.Martin JG, Duguet A, Eidelman DH. The contribution of airway smooth muscle to airway narrowing and airway hyperresponsiveness in disease. Eur Respir J. 2000;16:349–354. doi: 10.1034/j.1399-3003.2000.16b25.x. [DOI] [PubMed] [Google Scholar]
- 6.Hofmann Bowman M, Wilk J, Heydemann A, Kim G, Rehman J, et al. S100A12 mediates aortic wall remodeling and aortic aneurysm. Circ Res. 2010;106:145–154. doi: 10.1161/CIRCRESAHA.109.209486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 8.Ghavami S, Kerkhoff C, Chazin WJ, Kadkhoda K, Xiao W, et al. S100A8/9 induces cell death via a novel, RAGE-independent pathway that involves selective release of Smac/DIABLO and Omi/HtrA2. Biochem Biophys Acta. 2008;1783:297–311. doi: 10.1016/j.bbamcr.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 9.Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 10.Wu J, Kobayashi M, Sousa EA, Liu W, Cai J, et al. Differential proteomic analysis of bronchoalveolar lavage fluid in asthmatics following segmental antigen challenge. Mol Cell Proteomics. 2005;4:1251–1264. doi: 10.1074/mcp.M500041-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Yang Z, Yan WX, Cai H, Tedla N, Armishaw C, et al. S100A12 provokes mast cell activation: a potential amplification pathway in asthma and innate immunity. J Allergy Clin Immunol. 2007;119:106–114. doi: 10.1016/j.jaci.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 12.Lorenz E, Muhlebach MS, Tessier PA, Alexis NE, Duncan Hite R, et al. Different expression ratio of S100A8/A9 and S100A12 in acute and chronic lung diseases. Respir Med. 2008;102:567–573. doi: 10.1016/j.rmed.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rorke S, Jennison S, Jeffs JA, Sampson AP, Arshad H, et al. Role of cysteinyl leukotrienes in adenosine 5′-monophosphate induced bronchoconstriction in asthma. Thorax. 2002;57:323–327. doi: 10.1136/thorax.57.4.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118:3546–3556. doi: 10.1172/JCI36130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brusasco V, Crimi E, Gianiorio P, Lantero S, Rossi GA. Allergen-induced increase in airway responsiveness and inflammation in mild asthma. J Appl Physiol. 1990;69:2209–2214. doi: 10.1152/jappl.1990.69.6.2209. [DOI] [PubMed] [Google Scholar]
- 16.Durham SR, Craddock CF, Cookson WO, Benson MK. Increases in airway responsiveness to histamine precede allergen-induced late asthmatic responses. J Allergy Clin Immunol. 1988;82:764–770. doi: 10.1016/0091-6749(88)90077-2. [DOI] [PubMed] [Google Scholar]
- 17.Martinez FD. Viral infections and the development of asthma. Am J Respir Crit Care Med. 1995;151:1644–1647. doi: 10.1164/ajrccm/151.5_Pt_1.1644. [DOI] [PubMed] [Google Scholar]
- 18.Rossi GA, Crimi E, Lantero S, Gianiorio P, Oddera S, et al. Late-phase asthmatic reaction to inhaled allergen is associated with early recruitment of eosinophils in the airways. Am Rev Respir Dis. 1991;144:379–383. doi: 10.1164/ajrccm/144.2.379. [DOI] [PubMed] [Google Scholar]
- 19.Kava T. Acute respiratory infection, influenza vaccination and airway reactivity in asthma. Eur J Respir Dis Suppl. 1987;150:1–38. [PubMed] [Google Scholar]
- 20.Buckner CK, Clayton DE, Ain-Shoka AA, Busse WW, Dick EC, et al. Parainfluenza 3 infection blocks the ability of a beta adrenergic receptor agonist to inhibit antigen-induced contraction of guinea pig isolated airway smooth muscle. J Clin Invest. 1981;67:376–384. doi: 10.1172/JCI110045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castleman WL, Sorkness RL, Lemanske RF, Jr, McAllister PK. Viral bronchiolitis during early life induces increased numbers of bronchiolar mast cells and airway hyperresponsiveness. Am J Pathol. 1990;137:821–831. [PMC free article] [PubMed] [Google Scholar]
- 22.Holgate ST, Howell JBL, Jongejan RC. Airway hyperresponsiveness and inflammation. Oxford: Blackwell Scientific Publications; 1989. [Google Scholar]
- 23.Halayko AJ, Ghavami S. S100A8/A9: a mediator of severe asthma pathogenesis and morbidity? Can J Physiol Pharmacol. 2009;87:743–755. doi: 10.1139/Y09-054. [DOI] [PubMed] [Google Scholar]
- 24.Kheradmand F, Corry DB. Discovery of novel markers in allergic lung inflammation through proteomic-based technologies. Expert Rev Proteomics. 2008;5:9–12. doi: 10.1586/14789450.5.1.9. [DOI] [PubMed] [Google Scholar]
- 25.Fuellen G, Nacken W, Sorg C, Kerkhoff C. Computational searches for missing orthologs: the case of S100A12 in mice. OMICS. 2004;8:334–340. doi: 10.1089/omi.2004.8.334. [DOI] [PubMed] [Google Scholar]
- 26.Hofmann Bowman MA, Heydemann A, Gawdzik J, Shilling RA, Camoretti-Mercado B. Transgenic expression of human S100A12 induces structural airway abnormalities and limited lung inflammation in a mouse model of allergic inflammation. Clin Exp Allergy. 2011;41:878–889. doi: 10.1111/j.1365-2222.2011.03714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freyer AM, Johnson SR, Hall IP. Effects of growth factors and extracellular matrix on survival of human airway smooth muscle cells. Am J Respir Cell Mol Biol. 2001;25:569–576. doi: 10.1165/ajrcmb.25.5.4605. [DOI] [PubMed] [Google Scholar]
- 28.D'Antoni ML, Torregiani C, Ferraro P, Michoud MC, Mazer B, et al. Effects of decorin and biglycan on human airway smooth muscle cell proliferation and apoptosis. Am J Physiol Lung Cell Mol Physiol. 2008;294:L764–L771. doi: 10.1152/ajplung.00436.2007. [DOI] [PubMed] [Google Scholar]
- 29.Hamann KJ, Vieira JE, Halayko AJ, Dorscheid D, White SR, et al. Fas cross-linking induces apoptosis in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2000;278:L618–L624. doi: 10.1152/ajplung.2000.278.3.L618. [DOI] [PubMed] [Google Scholar]
- 30.Calvo FQ, Fillet M, de Seny D, Meuwis MA, Maree R, et al. Biomarker discovery in asthma-related inflammation and remodeling. Proteomics. 2009;9:2163–2170. doi: 10.1002/pmic.200800643. [DOI] [PubMed] [Google Scholar]
- 31.Bozinovski S, Cross M, Vlahos R, Jones JE, Hsuu K, et al. S100A8 chemotactic protein is abundantly increased, but only a minor contributor to LPS-induced, steroid resistant neutrophilic lung inflammation in vivo. J Proteome Res. 2005;4:136–145. doi: 10.1021/pr049829t. [DOI] [PubMed] [Google Scholar]
- 32.Greenlee KJ, Corry DB, Engler DA, Matsunami RK, Tessier P, et al. Proteomic identification of in vivo substrates for matrix metalloproteinases 2 and 9 reveals a mechanism for resolution of inflammation. J Immunol. 2006;177:7312–7321. doi: 10.4049/jimmunol.177.10.7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yin LM, Li HY, Zhang QH, Xu YD, Wang Y, et al. Effects of S100A9 in a rat model of asthma and in isolated tracheal spirals. Biochem Biophys Res Commun. 2010;398:547–552. doi: 10.1016/j.bbrc.2010.06.116. [DOI] [PubMed] [Google Scholar]
- 34.Lim SY, Raftery MJ, Goyette J, Geczy CL. S-glutathionylation regulates inflammatory activities of S100A9. J Biol Chem. 2010;285:14377–14388. doi: 10.1074/jbc.M109.075242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carta S, Castellani P, Delfino L, Tassi S, Vene R, et al. DAMPs and inflammatory processes: the role of redox in the different outcomes. J Leukoc Biol. 2009;86:549–555. doi: 10.1189/jlb.1008598. [DOI] [PubMed] [Google Scholar]
- 36.Lim SY, Raftery MJ, Geczy CL. Oxidative modifications of DAMPs suppress inflammation: the case for S100A8 and S100A9. Antioxid Redox Signal. 2011;15:2235–2248. doi: 10.1089/ars.2010.3641. [DOI] [PubMed] [Google Scholar]
- 37.Ramasamy R, Yan SF, Schmidt AM. Receptor for AGE (RAGE): signaling mechanisms in the pathogenesis of diabetes and its complications. Ann N Y Acad Sci. 2011;1243:88–102. doi: 10.1111/j.1749-6632.2011.06320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. 1993;143:1699–1712. [PMC free article] [PubMed] [Google Scholar]
- 39.Demling N, Ehrhardt C, Kasper M, Laue M, Knels L, et al. Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res. 2006;323:475–488. doi: 10.1007/s00441-005-0069-0. [DOI] [PubMed] [Google Scholar]
- 40.Hori O, Brett J, Slattery T, Cao R, Zhang J, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–25761. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 41.Englert JM, Hanford LE, Kaminski N, Tobolewski JM, Tan RJ, et al. A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am J Pathol. 2008;172:583–591. doi: 10.2353/ajpath.2008.070569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan SF, Ramasamy R, Schmidt AM. The RAGE axis: a fundamental mechanism signaling danger to the vulnerable vasculature. Circ Res. 2010;106:842–853. doi: 10.1161/CIRCRESAHA.109.212217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hofmann Bowman MA, Schmidt AM. S100/calgranulins EN-RAGEing the blood vessels: implications for inflammatory responses and atherosclerosis. Am J Cardiovasc Dis. 2011;1:92–100. [PMC free article] [PubMed] [Google Scholar]
- 44.Yan SF, Ramasamy R, Schmidt AM. Soluble RAGE: therapy and biomarker in unraveling the RAGE axis in chronic disease and aging. Biochem Pharmacol. 2010;79:1379–1386. doi: 10.1016/j.bcp.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yonekura H, Yamamoto Y, Sakurai S, Watanabe T, Yamamoto H. Roles of the receptor for advanced glycation endproducts in diabetes-induced vascular injury. J Pharmacol Sci. 2005;97:305–311. doi: 10.1254/jphs.cpj04005x. [DOI] [PubMed] [Google Scholar]
- 46.Hudson BI, Carter AM, Harja E, Kalea AZ, Arriero M, et al. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008;22:1572–1580. doi: 10.1096/fj.07-9909com. [DOI] [PubMed] [Google Scholar]
- 47.Sukkar MB, Wood LG, Tooze M, Simpson JL, McDonald VM, et al. Soluble RAGE is deficient in neutrophilic asthma and COPD. Eur Respir J. 2012;39:721–729. doi: 10.1183/09031936.00022011. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe T, Asai K, Fujimoto H, Tanaka H, Kanazawa H, et al. Increased levels of HMGB-1 and endogenous secretory RAGE in induced sputum from asthmatic patients. Respir Med. 2011;105:519–525. doi: 10.1016/j.rmed.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 49.Hofmann MA, Drury S, Hudson BI, Gleason MR, Qu W, et al. RAGE and arthritis: the G82S polymorphism amplifies the inflammatory response. Genes Immun. 2002;3:123–135. doi: 10.1038/sj.gene.6363861. [DOI] [PubMed] [Google Scholar]
- 50.Brusasco V, Crimi E, Pellegrino R. Airway hyperresponsiveness in asthma: not just a matter of airway inflammation. Thorax. 1998;53:992–998. doi: 10.1136/thx.53.11.992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crimi E, Spanevello A, Neri M, Ind PW, Rossi GA, et al. Dissociation between airway inflammation and airway hyperresponsiveness in allergic asthma. Am J Resp Crit Care Med. 1998;157:4–9. doi: 10.1164/ajrccm.157.1.9703002. [DOI] [PubMed] [Google Scholar]
- 52.Hewitt M, Creel A, Estell K, Davis IC, Schwiebert LM. Acute exercise decreases airway inflammation, but not responsiveness, in an allergic asthma model. Am J Respir Cell Mol Biol. 2009;40:83–89. doi: 10.1165/rcmb.2008-0172OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakagome K, Dohi M, Okunishi K, To Y, Sato A, et al. Antigen-sensitized CD4+CD62Llow memory/effector T helper 2 cells can induce airway hyperresponsiveness in an antigen free setting. Respir Res. 2005;6:46. doi: 10.1186/1465-9921-6-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Torres R, Herrerias A, Serra-Pages M, Roca-Ferrer J, Pujols L, et al. An intranasal selective antisense oligonucleotide impairs lung cyclooxygenase-2 production and improves inflammation, but worsens airway function, in house dust mite sensitive mice. Respir Res. 2008;9:72. doi: 10.1186/1465-9921-9-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sont JK, Willems LN, Bel EH, van Krieken JH, Vandenbroucke JP, et al. Clinical control and histopathologic outcome of asthma when using airway hyperresponsiveness as an additional guide to long-term treatment. The AMPUL Study Group. Am J Respir Crit Care Med. 1999;159:1043–1051. doi: 10.1164/ajrccm.159.4.9806052. [DOI] [PubMed] [Google Scholar]
- 56.Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Eng J Med. 2009;360:973–984. doi: 10.1056/NEJMoa0808991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kariyawasam HH, Aizen M, Barkans J, Robinson DS, Kay AB. Remodeling and airway hyperresponsiveness but not cellular inflammation persist after allergen challenge in asthma. Am J Respir Crit Care Med. 2007;175:896–904. doi: 10.1164/rccm.200609-1260OC. [DOI] [PubMed] [Google Scholar]
- 58.Oliver MN, Fabry B, Marinkovic A, Mijailovich SM, Butler JP, et al. Airway Hyperresponsiveness, Remodeling, and Smooth Muscle Mass. Am J Respir Cell Mol Biol. 2007;37:264–272. doi: 10.1165/rcmb.2006-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baroffio M, Barisione G, Crimi E, Brusasco V. Noninflammatory mechanisms of airway hyper-responsiveness in bronchial asthma: an overview. Ther Adv Respir Dis. 2009;3:163–174. doi: 10.1177/1753465809343595. [DOI] [PubMed] [Google Scholar]
- 60.Liakakos P, Snell GI, Ward C, Johns DP, Bamford TL, et al. Bronchial hyperresponsiveness in lung transplant recipients: lack of correlation with airway inflammation. Thorax. 1997;52:551–556. doi: 10.1136/thx.52.6.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gibson PG, Saltos N, Borgas T. Airway mast cells and eosinophils correlate with clinical severity and airway hyperresponsiveness in corticosteroid-treated asthma. J Allergy Clin Immunol. 2000;105:752–759. doi: 10.1067/mai.2000.105319. [DOI] [PubMed] [Google Scholar]
- 62.Jarai G, Sukkar M, Garrett S, Duroudier N, Westwick J, et al. Effects of interleukin-1[beta], interleukin-13 and transforming growth factor-[beta] on gene expression in human airway smooth muscle using gene microarrays. Eur J Pharmacol. 2004;497:255–265. doi: 10.1016/j.ejphar.2004.06.055. [DOI] [PubMed] [Google Scholar]
- 63.Johnson PR, Burgess JK, Underwood PA, Au W, Poniris MH, et al. Extracellular matrix proteins modulate asthmatic airway smooth muscle cell proliferation via an autocrine mechanism. J Allergy Clin Immunol. 2004;113:690–696. doi: 10.1016/j.jaci.2003.12.312. [DOI] [PubMed] [Google Scholar]
- 64.Lin WN, Luo SF, Lee CW, Wang CC, Wang JS, et al. Involvement of MAPKs and NF-κappaB in LPS-induced VCAM-1 expression in human tracheal smooth muscle cells. Cell Signal. 2007;19:1258–1267. doi: 10.1016/j.cellsig.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 65.Hakonarson H, Grunstein MM. Autocrine regulation of airway smooth muscle responsiveness. Respir Physiol Neurobiol. 2003;137:263–276. doi: 10.1016/s1569-9048(03)00152-6. [DOI] [PubMed] [Google Scholar]
- 66.Damera G, Tliba O, Panettieri RA., Jr Airway smooth muscle as an immunomodulatory cell. Pulm Pharmacol Ther. 2009;22:353–359. doi: 10.1016/j.pupt.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.John M, Hirst SJ, Jose PJ, Robichaud A, Berkman N, et al. Human airway smooth muscle cells express and release RANTES in response to T helper 1 cytokines: regulation by T helper 2 cytokines and corticosteroids. J Immunol. 1997;158:1841–1847. [PubMed] [Google Scholar]
- 68.Freyer AM, Johnson SR, Hall IP. Effects of growth factors and extracellular matrix on survival of human airway smooth muscle cells. Am J Respir Cell Mol Biol. 2001;25:569–576. doi: 10.1165/ajrcmb.25.5.4605. [DOI] [PubMed] [Google Scholar]
- 69.Black JL, Roth M, Lee J, Carlin S, Johnson PR. Mechanisms of airway remodeling. Airway smooth muscle. Am J Respir Crit Care Med. 2001;164:S63–S66. doi: 10.1164/ajrccm.164.supplement_2.2106059. [DOI] [PubMed] [Google Scholar]
- 70.Sukkar MB, Xie S, Khorasani NM, Kon OM, Stanbridge R, et al. Toll-like receptor 2, 3, and 4 expression and function in human airway smooth muscle. J Allergy Clin Immunol. 2006;118:641–648. doi: 10.1016/j.jaci.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 71.Eder W, Klimecki W, Yu L, von Mutius E, Riedler J, et al. Association between exposure to farming, allergies and genetic variation in CARD4/NOD1. Allergy. 2006;61:1117–1124. doi: 10.1111/j.1398-9995.2006.01128.x. [DOI] [PubMed] [Google Scholar]
- 72.Finn PW, Bigby TD. Innate immunity and asthma. Proc Am Thorac Soc. 2009;6:260–265. doi: 10.1513/pats.200807-064RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cox G, Miller JD, McWilliams A, FitzGerald JM, Lam S. Bronchial thermoplasty for asthma. Am J Respir Crit Care Med. 2006;173:965–969. doi: 10.1164/rccm.200507-1162OC. [DOI] [PubMed] [Google Scholar]
- 74.Castro M, Rubin A, Laviolette M, Hanania NA, Armstrong B, et al. Persistence of effectiveness of bronchial thermoplasty in patients with severe asthma. Ann Allergy Asthma Immunol. 2011;107:65–70. doi: 10.1016/j.anai.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 75.Berthier S, Paclet MH, Lerouge S, Roux F, Vergnaud S, et al. Changing the conformation state of cytochrome b558 initiates NADPH oxidase activation: MRP8/MRP14 regulation. J Biol Chem. 2003;278:25499–25508. doi: 10.1074/jbc.M209755200. [DOI] [PubMed] [Google Scholar]
- 76.Hofmann Bowman MA, Gawdzik J, Bukhari U, Husain AN, Toth PT, et al. S100A12 in vascular smooth muscle accelerates vascular calcification in apolipoprotein E-null mice by activating an osteogenic gene regulatory program. Arterioscler Thromb Vasc Biol. 2011;31:337–344. doi: 10.1161/ATVBAHA.110.217745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vij N. Nano-based theranostics for chronic obstructive lung diseases: challenges and therapeutic potential. Expert Opin Drug Deliv. 2011;8:1105–1109. doi: 10.1517/17425247.2011.597381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sammani S, Moreno-Vinasco L, Mirzapoiazova T, Singleton PA, Chiang ET, et al. Differential effects of sphingosine 1–phosphate receptors on airway and vascular barrier function in the murine lung. Am J Respir Cell Mol Biol. 2010;43:394–402. doi: 10.1165/rcmb.2009-0223OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Camoretti-Mercado B. Targeting the airway smooth muscle for asthma treatment. Transl Res. 2009;154:165–174. doi: 10.1016/j.trsl.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xia Y, Redhu NS, Moir LM, Koziol-White C, Ammit AJ, et al. Regulation of the pro-inflammatory and immunomodulatory functions of airway smooth muscle: Emerging Concepts. Pulm Pharmacol Ther. 2012 doi: 10.1016/j.pupt.2012.05.006. Available online: 24-MAY-2012. Epub ahead of print. [DOI] [PubMed] [Google Scholar]