

Summary
Accumulation of Aβ peptide fragments of the APP protein and neurofibrillary tangles of the microtubule-associated protein tau are the cellular hallmarks of Alzheimer’s disease (AD). To investigate the relationship between APP metabolism and tau protein levels and phosphorylation, we studied human-stem-cell-derived forebrain neurons with genetic forms of AD, all of which increase the release of pathogenic Aβ peptides. We identified marked increases in intracellular tau in genetic forms of AD that either mutated APP or increased its dosage, suggesting that APP metabolism is coupled to changes in tau proteostasis. Manipulating APP metabolism by β-secretase and γ-secretase inhibition, as well as γ-secretase modulation, results in specific increases and decreases in tau protein levels. These data demonstrate that APP metabolism regulates tau proteostasis and suggest that the relationship between APP processing and tau is not mediated solely through extracellular Aβ signaling to neurons.
Graphical Abstract

Highlights
-
•
Neurons from different genetic forms of Alzheimer’s disease differ in APP processing
-
•
APP mutations increase total and phosphorylated tau; PSEN1 mutations do not
-
•
Pharmacological manipulation of APP processing changes tau protein levels
-
•
APP regulation of tau proteostasis is not solely mediated through extracellular Aβ
Moore et al. use neurons made from familial Alzheimer’s disease stem cells to reveal how three proteins involved in the disease are linked in a pathway that controls disease progression. They show that drugs that target this pathway change levels of a protein involved in neurodegeneration, microtubule-associated protein tau, opening up a potential therapeutic pathway.
Introduction
Accumulation of Aβ peptide fragments of the APP protein and neurofibrillary tangles of the microtubule-associated protein tau are the cellular hallmarks of Alzheimer’s disease (AD). However, the molecular mechanisms linking APP metabolism; extracellular Aβ peptides; and changes in tau expression, phosphorylation, and cellular localization are currently unclear. Understanding of the genetics underlying monogenic familial Alzheimer’s disease (fAD) has provided several insights into disease pathogenesis (Blennow et al., 2006). The majority of known fAD mutations are autosomal dominant and affect APP or the catalytic components of the γ-secretase APP-processing complex, presenilin (PSEN) 1 and 2 (Bertram and Tanzi, 2008). Early onset AD also occurs in individuals with increased APP gene dosage due either to trisomy of chromosome 21 (Ts21) or duplication of the APP locus (APP (dup)) (Rovelet-Lecrux et al., 2006). The identification of the mutations involved in fAD, the discovery of Aβ42 as the primary component of cerebrovascular amyloid (Glenner and Wong, 1984a) and amyloid plaques (Masters et al., 1985) in late-onset sporadic AD, and the identification of the same peptide in amyloid plaques in Down syndrome (Glenner and Wong, 1984b) led to the development of the amyloid hypothesis for AD (Hardy and Allsop, 1991).
The amyloid hypothesis postulates that accumulation of Aβ42 is central to AD initiation and subsequently leads to changes in neuronal function, tau pathology, and ultimately cell death (Hardy and Allsop, 1991). Increased production of aggregation-prone Aβ monomers during AD initiation results in the formation of soluble extracellular oligomers that are proposed to signal via several different specific cell surface receptors or perturb membrane integrity in a non-specific manner, resulting in neuronal dysfunction (Benilova et al., 2012). Changes in APP processing and the generation of intracellular APP fragments have also been proposed to be involved in AD pathogenesis (Pimplikar et al., 2010). The β-secretase-generated C-terminal fragment of APP, referred to as β-CTF or C99, has been shown to be toxic in cultured cells (Yankner et al., 1989) and cause neurodegeneration and defects in synaptic plasticity in transgenic mouse models (Lauritzen et al., 2012). Similarly, cleavage of APP-C99 by γ-secretase releases the APP intracellular domain (AICD), which is proposed to contribute to neurodegeneration (Pimplikar et al., 2010).
We and others have previously shown that neurons generated from induced pluripotent stem cells (iPSCs) from genetic forms of AD recapitulate aspects of the disease, including increased Aβ peptide production in Down syndrome (Shi et al., 2012a) and APP duplication (Israel et al., 2012). Altered Aβ40:42 ratios have also been observed in PSEN1 and APP mutant neurons (Muratore et al., 2014; Yagi et al., 2011). Here, we investigate the relationship between APP processing and tau protein levels and phosphorylation by analysis of iPSC-derived cortical neurons with different genetic forms of AD and pharmacological manipulation of β-secretase and γ-secretase.
Results
Distinct Genetic Forms of Alzheimer’s Disease All Increase Aβ42 Generation
APP processing and generation of Aβ peptides in different genetic forms of AD was studied by generating cortical excitatory neurons from patient iPSCs (Shi et al., 2012c) harboring PSEN1 mutations (Y115C, M146I, and intron 4), an APP mutation (V717I), and APP duplication (APPdup) (Israel et al., 2012; Figures 1A–1C and S1). Over 3 months, neurons generated from monolayers of cortical progenitor cells formed dense 3D, electrically active neural networks that spanned >200 μm in thickness (Figure 1D; Movies S1 and S2).
Figure 1.

Altered APP Processing and Aβ Peptide Production in Stem Cell Models of Genetic Forms of Alzheimer’s Disease
(A–C) Representative immunohistochemistry of neurons (β3-tubulin positive, blue) generated from familial Alzheimer’s disease (fAD) (PSEN1 intron 4, Y115C, and APP V717I) iPSCs, expressing transcription factors restricted to layer 6 (Tbr1, red) or layer 5 (CTIP2, green) cortical projection neurons.
(D) 3D nature of stem-cell-derived cortical cultures. Cultures cleared by passive CLARITY and immunostained for neurons (tau, red) and nuclei (DAPI, blue). Single-plane XY, XZ, and YZ (right, top, and left panels, respectively) projections of control neurons 100 days post-induction. The scale bar represents 100 μm.
(E) Generation of Aβ peptides. APP is first cleaved by β-secretase to generate membrane-bound APP-C99. This is followed by the initial γ-secretase cleavage of APP-C99, termed ε-cleavage, to generate Aβ peptides of either 48 or 49 amino acids. Aβ peptides are then subject to sequential γ-secretase carboxypeptidase cleavages, leading to extracellular release of Aβ42, 40 and 38.
(F) Neurons with three different PSEN1 mutations generate equivalent amounts of total extracellular Aβ peptides over 40 days in culture as three different healthy control lines. APP V717I neurons also do not significantly alter the production of total Aβ peptides compared to controls. This is in contrast with APP (dup) neurons, which significantly increase Aβ production. Error bars, SD; n = 3 cultures for each genotype; ∗∗p < 0.01.
(G) PSEN1 and APP V717I mutant neurons have significantly reduced Aβ40:Aβ42 ratios compared with both control and APP (dup) neurons at all time points studied, reflecting a relative increase in the generation of Aβ42. Error bars, SEM; ∗∗p < 0.01.
(H) PSEN1 and APP V717I mutant neurons exhibit a relative increase in Aβ42 compared to Aβ40 at day 80, whereas Ts21 and APP (dup) do not. All data produced from three independent cultures. Error bars, SD; ∗∗p < 0.01.
(I) Comparing ratios of Aβ40 to the sum of Aβ38 and Aβ42 at day 80, as an indicator of ε-cleavage and processing pathway choice, reveals that neither PSEN1 mutants nor increased APP dosage affects APP-C99 cleavage. By contrast, APP V717I mutants significantly bias the ε-cleavage APP-C99 to Aβ48, which is processed to both Aβ42 and Aβ38. Error bars, SD.
(J) PSEN1 mutant neurons have reduced Aβ38:Aβ42 ratios, in contrast with all other genotypes analyzed, suggesting reduced γ-secretase processivity. Error bars, SD; ∗∗p < 0.01.
(K) Proposed alternative processing pathway of APP in the presence of γ-secretase inhibitors. APP can be sequentially cleaved by β- and then γ-secretase epsilon cleavage followed by α-secretase to generate Aβ14/15/16 and AICD.
(L) Percentage of total Aβ peptides that are generated from the proposed alternative pathway (K; Aβ14/15/16) as an indicator of processivity, detected and quantified by IP-MALDI. PSEN1 mutant neurons significantly increase the percentage of Aβ peptides made up of the sum of Aβ1-14, 1-15, and 1-16, compared with control and APP V717I neurons. Error bars, SD; ∗∗p < 0.01.
(M) Percentage of total Aβ peptides that is Aβ40 as an indicator of processivity. PSEN1 mutant neurons significantly decrease the percentage of Aβ40 peptides, compared with control and APP V717I neurons. Error bars, SD; ∗∗p < 0.01.
Production of extracellular Aβ peptides by neurons of each genotype was compared with that of three independent controls over the course of 90 days in culture. At all time points, PSEN1 and APP V717I neurons produced similar extracellular concentrations of the sum of Aβ38, 40, and 42 peptides as healthy control neurons (Figures 1E and 1F). However, these mutants decreased the ratio of Aβ40:Aβ42 at each point assessed (Figures 1F and 1G), reflecting an absolute and relative increase in Aβ42 production compared with controls. In contrast with the other genotypes, APPdup neurons greatly overproduce Aβ peptides over time, in line with increased substrate dosage (Figure 1F), as previously found for Ts21 neurons (Shi et al., 2012a). Overproduction of Aβ peptides in APPdup neurons did not alter the relative amounts of Aβ40 and Aβ42 (Figures 1G and 1H), indicating that Aβ generation is limited by APP availability, rather than β- and γ-secretase capacity.
Comparing relative amounts of Aβ40 with the sum of Aβ38 and Aβ42 peptides enables inference about the initial ε-cleavage of APP-C99 by γ-secretase to either Aβ48 or Aβ49 that are then processed in largely separate pathways (Figure 1E; Chávez-Gutiérrez et al., 2012). APP V717I neurons exhibited a significant decrease in the Aβ40:Aβ38+Aβ42 ratio, which was not observed in APP dosage models or PSEN1 mutants (Figure 1I), consistent with the V717I mutation biasing the initial ε-cleavage of APP to Aβ48, which is processed to both Aβ42 and Aβ38 (Figure 1E).
Multiple PSEN1 mutations resulted in a decreased Aβ38:Aβ42 ratio (Figure 1J), consistent with a hypomorphic loss of γ-secretase function (Chávez-Gutiérrez et al., 2012). In support of this, PSEN1 mutants significantly increased the release of Aβ14, Aβ15, and Aβ16 (Figures 1K, 1L, and S2), which are thought to be produced by sequential cleavage of APP by β- and then α-secretase in the context of reduced γ-secretase processivity (Portelius et al., 2011). This was accompanied by a reduction in Aβ40, reflecting the shift in production to shorter Aβ forms (Figure 1M), indicating that these hypomorphic PSEN1 mutations reduce γ-secretase’s carboxypeptidase activity.
Increased APP Gene Dosage and APP V717I Specifically Increase Neuronal Tau Protein Levels
Intracellular levels of total and phosphorylated tau were increased in APP V717I and APPdup neurons, compared with controls (Figures 2A, 2C–2E, and S3A; n = 2 independent inductions from each iPSC line). The changes in tau protein levels did not reflect an increase in the relative numbers of neurons carrying APP duplications or mutations, assessed by the levels of the neuron-specific β3-tubulin protein (Figure 2B). Neurons from two different PSEN1 mutations (Y115C and intron 4) did not exhibit increased total or phosphorylated tau levels, compared to controls (Figures 2A, 2C–2E, and S3A). Thus, intracellular tau levels do not correlate with the extracellular Aβ40:Aβ42 ratio, as PSEN1 mutant neurons exhibited a comparable ratio to APP V717I neurons (Figures 1G and 1H). MAPT transcription was assessed by RT-PCR, demonstrating no difference in mRNA expression between neurons of each genotype (Figure S3B) and suggesting that the increase in tau protein observed in APP V717I and APPdup neurons is post-transcriptional and a result of altered tau proteostasis.
Figure 2.

Increased APP Copy Number and the V717I Mutation Lead to Increases in Intracellular Tau Protein Levels, whereas PSEN1 Mutations Do Not
(A) Total tau levels are increased in APP (dup) and APP V717I neurons (90 days post-neural induction) but are not altered in PSEN1 mutants.
(B) Altered tau levels are not accompanied by changes in neuronal number or mass, assessed by β3-tubulin levels.
(C) Tau phosphorylation at S202/T205, S396, and S404 is increased in APP (dup) and APP V717I neurons, but not PSEN1 mutants, compared to controls (AT8 and pS396 share actin loading control; for clarity this appears beneath both western blots).
(D and E) Quantification of the data presented in (A), (C), and Figure S3 demonstrating that PSEN1 mutants do not exhibit increases in tau expression or phosphorylation by western blot analysis. Error bars, SEM.
Pharmacological Manipulation of APP Processing Regulates Tau Proteostasis in Neurons
As PSEN1 and APP V717I mutant neurons displayed strikingly different intracellular tau protein levels but comparable levels of total extracellular Aβ and Aβ40:Aβ42 ratios, we hypothesized that membrane-bound or intracellular products of APP processing might regulate tau proteostasis. Therefore, we compared the effects of acute γ-secretase or β-secretase inhibition on tau protein levels in control neurons. As expected, inhibition of either γ- or β-secretase significantly reduced extracellular Aβ peptides (Figure 3A). However, γ-secretase inhibition led to an increase in tau and a marked accumulation of APP-C83/C99. Tau levels were decreased by β-secretase inhibition (Figure 3B), a treatment that reduces APP-C99 generation. These data indicate a link between APP processing and tau proteostasis that is regulated by γ- and β-secretase activity, independent of extracellular Aβ38, Aβ40, and Aβ42.
Figure 3.

γ-Secretase Processing of APP Is Coupled to Tau Proteostasis
(A) Multiplexed ELISA quantification of extracellular Aβ peptides from healthy control neurons following treatment with the γ-secretase inhibitor, DAPT (GSI), shows significant reduction in the production of Aβ38, Aβ40, and Aβ42, compared to vehicle controls (Ctrl). Similarly, the β-secretase inhibitor LY2886721 (BSI) significantly reduces extracellular Aβ. Neurons treated between days 64 and 70 with the indicated compounds at a final concentration of 1 μM. Error bars, SD. n = 3 cultures for each treatment group; ∗∗p < 0.01.
(B) Immunoblot detection of APP-C83/99 peptides, total tau, and β3-tubulin extracted from healthy control neurons following treatment with DMSO (Ctrl), BSI, and GSI. BSI treatment reduces tau protein levels with no detectable changes in APP-C83/99, compared to DMSO-treated controls. By contrast, GSI treatment markedly increases both APP-C83/99 and tau in control neurons.
(C–E) Ts21 neurons treated with DAPT (GSI) for a 30-day period between days 60 and 90 post-induction exhibit a dose-dependent increase in both APP-C83/99 and tau protein. Treatment with the γ-secretase modulator E2012 (GSM) reduced tau levels in Ts21 neurons in a dose-dependent manner, with no detectable changes in APP-C83/C99.
We compared the effects of the γ-secretase inhibitor (GSI) DAPT with the imidazole-based γ-secretase modulator E2012 (GSM) on Ts21 neurons, which exhibit increased tau protein levels and phosphorylation compared with euploid controls, providing a sensitive background on which to detect changes in tau protein. Ts21 neurons treated with DAPT over a 30-day period exhibited a dose-dependent increase in tau protein levels, accompanied by corresponding increases in APP-C83/C99 (Figures 3C and 3E). However, γ-secretase modulation resulted in a dose-dependent decrease in tau protein levels in Ts21 neurons but did not increase APP-C83/C99 (Figures 3D and 3E).
γ-Secretase Modulation Reduces Intracellular Tau in fAD Neurons
We investigated the effect of manipulating γ-secretase activity on tau proteostasis in different genetic forms of AD. To do so, we compared the effects of γ-secretase inhibition and modulation on APP processing, Aβ peptide production, and tau protein levels in Ts21, PSEN1, and APP V717I neurons, representing AD initiation due to increased APP copy number, reduced γ-secretase carboxypeptidase processivity, and altered ε-cleavage of APP, respectively.
Inhibition of γ-secretase with DAPT significantly reduced the production of extracellular Aβ38, Aβ40, and Aβ42 in neurons of all genotypes (Figures 4A and S4A–S4C; n = 2 independent experiments for each genotype). Moreover, IP-MALDI analysis of extracellular DAPT-treated samples revealed a loss of all longer Aβ peptides and a significant increase in Aβ14, 15, and 16 (Figure S4E). By contrast, E2012 reduced the absolute amount of Aβ40 and Aβ42 in all genotypes assessed and resulted in a marked increase of Aβ37 and Aβ38 (Figures S4A–S4C and S4F), causing increased Aβ40/Aβ42 and Aβ38/Aβ42 ratios for E2012-treated neurons, compared to vehicle controls (Figures 4B and 4C). Notably, E2012 treatment increased Aβ40/Aβ42 and Aβ38/Aβ42 ratios in PSEN1 mutant neurons, demonstrating that GSMs can act on neurons carrying PSEN1 mutations. The magnitude of this effect was dependent on the nature of the mutation, with PSEN1 intron 4 mutations being least responsive to γ-secretase modulation by E2012 (Figures 4A–4D and S4A–S4C).
Figure 4.

Manipulation of γ-Secretase Activity Alters Aβ Peptide Production, Tau Expression, and Phosphorylation Status in Genetic Forms of AD
(A) DAPT (GSI) prevents the production of Aβ38, Aβ40, and Aβ42 observed in DMSO-treated controls (Ctrl). In contrast, E2012 (GSM) reduces total Aβ peptide production by approximately one third. All compounds used at 1 μM and extracellular media analyzed at day 80 post-neural induction, after 20 days of drug treatment. Error bars, SD. Neurons of each genotype are as marked; n = 3 cultures for each treatment group; ∗∗p < 0.01.
(B–D) Changes in relative Aβ peptide production in response to each compound are reflected in the ratios between the different Aβ species. E2012 (GSM) has particularly marked effects in reducing the concentration of Aβ42 relative to Aβ40 (B) and in increasing the concentration of Aβ38 relative to Aβ42 (C). Error bars, SD; ∗∗p < 0.01.
(E) Western blots performed on soluble protein extracts of neurons following treatment with the indicated compounds. Protein extraction performed at day 90 post-neural induction, after 30 days of drug treatment. DAPT treatment (GSI) increases both total and phosphorylated tau expression in the majority of genotypes, assessed at multiple epitopes. By contrast, E2012 (GSM) reduces total tau expression and its phosphorylation in all genotypes assayed, with particularly pronounced effects in Ts21 neurons. Error bars, SD; n = 2 for each treatment group; representative western blots shown, with additional data in Figure S4.
(F) GSI with DAPT increased APP-C83/99 in neurons of all genotypes, with differing efficacy in some PSEN1 mutants, whereas GSM (E2012) had no effect on APP-C83/99. Drug treatments had no effect on neuronal number, as reflected in the amount of neuron-specific β3-tubulin.
In agreement with our analyses of control and Ts21 neurons, DAPT increased total tau levels and APP-C83/99 in neurons of all genotypes (Figures 4E, 4F, and S4). This was accompanied by increases in site-specific phosphorylation of tau (pS202/T205 [AT8], pS396, and pS404; Figures 4E and S4E). E2012 significantly reduced tau protein levels and phosphorylation in neurons of each genotype, with the exception of PSEN1 intron 4 (Figures 4E and S4G). Ts21 neurons, which displayed the most marked changes in APP processing in response to E2012, similarly had the largest increase in both tau protein expression and phosphorylation (Figures 4E and S4G). Given that manipulating γ-secretase could affect neurogenesis and neuronal differentiation via Notch signaling, we measured levels of neuron-specific β3-tubulin in neurons of all genotypes following treatment with GSI and GSM (Figure 4F). Neither drug treatment had any effect on β3-tubulin levels (Figure 4F) and thus did not affect neuronal number or mass.
Discussion
To investigate the relationship between APP metabolism and tau protein levels and phosphorylation, we have utilized human-stem-cell-derived excitatory cortical neurons from representative genetic forms of AD with mutations in either APP or PSEN1 that are predicted to affect APP metabolism. In addition to identifying different classes of altered APP processing and Aβ peptide production under physiological conditions in human forebrain neurons with different genetic forms of AD, we identified regulation of tau proteostasis by metabolism of APP. Marked increases in tau protein levels were observed in genetic forms of AD that changed APP dosage or affected the ε-cleavage site of APP. Furthermore, pharmacological manipulation of APP metabolism changed tau protein levels in human forebrain neurons in a dose-dependent manner. These findings point to a potentially important pathogenic mechanism in AD, linking APP metabolism to tau protein levels. The pathological significance of tau protein levels is clear from the small number of identified individuals with frontotemporal dementia and MAPT duplications (Hooli et al., 2014; Rovelet-Lecrux et al., 2010).
Recent in vitro studies of AD employing overexpression of transgenes encoding mutant forms of PSEN1 and APP in human neurons or iPSC-derived APP V717I neurons have demonstrated an association between APP processing and tau pathology that is dependent on extracellular Aβ peptides (Choi et al., 2014; Muratore et al., 2014). Consistent with previous studies, we found that neurons from all fAD lines in this study led to an increase in extracellular Aβ42. However, only APP duplication, Ts21, and APP V717I resulted in increased tau protein levels and phosphorylation. Elevated tau protein levels were not due to an increase in MAPT mRNA expression, suggesting a post-transcriptional mechanism.
Given the similarity in extracellular total Aβ levels and specifically Aβ42, among different PSEN1 and APP mutant neurons, we hypothesized that the increase in intracellular tau protein seen in a subset of genetic forms of AD may be regulated by factors in addition to extracellular Aβ. As the three genotypes in which increased tau occurs either affect total APP levels (Ts21; APP duplication) or the initial ε-cleavage of APP by γ-secretase (APP V717I), our studies focused on the initial intracellular processing of APP to generate APP-C83/99. Therefore, we compared the effects of β-secretase and γ-secretase inhibition on APP processing and tau proteostasis in human cortical neurons. Both compounds greatly reduce extracellular Aβ peptide production but at different stages of the APP-processing pathway (De Strooper, 2010). β-secretase inhibition prevents generation of APP-C99 from full-length APP, whereas γ-secretase inhibition blocks the proteolysis of APP-C83/99, resulting in accumulation of APP-C83/99. In support of a role for APP metabolism and APP-C83/99 in regulating tau levels, we found that γ-secretase inhibition increased intracellular tau protein levels, whereas β-secretase inhibition reduced intracellular tau protein.
The strategy of γ-secretase modulation reduced tau protein in different genetic forms of AD, suggesting that this approach to reducing tau levels may be a useful therapeutic strategy in different forms of Alzheimer’s disease. The reduction in tau protein by γ-secretase modulation with E2012 had differential effects depending on the specific mutation, with the degree of tau reduction correlated with the magnitude of the change in APP processing in each genotype. For example, E2012 had the most-pronounced effects on APP processing and tau levels in Ts21/Down syndrome neurons and the least effects on both APP processing and tau protein levels in PSEN1 intron 4 neurons. Finally, γ-secretase modulation also reduced tau protein levels in healthy controls, indicating that the link between APP metabolism and tau proteostasis is a feature of neuronal biology in health and disease.
Overall, our data support a link between APP metabolism and tau proteostasis that is mediated by β- and γ-secretase. The lack of changes in tau protein in PSEN1 mutant neurons indicates that a simple lack of carboxypeptidase processivity does not lead to altered tau proteostasis. Instead, the data reported here suggest the changes in tau levels are related to altered ε-cleavage of APP by endopeptidase activity of γ-secretase and point to a possible role for APP-C99 in regulating tau proteostasis. A key question for further study is how intracellular metabolism of APP may input into controlling tau proteostasis, given the potential importance of this pathway for AD progression.
Experimental Procedures
Generation of Familial Alzheimer’s Disease iPSCs and Cerebral Cortex Neurons
PSEN1 Y115C, M146I, intron 4, and APP V717I mutant fibroblasts were sourced as described (Wray et al., 2012). Fibroblasts were reprogrammed at the Cambridge Biomedical Centre using the standard four-factor method, delivered by lentiviruses (Takahashi et al., 2007). Each mutation was sequenced in reprogrammed clones, and pluripotency was determined by differentiation to each germ layer from embryoid bodies (Figure S1). Healthy control cell lines were NDC (Israel et al., 2012), NAS6 (Devine et al., 2011), and the H9 ES (WiCell Research Institute); additional disease lines were APP duplication (Israel et al., 2012) and Ts21 iPSCs (Park et al., 2008). Pluripotent cells were cultured by standard methods (see Supplemental Information for details).
Directed differentiation of hESCs and iPSCs to cerebral cortex was carried out as described, with minor modifications (Shi et al., 2012b, c). For drug treatment, all compounds were dissolved in DMSO at the concentrations noted, and DMSO was the vehicle control in all experiments. Compounds were added every 48 hr during treatment period: γ-secretase inhibitor, DAPT (Sigma); γ-secretase modulator, E2012 (ChemExpress); and β-secretase inhibitor LY2886721 (Selleck).
Immunocytochemistry and Imaging
Fixed and immunostained cultures were imaged on an Olympus FV1000 inverted confocal microscope (see Supplemental Information for details). For optical clearing, fixed cultures were processed according to the method described (Yang et al., 2014).
Protein Analysis
Quantification of Aβ42, Aβ40, and Aβ38 were carried out with multiplexed MesoScale Discovery assay kits on a Quickplex SQ120 instrument (MesoScale Discovery) using 25 μl of cell culture supernatant. All statistical comparisons were between the entire set of controls samples and all samples of each genotype, using Student’s t test with the Bonferroni correction for multiple testing. Three independent cultures of neurons derived from each clone were used for all measurements, except where noted. Cellular protein extraction and western blot analysis were carried out as described (Supplemental Information).
IP-MALDI Analysis of Aβ Peptides
IP of cell media was performed using a KingFisher magnetic particle processor (Thermo Scientific) as described (Portelius et al., 2007). See Supplemental Information for further details.
Acknowledgments
The authors thank individual donors for providing skin biopsies; Dr. Nathalie Ryan, Prof. Nick Fox, and Prof. Martin Rossor for patient characterization and collection of patient material; Dr. Selina Wray for provision of dermal fibroblasts; Ayiba Momoh and Laura Brightman for technical support; and members of the F.J.L. lab for critical reading of the manuscript. This research was supported by grants from Alzheimer’s Research UK and the Wellcome Trust (to F.J.L.) and core funding to the Gurdon Trust from the Wellcome Trust and Cancer Research UK. N.S. was supported by a Woolf-Fisher Trust (NZ) PhD studentship. H.Z. was supported by the Wolfson Centre at UCL, and the UCLH Dementia BRU provided financial support for the collection of patient materials. F.J.L. is a Wellcome Trust Senior Investigator, K.B. is a Torsten Söderberg Academy Professor, and H.Z. is a Wallenberg Academy Fellow.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information
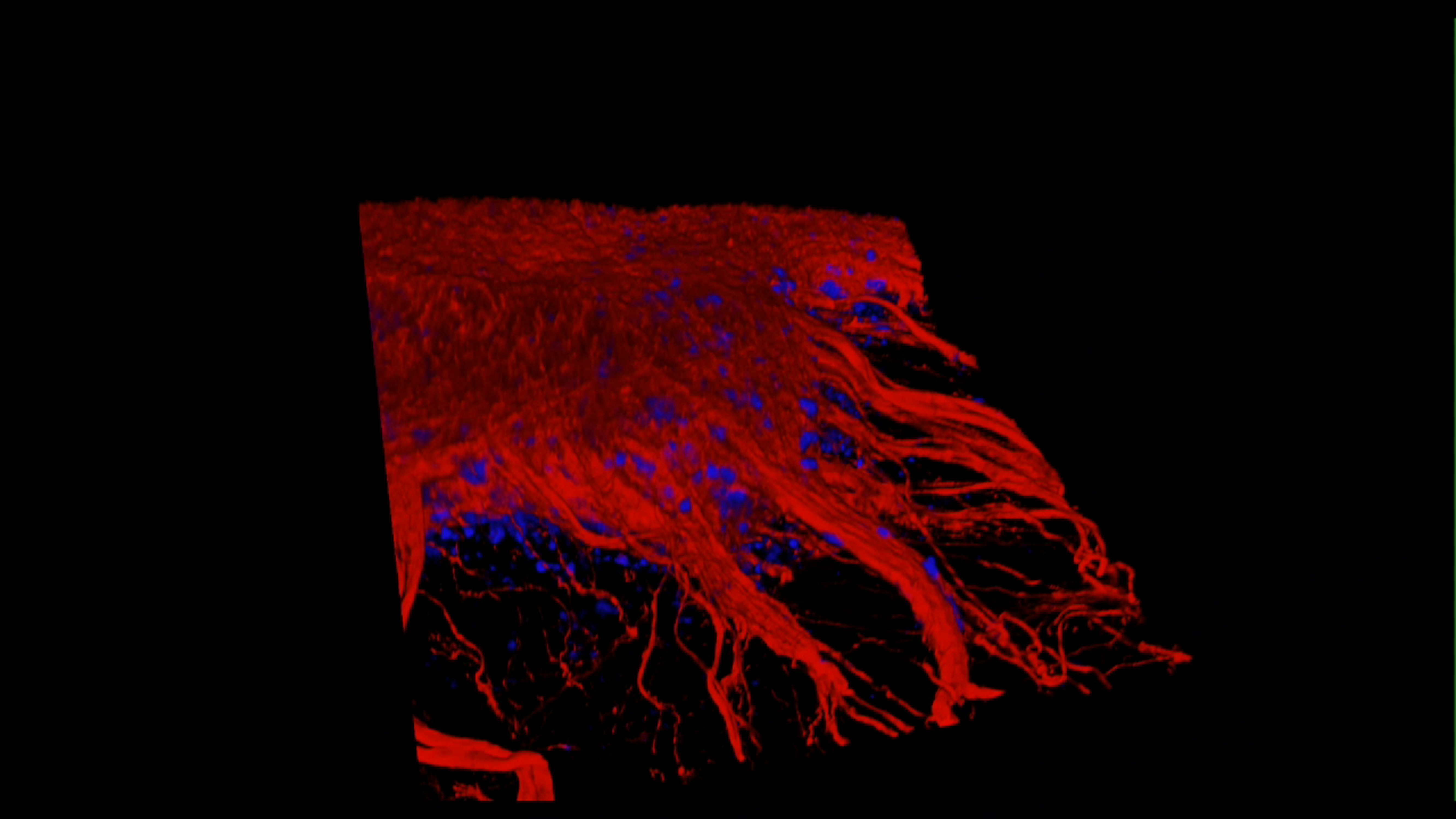
(A) 3D rendering of a control 1 neuronal culture at day 100 post-induction following tissue clearing and immunostaining for tau (red) and DNA (DAPI, blue). Removal of tau-positive neuronal processes reveals the cellular density achieved by this system after months in culture. Z = 145 μm.
(B) Serial Z sections of control 2 neurons at day 100 post-induction following tissue clearing and immunostaining for tau (red) and DNA (DAPI, blue). Z = 118 μm captured in 2 μm steps; scale bar represents 62 μm.
{kind=link}
Live imaging of neuronal cultures loaded with Oregon Green BAPTA to visualize calcium transients. Control 2 neurons (day 61 post-induction; A) demonstrate comparable network activity to PSEN1 intron 4 (day 64 post-induction; B), PSEN1 Y115C (day 55 post-induction; C), and APP V717I neurons (day 61 post-induction; D).
{kind=link}
References
- Benilova I., Karran E., De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Bertram L., Tanzi R.E. Thirty years of Alzheimer’s disease genetics: the implications of systematic meta-analyses. Nat. Rev. Neurosci. 2008;9:768–778. doi: 10.1038/nrn2494. [DOI] [PubMed] [Google Scholar]
- Blennow K., de Leon M.J., Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- Chávez-Gutiérrez L., Bammens L., Benilova I., Vandersteen A., Benurwar M., Borgers M., Lismont S., Zhou L., Van Cleynenbreugel S., Esselmann H. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012;31:2261–2274. doi: 10.1038/emboj.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.H., Kim Y.H., Hebisch M., Sliwinski C., Lee S., D’Avanzo C., Chen H., Hooli B., Asselin C., Muffat J. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature. 2014;515:274–278. doi: 10.1038/nature13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B. Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol. Rev. 2010;90:465–494. doi: 10.1152/physrev.00023.2009. [DOI] [PubMed] [Google Scholar]
- Devine M.J., Ryten M., Vodicka P., Thomson A.J., Burdon T., Houlden H., Cavaleri F., Nagano M., Drummond N.J., Taanman J.W. Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat. Commun. 2011;2:440. doi: 10.1038/ncomms1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner G.G., Wong C.W. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Glenner G.G., Wong C.W. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984;122:1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- Hardy J., Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- Hooli B.V., Kovacs-Vajna Z.M., Mullin K., Blumenthal M.A., Mattheisen M., Zhang C., Lange C., Mohapatra G., Bertram L., Tanzi R.E. Rare autosomal copy number variations in early-onset familial Alzheimer’s disease. Mol. Psychiatry. 2014;19:676–681. doi: 10.1038/mp.2013.77. [DOI] [PubMed] [Google Scholar]
- Israel M.A., Yuan S.H., Bardy C., Reyna S.M., Mu Y., Herrera C., Hefferan M.P., Van Gorp S., Nazor K.L., Boscolo F.S. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen I., Pardossi-Piquard R., Bauer C., Brigham E., Abraham J.-D., Ranaldi S., Fraser P., St-George-Hyslop P., Le Thuc O., Espin V. The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J. Neurosci. 2012;32:16243–16255a. doi: 10.1523/JNEUROSCI.2775-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters C.L., Simms G., Weinman N.A., Multhaup G., McDonald B.L., Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratore C.R., Rice H.C., Srikanth P., Callahan D.G., Shin T., Benjamin L.N.P., Walsh D.M., Selkoe D.J., Young-Pearse T.L. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum. Mol. Genet. 2014;23:3523–3536. doi: 10.1093/hmg/ddu064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I.H., Arora N., Huo H., Maherali N., Ahfeldt T., Shimamura A., Lensch M.W., Cowan C., Hochedlinger K., Daley G.Q. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimplikar S.W., Nixon R.A., Robakis N.K., Shen J., Tsai L.-H. Amyloid-independent mechanisms in Alzheimer’s disease pathogenesis. J. Neurosci. 2010;30:14946–14954. doi: 10.1523/JNEUROSCI.4305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portelius E., Tran A.J., Andreasson U., Persson R., Brinkmalm G., Zetterberg H., Blennow K., Westman-Brinkmalm A. Characterization of amyloid beta peptides in cerebrospinal fluid by an automated immunoprecipitation procedure followed by mass spectrometry. J. Proteome Res. 2007;6:4433–4439. doi: 10.1021/pr0703627. [DOI] [PubMed] [Google Scholar]
- Portelius E., Price E., Brinkmalm G., Stiteler M., Olsson M., Persson R., Westman-Brinkmalm A., Zetterberg H., Simon A.J., Blennow K. A novel pathway for amyloid precursor protein processing. Neurobiol. Aging. 2011;32:1090–1098. doi: 10.1016/j.neurobiolaging.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Rovelet-Lecrux A., Hannequin D., Raux G., Le Meur N., Laquerrière A., Vital A., Dumanchin C., Feuillette S., Brice A., Vercelletto M. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- Rovelet-Lecrux A., Hannequin D., Guillin O., Legallic S., Jurici S., Wallon D., Frebourg T., Campion D. Frontotemporal dementia phenotype associated with MAPT gene duplication. J. Alzheimers Dis. 2010;21:897–902. doi: 10.3233/JAD-2010-100441. [DOI] [PubMed] [Google Scholar]
- Shi Y., Kirwan P., Smith J., Maclean G., Orkin S.H., Livesey F.J. A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci. Transl. Med. 2012;4:124ra29. doi: 10.1126/scitranslmed.3003771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Kirwan P., Smith J., Robinson H.P., Livesey F.J. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci. 2012;15:477–486. doi: 10.1038/nn.3041. S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Kirwan P., Livesey F.J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 2012;7:1836–1846. doi: 10.1038/nprot.2012.116. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Wray S., Self M., Lewis P.A., Taanman J.W., Ryan N.S., Mahoney C.J., Liang Y., Devine M.J., Sheerin U.M., Houlden H., NINDS Parkinson’s Disease iPSC Consortium. NINDS Huntington’s Disease iPSC Consortium. NINDS ALS iPSC Consortium Creation of an open-access, mutation-defined fibroblast resource for neurological disease research. PLoS ONE. 2012;7:e43099. doi: 10.1371/journal.pone.0043099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T., Ito D., Okada Y., Akamatsu W., Nihei Y., Yoshizaki T., Yamanaka S., Okano H., Suzuki N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum. Mol. Genet. 2011;20:4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- Yang B., Treweek J.B., Kulkarni R.P., Deverman B.E., Chen C.-K., Lubeck E., Shah S., Cai L., Gradinaru V. Single-cell phenotyping within transparent intact tissue through whole-body clearing. Cell. 2014;158:945–958. doi: 10.1016/j.cell.2014.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankner B.A., Dawes L.R., Fisher S., Villa-Komaroff L., Oster-Granite M.L., Neve R.L. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science. 1989;245:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) 3D rendering of a control 1 neuronal culture at day 100 post-induction following tissue clearing and immunostaining for tau (red) and DNA (DAPI, blue). Removal of tau-positive neuronal processes reveals the cellular density achieved by this system after months in culture. Z = 145 μm.
(B) Serial Z sections of control 2 neurons at day 100 post-induction following tissue clearing and immunostaining for tau (red) and DNA (DAPI, blue). Z = 118 μm captured in 2 μm steps; scale bar represents 62 μm.
Live imaging of neuronal cultures loaded with Oregon Green BAPTA to visualize calcium transients. Control 2 neurons (day 61 post-induction; A) demonstrate comparable network activity to PSEN1 intron 4 (day 64 post-induction; B), PSEN1 Y115C (day 55 post-induction; C), and APP V717I neurons (day 61 post-induction; D).