

Abstract
Objective
The authors conducted the first genetic linkage study of families that segregate both autism and specific language impairment to find common communication impairment loci. The hypothesis was that these families have a high genetic loading for impairments in language ability, thus influencing the language and communication deficits of the family members with autism. Comprehensive behavioral phenotyping of the families also enabled linkage analysis of quantitative measures, including normal, subclinical and disordered variation in all family members for the three general autism symptom domains: social, communication, and compulsive behaviors.
Method
The primary linkage analysis coded persons with either autism or specific language impairment as “affected” with language impairment. The secondary linkage analysis consisted of quantitative metrics of autism-associated behaviors capturing normal to clinically severe variation, measured in all family members.
Results
Linkage to language phenotypes was established at two novel chromosomal loci, 15q23-26 and 16p12. The secondary analysis of normal and disordered quantitative variation in social and compulsive behaviors established linkage to two loci for social behaviors (at 14q and 15q) and one locus for repetitive behaviors (at 13q).
Conclusion
These data indicate shared etiology of autism and specific language impairment at two novel loci. Additionally, non-language phenotypes based on social aloofness and rigid personality traits showed compelling evidence for linkage in this sample. Further genetic mapping is warranted at these loci.
Keywords: genome scan, gene mapping, complex traits, genomewide association, linkage analysis
Introduction
Autism is a severe neurodevelopmental disorder characterized by altered functioning in three domains: 1) social interaction, 2) communication, and 3) stereotyped behavior and/or restricted interests and activities. Separately, there is a well-known classification in language disorders for children who have difficulties in acquiring language but are otherwise neurologically and psychologically normal, known as specific language impairment or SLI. Given that both disorders have a language component to their diagnosis, previous work has suggested that SLI and autism could have shared genetic contributors. This hypothesis is supported by a series of genetic mapping studies examining the relationship between autism and language impairment in complementary ways (1-12).
Autism linkage mapping studies have examined the relationship of language and autism with two paradigms. The first paradigm used language delay status of autism spectrum disorder (ASD) probands to stratify families into two groups, most often based on presence/absence of phrase speech by 36 months of age. Stratification on phrase speech delay postulates that there are subgroups of autism that can be genetically differentiated by language status. The language stratification paradigm has yielded autism findings on chromosomes 2q24-32 (5, 6), 7q22-32 (4) and 13q21-22 (4); this 13q region is also linked to SLIin non-ASD samples (2, 3, 7). At all three locations, stratifying ASD families on phrase speech delay in an a priori design nearly perfectly separated families that were linked to the given locus versus families that were not. Some loci were linked only in families including a person with autism and phrase speech delay and other loci were only linked in families without phrase speech delay. In contrast, a replication study used both stratification of families and determination of affection status in non-ASD parents based on self-report of language impairment history in childhood (8). This study found that the coding of parents as affected based on a history of language intervention or language delay minimally impacted the results, and while the group with phrase speech delay did have increased evidence of linkage, the chromosomes 2, 7 and 13 findings were not replicated.
The second paradigm used language phenotypes to directly map language quantitative trait loci or language impairment as a dichotomous trait within families with autism. Genomewide language quantitative trait locus mapping in ASD was performed using two items from the Autism Diagnostic Interview- Revised (ADI-R)(9), “age in months of onset of first word” and “age in months of onset of phrase speech”; both showed linkage to 7q36 (10, 11). CNTNAP2, a gene also associated with both SLI and normal language development, was later shown to be the most likely candidate responsible for this linkage (12), thus providing evidence that genetic variation relevant to both ASD and SLI can occur within the same gene.
This is the first linkage/association study to present a novel paradigm that complements both stratification and quantitative trait locus approaches, described above, in order to better understand the relevance of language variation in ASD. We selected families for the presence of both autism and SLI under the hypothesis that if autism and SLI are etiologically related then this sampling scheme will enrich our sample for loci related to language difficulties in autism and also reduce the genetic heterogeneity of ASD. Within these families, we collected state-of–the-art language phenotypes on relatives, and whenever possible, individuals with ASD. This study thus represents the most comprehensive family-based language phenotyping in a molecular genetic study of ASD.
We tested the hypothesis of genetic overlap by performing our analysis assuming that ASD and language impairments have the same underlying genetic etiology (i.e., we considered phenotypes from both disorders as equally affected) and then performed genome-wide linkage scans. Our analyses coded individuals with oral or written language impairments without ASD, as well as individuals with similar types of oral and written language impairments and ASD, as “affected.” Additionally, persons with ASD who could not be evaluated using quantitative language measures were incorporated into the analysis using a method for censored data (i.e., systematically missing data), which in this case assumed the censoring was due to low language ability (14) (see also Supplemental Methods). Our study design is most effective for mapping loci that are etiologically relevant to both ASD and language impairment. If ASD and language impairment are genetically unrelated in these families, then coding both disorders as “affected” in the same analysis will reduce evidence for linkage/association. Positive findings were formally tested to determine if autism or SLI or both jointly contributed to detected linkage signals.
The phenotypic battery also included seventeen quantitative population-normed language assessments and quantitative measures of social and compulsive behaviors. As a secondary objective, we performed linkage analysis using these quantitative data to capture both normal variation and clinically severe variation on the same scale, an analysis generally considered to be more powerful than analysis of only affected/unaffected. These analyses included the first use of the Yale-Brown Obsessive Compulsive Scale in a molecular genetic study of autism.
Methods
Overview of Design
Our primary goal was to find genetic variation relevant to both language impairments and ASD, using a set of previously described families recruited for the presence of persons with autism and separate individuals with SLI (15). To accomplish our goal, we created subgroups from 79 families (Table S1) according to phenotypic characteristics. We established three groups of families that we denoted Tier I-III. Tier I (N=46) consisted of families with both an autism proband and a different proband with SLI, as defined in this sample previously (15), or in a few cases, one autism proband and one ASD proband with low language often called “autism language impaired” in the literature (16, 17). Autism language impaired individuals are contrasted with ASD probands who are language normal (“autism language normal”), and ASD probands who are nonverbal ( “autism nonverbal”), with this last category often ignored in the literature since language cannot be quantitatively assessed.
Tier I had an internal contrast since any observed linkage could be further examined by excluding either the SLI proband or the autism proband to understand the connection between SLI and autism to the linkage signal on a locus-by-locus basis. The other two tiers included autism families that did not have an SLI or autism language impaired proband after direct testing. Tier II (N=15) consisted of multiplex ASD families without an SLI proband where neither ASD proband were operationally defined as language impaired (neither normal structural language deficits nor nonverbal). Tier III contained families (N=9) with an ASD proband and at least one relative who scored in the impaired range on either the Social Responsiveness Scale, a well-studied inventory of social functioning (18, 19), or the Yale-Brown Obsessive Compulsiveness Scale, used to evaluate OCD in psychiatric evaluations (20-23). For association analysis, an additional nine autism trios (N=9) were added to Tier III.
Prior to behavioral testing all subjects gave informed consent conforming to the guidelines for treatment of human subjects at Rutgers University. All family members as well as higher functioning family members with ASD received age appropriate measures of language and reading (Table S2). Descriptive statistics by diagnostic group, SLI, autism and other, have been previously published (15). Observed correlations between measures are in the Supplement (Table S3).
For the purpose of categorical phenotype linkage/association analysis, we define oral language impairment, called “LI” in our previous papers (2, 3, 7), as either an age appropriate Clinical Evaluation of Language Fundamentals – Fourth Edition (CELF-4) core standard score of <= 85, or at least 1 SD below peers on >= 60% of all oral language subtest scores and a significant history of language/reading difficulties, defined as >2 years of intervention and/or childhood diagnosis of language and/or reading impairment. For purpose of finding loci that jointly influence oral language impairment and autism, we define “LI*” as a phenotype that includes as affected persons affected with our definition of language impairment as well as persons affected with ASD (i.e., etiological equivalence).
In our previous studies of multiplex language impairment families, we observed many instances of semi-compensated adults with a childhood diagnosis of language problems and currently presenting with weak language skills who did not meet the cut-off for language impairment but did meet the cut-off for reading impairment (2, 3, 7). Based on our prior successful mapping of an SLI locus with that reading impairment phenotype (2, 3, 7), we defined written language (reading) impairment, called “RI” in our previous publications (2, 3, 7, 15), as >=1 SD below the population mean on 60% of all reading tests and subtests. For purpose of finding shared loci that jointly influence written language impairment and autism, we define “RI*” as a phenotype that includes as affected persons affected for our definition of reading impairment as well as persons affected with ASD (i.e., etiological equivalence). Throughout this paper, LI* and RI* refer to our specific diagnostic definitions of language impairment and/or autism and reading impairment and/or autism, respectively, while the term language impairments is meant in a more general sense to apply to oral and/or written language impairments, in context.
Genotyping
Affymetrix Axiom 1.0 arrays were used to generate 567,893 SNP genotypes on 440 individuals from the 79 families. Quality control on SNP genotypes was conducted as described previously (7), with additional details included in the Supplementary Methods, based on individual/SNP genotype completion, relationship checking, Mendelian errors and ancestry. A subset of 8086 SNPs was chosen for linkage analysis to minimize marker-to-marker LD and retain high minor allele frequency to provide suitable genomic coverage of recombination events in the pedigrees. Association analysis used all SNPs that met quality control standards and had a minor allele frequency > 0.05, yielding 529,874 SNPs. Validation genotyping was conducted on a Luminex 200 machine using a custom oligonucleotide ligation assay (24), with allele calling and quality control as described elsewhere (7, 25).
Statistical Analysis
Overall data analysis plan
We first conducted genomewide linkage scans with follow-up association analysis in the linkage regions. We also conducted a genomewide association analysis over the remainder of the genome. Given the depth of phenotyping, it was not considered reasonable to perform univariate analyses of all 21 cognitive measures on a genomewide basis due to the difficulty of interpreting results from analyses of many correlated traits. Instead, we opted for a mix of empirically and theoretically driven phenotypes. We used two categorical phenotypes, LI* for oral language impairment and RI* for written language (reading) impairment, the latter being a strong indicator of an unresolved oral language impairment in multiplex SLI pedigrees (reading deficits caused by an underlying language deficit) based on our previous studies (2, 3, 7). We also derived three quantitative traits using a factor analysis (see Supplemental Methods) to reduce the phenotypic data, which we called factors one through three (F1, F2, and F3; factor loadings in Table S4). To elucidate possible shared etiology in language phenotype linked regions, follow-up analyses were conducted to assess if ASD or language impairment or both were required to detect the linkage peak. The non-language traits were analyzed as quantitative traits, which included the Yale-Brown Obsessive Compulsiveness and the Social Responsiveness Scale (SRS-QT). The Social Responsiveness Scale was also analyzed as a dichotomous trait (SRS-DT) using a mild impairment threshold (see Supplement).
Linkage/association analysis methods
Linkage and association analyses were conducted with the Kelvin 2.3.3 package (http://kelvin.mathmed.org/;(26)). Kelvin implements the posterior probability of linkage (PPL) metric to measure the probability that a genetic location is linked with the trait of interest and the combined posterior probability of linkage disequilibrium (cPPLD) metric to measure the probability that a single nucleotide polymorphism (SNP) is in linkage disequilibrium (LD) with the trait of interest conditional upon the evidence for linkage at a given locus. It is important to note that the PPLD uses a pedigree likelihood that explicitly accounts for family structure while assessing the evidence that LD is present between the SNP and the disease (27, 28). For quantitative trait analysis where some ASD subjects lacked data due to inability to participate in some cognitive tests, the measures for those individuals were treated as censored data, meaning the “true scores” were unknown but known to be below a threshold. These analyses were handled using the PPL model for censored data (29) as described in the Supplemental Methods. The sex-averaged marker map for linkage was obtained from the Rutgers Combined Linkage-Physical Map of The Human Genome (30).
Primary linkage analysis of language phenotypes was conducted on each of the three tiers separately and the linkage evidence was sequentially updated across the three tiers to provide a single metric for linkage evidence. Follow-up family-based association analysis was conducted similarly; however, families that contained persons that were not of European ancestry (N=1 in Tier III) were dropped from the association analysis, since combining samples with different genetic ancestries can generate false positive results (25). For non-language phenotypes, all families were run as a single dataset.
Statistical Correction for Multiple Phenotypes
In order to assess the effect of performing multiple genome-scans using correlated phenotypic traits, we simulated 3000 genomes without regard to phenotypes to create an empirical null distribution for estimating p-values. We simulated chromosomes using the same SNP allele frequency and genetic distances as our linkage dataset. For each simulated genome, we conducted analysis with the five phenotypes, saving the overall maximum PPL per replicated genome. This list of maximum PPLs is the null distribution accounting for analysis with our five phenotypes, using our specific pedigree configuration and patterns of missing data etc. After correcting for multiple phenotypes, a PPL of 0.347 or greater retains a genome-wide error rate consistent with p<.001, a PPL of 0.269 corresponds to p<0.01, and a PPL of 0.104 to p<0.05. Note these threshold values are slightly higher than two previous studies of the false positive rate of the PPL (3, 31) due to correcting for multiple phenotypes in the present study.
Results
Initial Linkage Results for Language
We performed genomewide linkage analysis using two categorical definitions of language impairment (affected/unaffected status) and three quantitative language scores from a factor analysis of all 21 language measures, which we denote F1, F2 and F3 (see Supplement). The first trait, abbreviated LI*, defined individuals as affected if they possessed either an oral language impairment oran ASD. The second trait, abbreviated RI*, defined individuals as affected if they possessed either a written language impairment or an ASD. LI* produced clear evidence of linkage to chromosome 15 and RI* to chromosome 16, as shown in Figure 1 (with a summary of all large linkage peaks given in Table 1). The posterior probability of linkage (PPL) is scaled such that genomewide plots show very clear signal-to-noise ratios as seen in the figure. The magnitudes of both signals (> 35%) have Type I error rates appropriate for establishing linkage in a genome-wide scan, even after accounting for testing of multiple phenotypes (see Methods). Table 1 also includes the fully maximized LOD score (or MOD score) to allow comparison of the PPL to a statistic with a more commonly used scale; the estimated disease gene frequency and risk probabilities by genotype (penetrances) for the linkage peak are also included in the table including the estimate of the proportion of families linked to a given locus (α).
Figure 1.
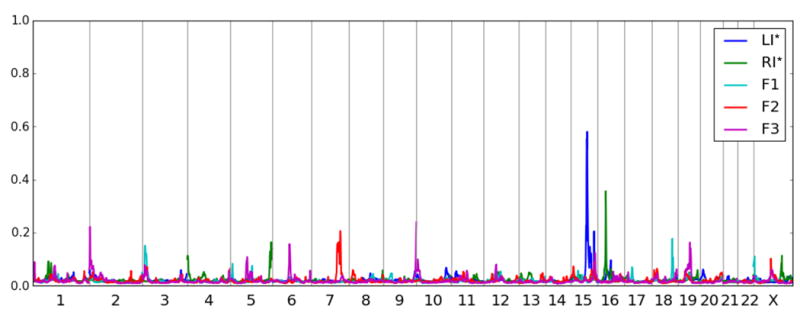
Genomewide linkage analysis of the 5 language-related traits. The posterior probability of linkage (PPL) is scaled such that PPL values < 2% represent evidence against linkage to that location while values > 2% represent evidence for linkage to that location. A PPL value of exactly 2% indicates that the data are not informative for linkage. The peaks on chromosomes 15 and 16, which represent oral language impairment and/or ASD (LI*) and written language (reading) impairment and/or ASD (RI*), respectively, clearly stand out from the rest of the genome, and overall the PPL displays a high signal to noise ratio for linkage mapping. While the three factor scores derived from 21 standardized measures of language (F1, F2 and F3) lack strong peaks, several regions of potential interest are identified.
Table 1. Linkage Peaks with PPL > 0.35 from All Analyses.
| ------Maximizing Model------ | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Phenotype | Chr | cM | PPL | Band range | Width (Mb) | LOD | -/- | +/- | +/+ | DGF | α |
| YBOCS | 13 | 58 | 0.36 | q14.3-21.3 | 17.3 | 4.2 | -3.0 | 0.0 | 3.0 | 0.300 | 0.7 |
| SRS-DT | 14 | 117 | 0.37 | q32.2-32.23 | 7.6 | 3.5 | 0.00 | 0.10 | 0.80 | 0.100 | 1.0 |
| LI* | 15 | 83 | 0.57 | q23-26.2 | 24.2 | 4.1 | 0.00 | 0.70 | 0.99 | 0.001 | 1.0 |
| SRS-QT | 15 | 120 | 0.52 | q26.2-26.3 | 6.2 | 4.5 | -2.0 | 1.0 | 2.0 | 0.100 | 0.9 |
| RI* | 16 | 43 | 0.36 | p12.1-12.3 | 8.9 | 4.6 | 0.00 | 0.00 | 0.90 | 0.200 | 1.0 |
PPL is the posterior probability of linkage. LOD is the fully maximized LOD score, sometimes referred to as a MOD score. -/-, +/-, and +/+ are the estimated genotypic effects for the locus; for categorical analysis these quantities are penetrances and for quantitative traits they are genotypic means on a z-score scale. DGF is the disease gene frequency and α is the heterogeneity parameter in the admixture likelihood at the maximizing model.
Phenotypes are: YOBCS=Yale-Brown Obsessive Compulsive Scale, SRS-DT=Social Responsiveness Scale- Dichotomous trait, LI*=Oral Language Impairment and/or ASD, SRS-QT=Social Responsiveness Scale-Quantitative Trait, RI*= Written Language (Reading) Impairment and/or ASD.
Chromosome 15q23-26.2 was linked to LI* with a maximum PPL of 57% and implicating a region of 24.1 Mb from 73 cM to 106 cM. As can be seen in Figure 2A, the linked region has a 15 cM high confidence linkage region with a much larger low confidence region (to the right) that accounts for about half of the total implicated region. Non-verbal IQ was not linked at this locus, indicating a dissociation of language and intelligence at this locus, as expected, since SLI does not include deficits in intelligence. We next wanted to asses show the level of language impairment in the ASD subjects (autism language impaired, autism language normal, and autism nonverbal) in each family modulates the linkage signal. We therefore defined a metric to quantify the relative contribution of the three language levels (autism with impaired language, autism with normal language, and autism nonverbal) , which indicated homogeneous contributions from all three groups to the linkage at this peak (see Assessing the relative contribution of the three proband types to the final PPL in the Supplement).
Figure 2.
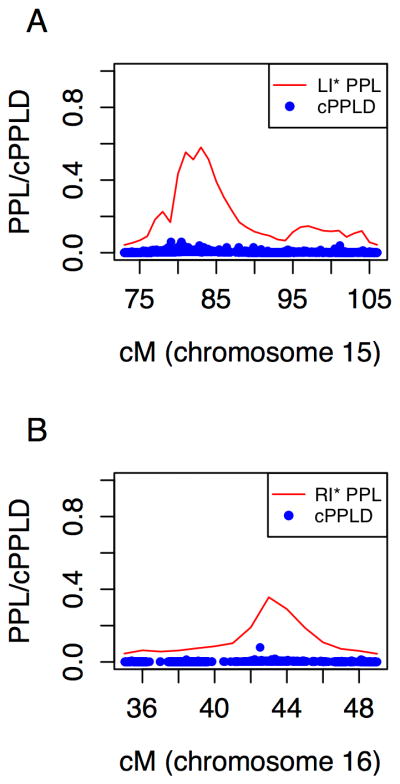
Follow-up association analysis ofSNPs under the language-related linkage peaks for oral language impairment and/or ASD, LI* (A) and written language (reading) impairment and/or ASD, RI* (B) phenotypes.
Family-based association analysis of all available SNPs with minor allele frequency > 0.05 in the linked region yielded only weak evidence of association, with a maximum combined posterior probability of linkage disequilibrium (cPPLD) of 6%. These results are not consistent with strong evidence for association that would account for the observed linkage. However, the Axiom array SNPs in the region successfully haplotype tag only 48% of the common variation (as described in the Supplemental Methods with results in Table S5); thus, follow-up of the region based on this SNP genotyping platform should be considered incomplete.
Chromosome 16p12.1-12.3 was linked to RI* over 8.9 Mb from 35 cM to 49 cM, with a maximum PPL of 36% (Figure 2B). All three ASD language levels contributed equally to the PPL (see Assessing the relative contribution of the three proband types to the final PPL in the Supplement). Similar to the chromosome 15 results, analysis of non-verbal IQ as a quantitative trait yielded evidence against linkage at this locus (PPL=1.7%, or below the prior probability of linkage, which is 2%), indicating a dissociation of language and intelligence at this locus as well. Follow-up cPPLD analysis to find SNPs that accounted for the linkage signal yielded a maximum cPPLD of 8% at a single SNP (next highest cPPLD=1.7%). Genotyped SNPs in this region only successfully tagged 55% of the common variation (Table S5).
Additional language-related peaks of interest (PPL > 20%) were observed on chromosome 7 with F2 (the second trait defined from the factor analysis of all 21 language tests) and chromosomes 2 and 9 with F3 (the third trait defined from our factor analysis). The linkage to chromosome 7 (PPL=21%), located over the region containing CNTNAP2, has been replicated in both ASD and SLI. This PPL may be considered appropriately large to replicate the CNTNAP2 locus.
Further Characterizing the Role of Language and Autism at Linked Loci
We sought to define the relative contribution of autism versus SLI to the linkage results on 15q and 16p, i.e., to assess if each disorder contributes equally to those linkage findings as an indication of the equivalence of the disorders. We restricted these analyses to Tier I since only this subset of families contains both autism and SLI probands in each pedigree. Each locus was assessed separately. We assessed the specificity of chromosome 15 for language versus autism by removing the autism proband from each pedigree and repeating the linkage analysis, then doing a separate and equivalent analysis removing the SLI proband while retaining the ASD proband. In both cases, the linkage signal was greatly reduced (PPLs dropped to 2% and 4%). Similarly, on chromosome 16 we removed the autism proband from each pedigree yielding a PPL of 3%, and removed one non-ASD RI* subject from the linkage analysis, giving a PPL of 6%. Therefore, both the 15q and 16p loci are sensitive to the presence of both autism and SLI. However, the reduction in PPL signals could have been the result of lower power from including fewer affected individuals in the analysis or due to loss of specific and relevant disease information. To rule out the former, we then used a permutation study to assess if removing language-impaired and/or ASD subjects from the analysis induced a greater average drop in the PPL than removing subjects randomly (see also Assessing the relative contributions of language impairment and ASD within families to the final PPL in the Supplement). For chromosome 15, the permutation test was significant for an effect of SLI and ASD having more dramatic effects on the PPL than other combinations (p<.01). This was not the case for chromosome 16, where the test was not significant, indicating that low power cannot be ruled out as a confound when interpreting the contribution of reading impairment and autism to this linkage peak.
To test whether our phenotypic definitions of LI* and RI* were too restricted, we also repeated the linkage analysis on chromosomes 15 and 16 using a combined phenotypic definition of both oral and written language impairment, where persons were defined as affected if they were either LI* or RI*(or both). For both chromosomes, the PPL was attenuated to less than half the original linkage signal (dropped to 19% and 12%) and the linkage region was greatly broadened (data not shown).
Linkage Analysis of Non-Language Phenotypes
All families were ascertained only for language phenotypes in the non-ASD individuals, not for any non-language characteristics of the broad autism phenotype. However, we quantitatively assessed social responsiveness and aspects of obsessive compulsiveness in all pedigree members able to participate in an assessment and used the phenotypes for linkage analysis. Results are summarized in Figure 3. We analyzed the Yale–Brown Obsessive Compulsive Scale (YBOCS) as a quantitative trait yielding a 36% PPL on 13q14.3-21.33 (55cM to 63 cM) with the peak directly over PCDH20, close to previous linkage studies of ASD and studies of SLI (2-4). The linked region is 17.3 Mb in size. Analysis of all available SNPs with minor allele frequency >0.05 did not yield any cPPLD results over 2% (Figure 4A). Two smaller peaks of interest occurred on chromosomes 8 (PPL = 22%) and 21 (PPL=22%) with no cPPLDs under those peaks greater than 3%.
Figure 3.
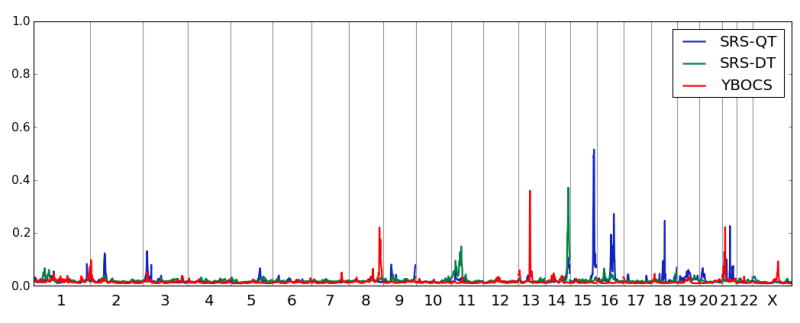
Genomewide linkage analysis of the 3 non-language-related traits. The largest signals with each of the three traits are on chromosomes 13, 14 and 15. No overlap was observed between the social assessment scale, Social Responsiveness Scale (SRS) for both quantitative (SRS-QT) and dichotomous (SRS-DT) trait analyses.
Figure 4.
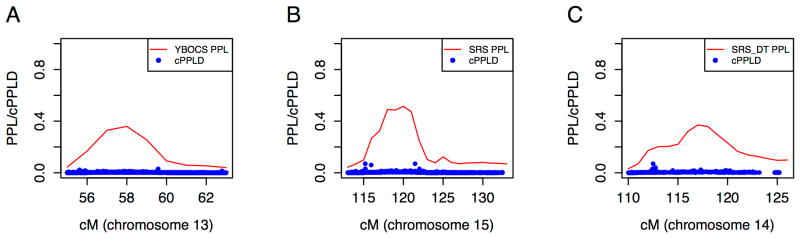
Follow-up analysis of non-language-related linkage peaks. No combined posterior probability of linkage disequilibrium (cPPLD) values were observed that could account for the linkage signals with the Yale-Brown Obsessive Compulsive Scale (Y-BOCS) (A) or the Social Responsiveness Scale (SRS) as a quantitative trait (B) or dichotomous trait (C).
Our analysis of the Social Responsiveness Scale was conducted two ways. The first was as a quantitative trait (SRS-QT), using the numerical scores generated by the test. This yielded a linkage peak PPL of 52% on chromosome 15q26.2-26.3 (113cM to 133 cM) that does not overlap with the LI* peak described above. We observed three SNPs spanning 3 Mb (rs12440787, rs7170868, rs9672677) with cPPLD=7% under this peak, but none are in linkage disequilibrium with each other and so they do not represent a single coherent signal. In addition, we observed two smaller peaks on chromosomes 16 (PPL=27%) and 18 (PPL=25%). The second was using the “mild” impairment threshold (see supplement) to dichotomize the Social Responsiveness Scale for analysis (SRS-DT). We chose to create this simple mild impairment v. unimpaired distinction since this distinction is commonly used in the broader autism phenotype literature. We saw the peak PPL of 37% on chromosome 14q32.2-32.33 (110-126cM) encompassing 7.7 Mb. No cPPLD from available SNPs in these regions was greater than 2%.
Genomewide Association Analysis
Linkage analysis only requires a limited number of SNPs to attain essentially full information and thus utilized only 8,086 of the 529,874 SNPs that passed quality control filtering. We conducted association analyses on the 529,874 genomewide SNPs, yielding 19 SNPs with PPLD > 10% (see Table S6 SNP names). We then performed follow-up genotyping, using a different platform, of these SNPs or SNPs in strong LD (r2 > 0.95) for validation, and cPPLD analysis using both linkage information from the families as well as LD with the trait. The highest cPPLD of 20% was located at rs3792495 for the F2 trait.
Discussion
We identified two linkage peaks for language impairment in families with both language impairment and autism (15q25.1 and 16p12.3) that do not overlap with previously discovered autism or language impairment loci. The two linkage signals showed specificity for oral language impairments for 15q and for written language impairment for 16p. This specificity was evidenced by attenuation of the linkage signals when a combined oral/written language impairment phenotype was applied suggesting there is a subset of individuals with reading problems who do not have comorbid oral language deficits. Additionally, there was no evidence that either locus is primarily related to SLI alone or autism alone, rather, it appears each locus is jointly related to both SLI and ASD. These findings are in keeping with the goal of the study to find genetic variation that is relevant to both disorders. This is the first molecular genetic study of families that segregate both SLI and autism, and we hypothesize that these families have a high genetic loading for impairments in language ability, further influencing the language and communication deficits of the autism probands. In our previous study (17), we also observed that the average scores on a standardized test of the social use of language (pragmatics) were similar in the SLI and ASD subjects in these families. Pragmatic impairment is not part of the defined deficits in SLI but is commonly seen in ASD, suggesting that the two disorders, as presenting in the families selected by our ascertainment criteria and recruitment methods, may be on an etiological continuum.
There were also several compelling linkage peaks for non-language traits even though the sample ascertainment scheme did not include any requirements of non-language traits beyond the autism proband. The Social Responsiveness Scale peak on 15q26.3 was the second largest in the study (PPL=52%) and had the narrowest linkage region (6.2 Mb). This region was implicated in a meta-analysis of ASD and schizophrenia (32). An additional Social Responsiveness Scale peak was noted when using a mild cut-off to create a categorical affection status (14q32; PPL=37%)., The 14q32 region has been associated with autism through cytogenetic abnormalities and copy number variation (33, 34). While social skills and communication are fundamentally related, it is unclear from our data if ascertainment for performance language assessments increased power to detect social behavior (35-38) due to that relationship. A conservative interpretation is that the deep phenotyping performed here was simply more likely to find multiple strong effects across phenotypic domains relative to other studies with less phenotypic data. The Social Responsiveness Scale is a good quantitative metric for mapping autism loci as it has yielded strong findings in other studies (39, 40). However, this was the first gene mapping study to also use a mild cut-off with the Social Responsiveness Scale, which is more analogous to the broader autism phenotype literature where affected/unaffected distinctions are commonly applied. We also believe this is the first use of the YBOCS as a quantitative trait in genome wide analysis of ASD. The empirical performance appears quite good based on our data, suggesting that wider use of this measure may be warranted in ASD research, especially in family genetic studies where specific behaviors may be apparent in some family members but not severe enough to impair activities of daily-living. Our specific finding on 13q does not coincide with OCD studies but does align with previous autism genetic studies (4, 41, 42).
While the linkage analysis showed several strong peaks with different language and non-language related traits, there were no strong association signals either under the peaks or across the remaining genome. As mentioned in the results section, the regions under the linkage peaks are not adequately tagged for comprehensive association analysis by the Axiom 1.0 array, as is the case for much of the genome, thus greatly decreasing the chance of observing associations. The modest sample size also limits power to detect common variants of small effect. Additionally, recent evidence suggests autism has significant allelic heterogeneity, and association analyses have been generally not replicated except for variants with very small effects (43). Rare variant studies indicate widespread heterogeneity (44-47), though it remains unclear what proportions of genetic mechanisms for ASD involve rare, infrequent or common variants.
The issue of allelic heterogeneity across disorders is still uncharacterized from both theoretical and empirical points of view but quite relevant to the debate on genetic overlap between disorders. Until reasonably inferred functional alleles are associated in each disorder, it is not possible to directly address the issue of whether the same genetic variants within the same gene are relevant to both SLI and ASD (12, 48). Hence it is possible that SLI and ASD have some of the same key genes in pathogenesis but not the same underlying variants or molecular mechanisms. However, if functional variants are the same for both disorders, then it remains to be explained why some members of the family develop SLI and not ASD. It could be that individuals manifesting SLI have a smaller genetic load for such variants, or that SLI is a truly dissociable sub-component in at least some forms of ASD. We will continue susceptibility allele mapping in our autism-SLI pedigrees to disentangle these complicated mechanisms.
Supplementary Material
Acknowledgments
Most sincere thanks go out to all our study families who have taken time away from their very busy lives to help us try to answer the many questions about the genetic basis of autism. We also thank Jonathan Gray, Laszlo Szabo and Julie Griffith for their earlier work on the NJLAGS project, Joshua Pennino for database management, and all the language, autism, and cognitive specialists for their many hours of testing and Dr. John Constantino for a helpful discussion. We appreciate the expertise of Dr. Kapila Seshadri for her role in the identification and assessment of probands and the Interactive Autism network (IAN) for their help in family recruitment. This project was supported by the National Institute of Mental Health grants R01MH070366 and RC1MH088288 to LMB, and by the support and services provided by the NIMH Center for Collaborative Genomic Studies on Mental Disorders, funded by U24 MH068457. This work was also supported in part by an allocation of computing time from the Ohio Supercomputer Center Grant PCCR0001-2 to CWB.
References
- 1.Warburton P, Baird G, Chen W, Morris K, Jacobs BW, Hodgson S, Docherty Z. Support for linkage of autism and specific language impairment to 7q3 from two chromosome rearrangements involving band 7q31. American journal of medical genetics. 2000;96(2):228–34. doi: 10.1002/(sici)1096-8628(20000403)96:2<228::aid-ajmg20>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 2.Bartlett CW, Flax JF, Logue MW, Smith BJ, Vieland VJ, Tallal P, Brzustowicz LM. Examination of potential overlap in autism and language loci on chromosomes 2, 7, and 13 in two independent samples ascertained for specific language impairment. Hum Hered. 2004;57(1):10–20. doi: 10.1159/000077385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartlett CW, Flax JF, Logue MW, Vieland VJ, Bassett AS, Tallal P, Brzustowicz LM. A major susceptibility locus for specific language impairment is located on 13q21. Am J Hum Genet. 2002;71(1):45–55. doi: 10.1086/341095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradford Y, Haines J, Hutcheson H, Gardiner M, Braun T, Sheffield V, Cassavant T, Huang W, Wang K, Vieland V, Folstein S, Santangelo S, Piven J. Incorporating language phenotypes strengthens evidence of linkage to autism. American journal of medical genetics. 2001;105(6):539–47. [PubMed] [Google Scholar]
- 5.Buxbaum JD, Silverman JM, Smith CJ, Kilifarski M, Reichert J, Hollander E, Lawlor BA, Fitzgerald M, Greenberg DA, Davis KL. Evidence for a susceptibility gene for autism on chromosome 2 and for genetic heterogeneity. Am J Hum Genet. 2001;68(6):1514–20. doi: 10.1086/320588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shao Y, Raiford KL, Wolpert CM, Cope HA, Ravan SA, Ashley-Koch AA, Abramson RK, Wright HH, DeLong RG, Gilbert JR, Cuccaro ML, Pericak-Vance MA. Phenotypic homogeneity provides increased support for linkage on chromosome 2 in autistic disorder. Am J Hum Genet. 2002;70(4):1058–61. doi: 10.1086/339765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simmons TR, Flax JF, Azaro MA, Hayter JE, Justice LM, Petrill SA, Bassett AS, Tallal P, Brzustowicz LM, Bartlett CW. Increasing genotype-phenotype model determinism: application to bivariate reading/language traits and epistatic interactions in language-impaired families. Hum Hered. 2010;70(4):232–44. doi: 10.1159/000320367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spence SJ, Cantor RM, Chung L, Kim S, Geschwind DH, Alarcon M. Stratification based on language-related endophenotypes in autism: attempt to replicate reported linkage. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of. 2006;141B(6):591–8. doi: 10.1002/ajmg.b.30329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24(5):659–85. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- 10.Alarcon M, Yonan AL, Gilliam TC, Cantor RM, Geschwind DH. Quantitative genome scan and Ordered-Subsets Analysis of autism endophenotypes support language QTLs. Molecular psychiatry. 2005;10(8):747–57. doi: 10.1038/sj.mp.4001666. [DOI] [PubMed] [Google Scholar]
- 11.Alarcon M, Cantor RM, Liu J, Gilliam TC, Geschwind DH. Evidence for a language quantitative trait locus on chromosome 7q in multiplex autism families. Am J Hum Genet. 2002;70(1):60–71. doi: 10.1086/338241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alarcon M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, Sebat J, Wigler M, Martin CL, Ledbetter DH, Nelson SF, Cantor RM, Geschwind DH. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008;82(1):150–9. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomblin B. Co-morbidity of autism and SLI: kinds, kin and complexity. Int J Lang Commun Disord. 2011;46(2):127–37. doi: 10.1111/j.1460-6984.2011.00017.x. [DOI] [PubMed] [Google Scholar]
- 14.Hou L, Wang K, Bartlett CW. Evaluation of a bayesian model integration-based method for censored data. Hum Hered. 2012;74(1):1–11. doi: 10.1159/000342707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bartlett CW, Flax JF, Fermano Z, Hare A, Hou L, Petrill SA, Buyske S, Brzustowicz LM. Gene x gene interaction in shared etiology of autism and specific language impairment. Biological psychiatry. 2012;72(8):692–9. doi: 10.1016/j.biopsych.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Fosse L, Hodge SM, Makris N, Kennedy DN, Caviness VS, Jr, McGrath L, Steele S, Ziegler DA, Herbert MR, Frazier JA, Tager-Flusberg H, Harris GJ. Language-association cortex asymmetry in autism and specific language impairment. Annals of neurology. 2004;56(6):757–66. doi: 10.1002/ana.20275. [DOI] [PubMed] [Google Scholar]
- 17.Lindgren KA, Folstein SE, Tomblin JB, Tager-Flusberg H. Language and reading abilities of children with autism spectrum disorders and specific language impairment and their first-degree relatives. Autism research : official journal of the International Society for Autism Research. 2009;2(1):22–38. doi: 10.1002/aur.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Constantino JN, Todd RD. Intergenerational transmission of subthreshold autistic traits in the general population. Biological psychiatry. 2005;57(6):655–60. doi: 10.1016/j.biopsych.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 19.Constantino JN, Davis SA, Todd RD, Schindler MK, Gross MM, Brophy SL, Metzger LM, Shoushtari CS, Splinter R, Reich W. Validation of a brief quantitative measure of autistic traits: comparison of the social responsiveness scale with the autism diagnostic interview-revised. J Autism Dev Disord. 2003;33(4):427–33. doi: 10.1023/a:1025014929212. [DOI] [PubMed] [Google Scholar]
- 20.Goodman WK, Price LH, Rasmussen SA, Mazure C, Fleischmann RL, Hill CL, Heninger GR, Charney DS. The Yale-Brown Obsessive Compulsive Scale. I. Development, use, and reliability. Archives of general psychiatry. 1989;46(11):1006–11. doi: 10.1001/archpsyc.1989.01810110048007. [DOI] [PubMed] [Google Scholar]
- 21.Goodman WK, Price LH, Rasmussen SA, Mazure C, Delgado P, Heninger GR, Charney DS. The Yale-Brown Obsessive Compulsive Scale. II. Validity. Archives of general psychiatry. 1989;46(11):1012–6. doi: 10.1001/archpsyc.1989.01810110054008. [DOI] [PubMed] [Google Scholar]
- 22.Scahill L, Riddle MA, McSwiggin-Hardin M, Ort SI, King RA, Goodman WK, Cicchetti D, Leckman JF. Children's Yale-Brown Obsessive Compulsive Scale: reliability and validity. Journal of the American Academy of Child and Adolescent Psychiatry. 1997;36(6):844–52. doi: 10.1097/00004583-199706000-00023. [DOI] [PubMed] [Google Scholar]
- 23.Scahill L, McDougle CJ, Williams SK, Dimitropoulos A, Aman MG, McCracken JT, Tierney E, Arnold LE, Cronin P, Grados M, Ghuman J, Koenig K, Lam KS, McGough J, Posey DJ, Ritz L, Swiezy NB, Vitiello B. Children's Yale-Brown Obsessive Compulsive Scale modified for pervasive developmental disorders. Journal of the American Academy of Child and Adolescent Psychiatry. 2006;45(9):1114–23. doi: 10.1097/01.chi.0000220854.79144.e7. [DOI] [PubMed] [Google Scholar]
- 24.Bruse S, Moreau M, Azaro M, Zimmerman R, Brzustowicz L. Improvements to bead-based oligonucleotide ligation SNP genotyping assays. BioTechniques. 2008;45(5):559–71. doi: 10.2144/000112960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou L, Phillips C, Azaro M, Brzustowicz LM, Bartlett CW. Validation of a cost-efficient multi-purpose SNP panel for disease based research. PloS one. 2011;6(5):e19699. doi: 10.1371/journal.pone.0019699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vieland VJ, Huang Y, Seok SC, Burian J, Catalyurek U, O'Connell J, Segre A, Valentine-Cooper W. KELVIN: a software package for rigorous measurement of statistical evidence in human genetics. Hum Hered. 2011;72(4):276–88. doi: 10.1159/000330634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Y, Vieland VJ. Association statistics under the PPL framework. Genetic epidemiology. 2010;34(8):835–45. doi: 10.1002/gepi.20537. [DOI] [PubMed] [Google Scholar]
- 28.Yang X, Huang J, Logue MW, Vieland VJ. The posterior probability of linkage allowing for linkage disequilibrium and a new estimate of disequilibrium between a trait and a marker. Hum Hered. 2005;59(4):210–9. doi: 10.1159/000086699. [DOI] [PubMed] [Google Scholar]
- 29.Hou L, Wang K, Bartlett CW. Evaluation of a Bayesian Model-Integration-Based Method for Censored Data. Hum Hered. doi: 10.1159/000342707. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matise TC, Chen F, Chen W, De La Vega FM, Hansen M, He C, Hyland FC, Kennedy GC, Kong X, Murray SS, Ziegle JS, Stewart WC, Buyske S. A second-generation combined linkage physical map of the human genome. Genome research. 2007;17(12):1783–6. doi: 10.1101/gr.7156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Logue MW, Vieland VJ, Goedken RJ, Crowe RR. Bayesian analysis of a previously published genome screen for panic disorder reveals new and compelling evidence for linkage to chromosome 7. American journal of medical genetics Part B, Neuropsychiatric genetics : the official publication of. 2003;121B(1):95–9. doi: 10.1002/ajmg.b.20072. [DOI] [PubMed] [Google Scholar]
- 32.Chagnon YC. Shared susceptibility region on chromosome 15 between autism and catatonia. Int Rev Neurobiol. 2006;72:165–78. doi: 10.1016/S0074-7742(05)72010-9. [DOI] [PubMed] [Google Scholar]
- 33.Merritt JL, 2nd, Jalal SM, Barbaresi WJ, Babovic-Vuksanovic D. 14q32.3 deletion syndrome with autism. Am J Med Genet A. 2005;133A(1):99–100. doi: 10.1002/ajmg.a.30462. [DOI] [PubMed] [Google Scholar]
- 34.Qiao Y, Tyson C, Hrynchak M, Lopez-Rangel E, Hildebrand J, Martell S, Fawcett C, Kasmara L, Calli K, Harvard C, Liu X, Holden J, Lewis S, Rajcan-Separovic E. Clinical application of 2.7M Cytogenetics array for CNV detection in subjects with idiopathic autism and/or intellectual disability. Clin Genet. 2013;83(2):145–54. doi: 10.1111/j.1399-0004.2012.01860.x. [DOI] [PubMed] [Google Scholar]
- 35.Bolte S, Poustka F, Constantino JN. Assessing autistic traits: cross-cultural validation of the social responsiveness scale (SRS) Autism research : official journal of the International Society for Autism Research. 2008;1(6):354–63. doi: 10.1002/aur.49. [DOI] [PubMed] [Google Scholar]
- 36.Constantino JN, Gruber CP, Davis S, Hayes S, Passanante N, Przybeck T. The factor structure of autistic traits. Journal of child psychology and psychiatry, and allied disciplines. 2004;45(4):719–26. doi: 10.1111/j.1469-7610.2004.00266.x. [DOI] [PubMed] [Google Scholar]
- 37.Kamio Y, Inada N, Moriwaki A, Kuroda M, Koyama T, Tsujii H, Kawakubo Y, Kuwabara H, Tsuchiya KJ, Uno Y, Constantino JN. Quantitative autistic traits ascertained in a national survey of 22 529 Japanese schoolchildren. Acta psychiatrica Scandinavica. 2012 doi: 10.1111/acps.12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wigham S, McConachie H, Tandos J, Le Couteur AS. The reliability and validity of the Social Responsiveness Scale in a UK general child population. Research in developmental disabilities. 2012;33(3):944–50. doi: 10.1016/j.ridd.2011.12.017. [DOI] [PubMed] [Google Scholar]
- 39.Coon H, Villalobos ME, Robison RJ, Camp NJ, Cannon DS, Allen-Brady K, Miller JS, McMahon WM. Genome-wide linkage using the Social Responsiveness Scale in Utah autism pedigrees. Molecular autism. 2010;1(1):8. doi: 10.1186/2040-2392-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duvall JA, Lu A, Cantor RM, Todd RD, Constantino JN, Geschwind DH. A quantitative trait locus analysis of social responsiveness in multiplex autism families. The American journal of psychiatry. 2007;164(4):656–62. doi: 10.1176/ajp.2007.164.4.656. [DOI] [PubMed] [Google Scholar]
- 41.Barrett S, Beck JC, Bernier R, Bisson E, Braun TA, Casavant TL, Childress D, Folstein SE, Garcia M, Gardiner MB, Gilman S, Haines JL, Hopkins K, Landa R, Meyer NH, Mullane JA, Nishimura DY, Palmer P, Piven J, Purdy J, Santangelo SL, Searby C, Sheffield V, Singleton J, Slager S, et al. An autosomal genomic screen for autism. Collaborative linkage study of autism. American journal of medical genetics. 1999;88(6):609–15. doi: 10.1002/(sici)1096-8628(19991215)88:6<609::aid-ajmg7>3.3.co;2-c. [DOI] [PubMed] [Google Scholar]
- 42.Steele MM, Al-Adeimi M, Siu VM, Fan YS. Brief report: A case of autism with interstitial deletion of chromosome 13. J Autism Dev Disord. 2001;31(2):231–4. doi: 10.1023/a:1010759401344. [DOI] [PubMed] [Google Scholar]
- 43.Anney R, Klei L, Pinto D, Almeida J, Bacchelli E, Baird G, Bolshakova N, Bolte S, Bolton PF, Bourgeron T, Brennan S, Brian J, Casey J, Conroy J, Correia C, Corsello C, Crawford EL, de Jonge M, Delorme R, Duketis E, Duque F, Estes A, Farrar P, Fernandez BA, Folstein SE, Fombonne E, Gilbert J, Gillberg C, Glessner JT, Green A, Green J, Guter SJ, Heron EA, Holt R, Howe JL, Hughes G, Hus V, Igliozzi R, Jacob S, Kenny GP, Kim C, Kolevzon A, Kustanovich V, Lajonchere CM, Lamb JA, Law-Smith M, Leboyer M, Le Couteur A, Leventhal BL, Liu XQ, Lombard F, Lord C, Lotspeich L, Lund SC, Magalhaes TR, Mantoulan C, McDougle CJ, Melhem NM, Merikangas A, Minshew NJ, Mirza GK, Munson J, Noakes C, Nygren G, Papanikolaou K, Pagnamenta AT, Parrini B, Paton T, Pickles A, Posey DJ, Poustka F, Ragoussis J, Regan R, Roberts W, Roeder K, Roge B, Rutter ML, Schlitt S, Shah N, Sheffield VC, Soorya L, Sousa I, Stoppioni V, Sykes N, Tancredi R, Thompson AP, Thomson S, Tryfon A, Tsiantis J, Van Engeland H, Vincent JB, Volkmar F, Vorstman J, Wallace S, Wing K, Wittemeyer K, Wood S, Zurawiecki D, Zwaigenbaum L, Bailey AJ, Battaglia A, Cantor RM, Coon H, Cuccaro ML, Dawson G, Ennis S, Freitag CM, Geschwind DH, Haines JL, Klauck SM, McMahon WM, Maestrini E, Miller J, Monaco AP, Nelson SF, Nurnberger JI, Jr, Oliveira G, Parr JR, Pericak-Vance MA, Piven J, Schellenberg GD, Scherer SW, Vicente AM, Wassink TH, Wijsman EM, Betancur C, Buxbaum JD, Cook EH, Gallagher L, Gill M, Hallmayer J, Paterson AD, Sutcliffe JS, Szatmari P, Vieland VJ, Hakonarson H, Devlin B. Individual common variants exert weak effects on the risk for autism spectrum disorderspi. Human molecular genetics. 2012 doi: 10.1093/hmg/dds301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, Kendall J, Grabowska E, Ma B, Marks S, Rodgers L, Stepansky A, Troge J, Andrews P, Bekritsky M, Pradhan K, Ghiban E, Kramer M, Parla J, Demeter R, Fulton LL, Fulton RS, Magrini VJ, Ye K, Darnell JC, Darnell RB, Mardis ER, Wilson RK, Schatz MC, McCombie WR, Wigler M. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74(2):285–99. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, Cai G, Lihm J, Dannenfelser R, Jabado O, Peralta Z, Nagaswamy U, Muzny D, Reid JG, Newsham I, Wu Y, Lewis L, Han Y, Voight BF, Lim E, Rossin E, Kirby A, Flannick J, Fromer M, Shakir K, Fennell T, Garimella K, Banks E, Poplin R, Gabriel S, DePristo M, Wimbish JR, Boone BE, Levy SE, Betancur C, Sunyaev S, Boerwinkle E, Buxbaum JD, Cook EH, Jr, Devlin B, Gibbs RA, Roeder K, Schellenberg GD, Sutcliffe JS, Daly MJ. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242–5. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485(7397):246–50. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, Meyer KA, Bilguvar K, Mane SM, Sestan N, Lifton RP, Gunel M, Roeder K, Geschwind DH, Devlin B, State MW. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485(7397):237–41. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chapman NH, Estes A, Munson J, Bernier R, Webb SJ, Rothstein JH, Minshew NJ, Dawson G, Schellenberg GD, Wijsman EM. Genome-scan for IQ discrepancy in autism: evidence for loci on chromosomes 10 and 16. Human genetics. 2011;129(1):59–70. doi: 10.1007/s00439-010-0899-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.